

Abstract
There are very few antiviral drugs available to fight viral infections and the appearance of viral strains resistant to these antivirals is not a rare event. Hence, the design of new antiviral drugs is important. We describe the prediction of peptides with antiviral activity (AVP) derived from the viral glycoproteins involved in the entrance of herpes simplex (HSV) and influenza A viruses into their host cells. It is known, that during this event viral glycoproteins suffer several conformational changes due to protein-protein interactions, which lead to membrane fusion between the viral envelope and the cellular membrane. Our hypothesis is that AVPs can be derived from these viral glycoproteins, specifically from regions highly conserved in amino acid sequences, which at the same time have the physicochemical properties of being highly exposed (antigenic), hydrophilic, flexible, and charged, since these properties are important for protein-protein interactions. For that, we separately analyzed the HSV glycoprotein H and B, and influenza A viruses hemagglutinin (HA), using several bioinformatics tools. A set of multiple alignments was carried out, to find the most conserved regions in the amino acid sequences. Then, the physicochemical properties indicated above were analyzed. We predicted several peptides 12-20 amino acid length which by docking analysis were able to interact with the fusion viral glycoproteins and thus may prevent conformational changes in them, blocking the viral infection. Our strategy to design AVPs seems to be very promising since the peptides were synthetized and their antiviral activities have produced very encouraging results.
Keywords: Herpes simplex viruses, influenza A viruses, antiviral peptides (AVPs), gB, gH, HA, proteins
Background
Viral infections constitute a very important issue for human and animal health and there are very few available antiviral drugs to fight these infections. Even more, emerging and re-emerging viruses have further emphasized the efforts to accelerate antiviral drug discovery. The design of peptides to study the fusion process of the human immunodeficiency virus type 1 (HIV-1) corresponding to both heptad-repeat regions HR-1 and -2 of the HIV-1 gp41, demonstrated that this type of peptides were able to block viral entry and lead to the development of the first antiviral peptide (AVP) the Fuzeon®, approved by the FDA for treatment of HIV-1 infected patients [1]. This AVP approach has been extended to other viruses such as, SARSCoV, HRSV, HPIV-3, APV, NDV, SV-5, HMPV, Measles virus [2, 3].
Currently available drugs against HSV infections are nucleotide analogues, which block viral DNA replication; and even though antiviral resistant strains are rare in immunocompetent patients (0.1-0.7%), they are very common (7-14%) in immunocompromised hosts. Furthermore, drug-resistant HSV are becoming a threat especially for the immunocompromised population which has been increasing around the world, either due to viral infections such as AIDS or immunosuppression related to cancer, organ transplants, and chronic diseases treatments [4]. For influenza A virus infections, the only effective antivirals nowadays are the neuraminidase inhibitors zanamivir and oseltamivir, but in recent years the emergence of influenza A virus strains resistant to this type of inhibitors has been demonstrated [5].
These findings support the need to develop new antiviral drugs using other viral targets. The viral glycoproteins involved in the entry event to the cell are an attractive target, because they are essential proteins for viral penetration and require conformational changes mediated by protein-protein interactions to carry out their function. HSV entry requires several viral glycoproteins (gD, gH/gL, gB) and begins with the binding of gD to specific cell receptors. Receptor binding triggers displacement of the C-terminus of gD to expose a previously hidden region of gD, which has been proposed interacts with gH/gL. This interaction results in a conformational change in gH/gL that enables it to promote the fusogenic state of gB. Insertion of the gB fusion loops into the cellular membrane and an interaction between the ectodomains of gB and gH/gL results in fusion of the viral envelope with the cell membrane and releasing of the viral nucleocapsid into the cytoplasm of the host cell [6].
Influenza virus HA, is a homotrimeric protein that controls two critical aspects of viral entry: virus binding and membrane fusion. To carry out these functions, HA must undergo a proteolytic cleavage, to generate two distinct subunits (HA1 and HA2), which renders it fusion competent. The HA trimeric structure comprises a large membrane-distal, globular domain formed by HA1 that binds to glycan receptors on host cells, and an elongated membrane-proximal domain (stem region) dominated by intertwined and interconnecting α-helices. The stem region is the main component of the HA fusion machinery and is comprised of HA2 (F fusion subdomain) and the N- and C-terminal segments of HA1 (F´ fusion subdomain). The connecting loop between the antiparallel helices of HA2, the B loop, has a high propensity for a helical conformation and this loop-to-helix transition enables extension of the central coiledcoil and facilitates relocation of the fusion peptide toward the target membrane. Membrane fusion begins inside the endosome after a drop in lumenal pH leading to the release of influenza virus nucleocapsids into the cellular cytoplasm [7].
Despite the immense potential of this field, there is no virus specific AVP prediction algorithm [8]. Our hypothesis is that the viral glycoproteins required for the entry of enveloped viruses by a fusion mechanism, should have conserved amino acid sequences in their functional regions. Due to the nature of the membrane fusion process, we propose that the functional regions in these viral glycoproteins should be accessible (surface exposed), hydrophilic, flexible, and have an overall charge, since these properties have been found to be important for the protein-protein interactions, necessary for the activation of the fusion event. Therefore these regions could be used for AVPs design, and furthermore, we hypothesized that by analyzing different strains belonging to the same species, the derived AVPs should have antiviral activity against the different strains of the viral species analyzed. For AVPs design using bioinformatics tools we select the HSV viral glycoproteins gB and gH, and the HA of influenza A viruses.
Methodology
Protein sequences. The viral amino acid sequences for HSV-1 and HSV-2 gB, and gH proteins were downloaded from the GenBank NCBI data base (http://www.ncbi.nlm.nih.gov/); and for influenza A virus hemagglutinin (HA), the Influenza virus resource was used (http://www.ncbi.nlm.nih.gov/genomes/FLU/).
Multiple protein alignments:
The proteins gB, gH and HA (HA1 and HA2 subdomains) were separately analyzed, using a series of multiple alignments to search for conserved sequences. We started with ClustalX2 (http://www.clustal.org/), then, T-Coffee Expresso, which aligns protein sequences using structural information (http://tcoffee.crg.cat/apps/tcoffee/index.html), and PRALINE, which is a multiple sequence alignment program that integrates homology-extended and secondary structure (www.ibi.vu.nl/programs/pralinewww/). The results were evaluated by sum of pairs, minimum entropy, Coffee (Core and IRMSD-APDB).
Consensus sequence:
With the results of the multiple alignments a consensus amino acid sequence was obtained for each protein using WebLogo (http://weblogo.berkeley.edu/) or GeneDoc (http://www.nrbsc.org/gfx/genedoc/).
Physicochemical analysis:
Several physicochemical parameters were evaluated for each consensus sequence using a 10-20 amino acid window. 1) Hydrophobicity using ProtScale (http://web.expasy.org/protscale/) with the Kyte-Doolitle scale [9]. 2) Hydrophilicity using MPEx with the Wimley and White scale (http://blanco.biomol.uci.edu/mpex/) [10]. 3) Antigenicity using Antibody Epitope Prediction (http://tools.immuneepitope.org/tools/ElliPro/iedb_input) with Parker scale [11]. 4). Flexibility, with PROFbval (http://www.predictprotein.org); Average Flexibility using ProtScale available at ExPasy (http://web.expasy.org/protscale/). 5) Overall charge, with Charge EMBOSS (http://emboss.sourceforge.net/apps/cvs/emboss/apps/charge.html).
Peptide selection and structure prediction:
The regions of the protein that showed to be highly conserved in amino acid sequences (a minimum of 80% of conserved amino acids) and also, hydrophilic, exposed (antigenic), flexible and charged, were chosen for designing the AVPs and their 3D structures were predicted by using PEPFOLD (http://bioserv.rpbs.univ-paris-diderot.fr/PEP-FOLD/) [12].
Docking analysis:
To determine probable interactions among the 3D peptide structures and the viral glycoproteins involved in the fusion event, a docking analysis was carried out using ClusPro 2.0: protein-protein docking (http://cluspro.bu.edu/login.php) [13]. For the docking analysis with the influenza A AVPs we used as a target the HA PDB (3LZG). For the HSV AVPs we used HSV-1 or HSV-2 gB, gD and gH proteins available at PDB; and the missing proteins were modeled using the Modeller program [14].
The ClusPro server gave four different choices for docking results. We used the balanced coefficients considering that we did not know what forces dominate in our complex. Thus, of the generated models we selected those with the lowest ΔG (energetically favorable) and lowest cluster size complex (the most frequent interaction between receptor and ligand at the same site).
Discussion
Fusion between the viral envelope and the cellular membrane is a complicated and elegant process used by enveloped viruses to penetrate into their host cell. Fusion can be executed by one or more viral glycoproteins and different cellular receptors, which interact and undergo a series of conformational changes, before releasing the viral capsids into the cellular cytoplasm. Since this process is one of the initial steps of infection, it is an ideal target for the design of novel antivirals.
Several approaches have been used to design or identify AVPs capable of blocking virus entry into the cell. One strategy is based on peptides derived from the HR of viral glycoproteins involved in the fusion event for example, the Fuzeon® (enfurvitide). It is known that HIV-1 predominantly infects T cells carrying the CD4 antigen by binding the viral envelope glycoprotein gp120 to the CD4 cellular receptor. After this initial attachment, a conformational change in gp120 allows its further association with the cellular co-receptors CCR5 or CXCR4. Subsequently, a conformational change in the viral glycoprotein gp41 allows the HR2 domain to fold back on itself to interact with the HR1 domain. This process (known as gp41 zipping) leads to fusion of the viral and host-cell membranes. The enfuvirtide inhibits the fusion event by preventing the gp41 zipping due to its association with gp41 [1].
Other approach to find AVPs has been the screening of phage display libraries to identify peptides capable of inhibiting virus infectivity when they bind to viral particles. AVPs for several viruses such as West Nile virus [15], Hantavirus [16], or even influenza A virus [17] have been found using this type of libraries.
Our strategy to design AVPs derived from the viral membrane glycoproteins of HSV and influenza A viruses, is based on the search for highly conserved regions in the viral fusion proteins. For HSV, we used the gB and gH proteins which are known to be involved in HSV membrane fusion process [18]. For influenza A, we chose the HA which is known to be the viral fusogen. By a series of multiple alignments we identified the conserved regions among the 16 subtypes of HA of influenza A viruses since we hypothesized that the important functional regions of the HA should be conserved among the different influenza A subtypes. The same was done with the gB and gH proteins of HSV-1 and -2 strains. Therefore, we assumed that the derived AVPs should have antiviral activity against influenza A viruses, independently of their HA subtype; and in the case of HSV, the AVPs should be effective against HSV-1 as well as HSV-2.
For designing the AVPs we took only in account the regions of the protein whose amino acids were at least 80% conserved in the consensus sequence. Then, we performed an in silico physicochemical analysis looking for surface exposed, hydrophilic, flexible, and charged properties in the conserved regions of the protein, since these properties have been found to be important for protein-protein interactions. We chose probably exposed regions according to, hydrophilicity values lower than -1 of the Kyte & Doolitle scale (values <0 indicate hydrophilicity and the maximum value is -2.5). This property was corroborated using MPEX with the Wimley & White scale considering only values lower than -10 (which means hydrophilicity, the maximum value is -20). Since the hydrophilic regions in a protein are known to correlate with the antigenic sites identified by several immunological studies, we also used the Parker scale for the prediction of antibody epitopes and we selected those regions with values >4 (values >1 mean hydrophilicity). The flexibility was determined using PROFBval and only those regions with values >0.6 were considered (minimum value of flexibility is 0.4). We also looked for an overall charge in these regions. For each analyzed protein we detected several regions with a maximum length of 12-20 amino acid residues sharing all these properties. For HSV gB protein we found a total of 4 peptides corresponding to positions 126-145, 224-243, 413-432 and 467-486 of gB. They are different to other gB derived peptides previously reported [19]. In the case of HSV gH protein, 5 peptides corresponding to the positions 362-379, 569-586, 609-626, 646-664, and 706-723 were found. Figure 1 shows some of the in silico results for HSV gH protein.
Figure 1.
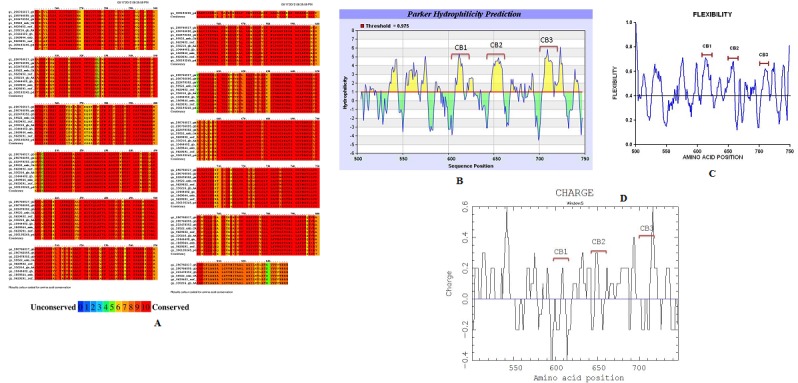
Multiple alignments for HSV gH protein and physicochemical parameters evaluated for the gH consensus sequence. A) Conserved amino acid residues identified with PRALINE; B) Parker hydrophilicity prediction determined with Antibody Epitope Prediction (the hydrophilic regions are indicated above the horizontal line); C) Flexibility determined with PROFbval (values >0.4 indicated flexibility), and D) overall charge determined with the CHARGE program. CB-1, CB-2 and CB-3 indicate the regions we have chosen to design the AVPs.
For influenza A HA, we found 6 peptides in the subunit HA1 corresponding to the position 10-24, 44-59, 26-40, 279-296, 299- 310, 270-285, and 3 peptides in the subunit HA2, located at the positions 410-421, 489-510, 517-537. Table 1 shows a summary of the interactions we found with the influenza A AVPs derived from the subunit HA1.
A similar approach was reported to design AVPs for SARSCoV, derived from the S protein which blocked the binding of S protein to the cellular receptor. These peptides were designed based on hydrophilicity, surface probability and chain flexibility of the S protein [1]. Other strategies, such as a Monte Carlo based computational method have been proposed to identify and optimize potential self-inhibitory peptides derived from viral envelope proteins, which inhibit the protein-protein interactions between viral an cellular proteins during the membrane fusion step [20]. But despite immense potential of this field, there is no virus specific AVP prediction algorithm, and even more, Thakur et al., [8] have collected the information of 1245 AVPs which were experimentally checked for their antiviral activity against different important viruses, and by analyzing amino acid composition and several physicochemical properties such as: secondary structure, overall charge, size, residue composition, hydrophobicity and amphiphilic character, have proposed the first web server for predicting effective AVPs (http://crdd.osdd.net/servers/avppred). However, it is necessary to gather more information about AVP derived from the viral fusion proteins to feed the AVP web server to develop an accurate predictive algorithm for AVPs, and our strategy seems to be a promising procedure to design AVPs. It is worth mentioning that the peptides we predicted have been synthesized and their antiviral activity has being tested and the results are very encouraging (manuscript in preparation).
Conclusion
We have successfully produced new AVPs 12-20 amino acid length derived from the fusion proteins of HSV gB and gH, and influenza A HA, which were designed using a strategy based on the bioinformatics analysis of those proteins, looking for highly conserved amino acids regions with properties of hydrophilicity, exposed surface, flexibility and charge.
Supplementary material
Acknowledgments
We thank CONACyT-SEP 99164, ICyTDF 2009/225: and Secretaría de Investigación y Posgrado, Instituto Politécnico Nacional for the financial support to carry out this work.
Footnotes
Citation:Jesús et al, Bioinformation 8(18): 870-874 (2012)
References
- 1.T Matthews, et al. Nat Rev Drug Discov. 2004;3:215. doi: 10.1038/nrd1331. [DOI] [PubMed] [Google Scholar]
- 2.T Ho, et al. Antiviral Res. 2006;69:70. doi: 10.1016/j.antiviral.2005.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.S Miller, et al. J Virol. 2007;81:141. [Google Scholar]
- 4.PD Griffiths, et al. J Clin Virol. 2009;46:3. [Google Scholar]
- 5.P Cheng, et al. Emerg Infect Dis. 2010;16:155. doi: 10.3201/eid1601.091304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.R Eisenberg, et al. Viruses. 2012;4:800E. [Google Scholar]
- 7.B Hamilton, et al. Viruses. 2012;4:1144. [Google Scholar]
- 8.N Thakur, et al. Nucleic Acids Res. 2012;40:W199. [Google Scholar]
- 9.J Kyte, RF Doolitle. J Mol Biol. 1982;157:105. doi: 10.1016/0022-2836(82)90515-0. [DOI] [PubMed] [Google Scholar]
- 10.C Snider, et al. Protein Sci. 2009;18:2624. doi: 10.1002/pro.256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.JM Parker, et al. Biochemistry. 1986;25:5425. doi: 10.1021/bi00367a013. [DOI] [PubMed] [Google Scholar]
- 12.P Thévenet, et al. Nucleic Acids Res. 2012;40:W288. [Google Scholar]
- 13.D Kozakov, et al. Proteins. 2010;78:3124. doi: 10.1002/prot.22835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.MA Marti-Renom, et al. Annu Rev Biophys Biomol Struct. 2000;29:291. doi: 10.1146/annurev.biophys.29.1.291. [DOI] [PubMed] [Google Scholar]
- 15.F Bai, et al. J Virol. 2007;81:2047. doi: 10.1128/JVI.01840-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.R Larson, et al. J Virol. 2005;79:7319. doi: 10.1128/JVI.79.12.7319-7326.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.J Jones, et al. J Virol. 2006;80:11960. doi: 10.1128/JVI.01678-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.S Connolly, et al. Nat Rev Microbiol. 2011;9:369. doi: 10.1038/nrmicro2548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.WF Walker, GE Trahey. J Acoust Soc Am. 2009;53:987. [Google Scholar]
- 20.Y Xu, et al. Proteins. 2012;80:2154. doi: 10.1002/prot.24105. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.