

The crystal structures of the far-red fluorescent proteins eqFP650 and eqFP670 have been solved at 1.8 and 1.6 Å resolution, respectively. This permitted identification of the structural elements responsible for the bathochromic shift in both considered far-red fluorescent proteins.
Keywords: far-red fluorescent proteins, cell imaging, tissue visualization, Katushka
Abstract
The crystal structures of the far-red fluorescent proteins (FPs) eqFP650 (λex max/λem max 592/650 nm) and eqFP670 (λex max/λem max 605/670 nm), the successors of the far-red FP Katushka (λex max/λem max 588/635 nm), have been determined at 1.8 and 1.6 Å resolution, respectively. An examination of the structures demonstrated that there are two groups of changes responsible for the bathochromic shift of excitation/emission bands of these proteins relative to their predecessor. The first group of changes resulted in an increase of hydrophilicity at the acylimine site of the chromophore due to the presence of one and three water molecules in eqFP650 and eqFP670, respectively. These water molecules provide connection of the chromophore with the protein scaffold via hydrogen bonds causing an ∼15 nm bathochromic shift of the eqFP650 and eqFP670 emission bands. The second group of changes observed in eqFP670 arises from substitution of both Ser143 and Ser158 by asparagines. Asn143 and Asn158 of eqFP670 are hydrogen bonded with each other, as well as with the protein scaffold and with the p-hydroxyphenyl group of the chromophore, resulting in an additional ∼20 nm bathochromic shift of the eqFP670 emission band as compared to eqFP650. The role of the observed structural changes was verified by mutagenesis.
1. Introduction
Far-red fluorescent proteins (FPs) are in high demand for whole-body imaging because of the intrinsic optical properties of living tissues. Such tissues are almost opaque when illuminated with wavelengths shorter than ~600 nm owing to the light-absorbing properties of hemoglobin and melanin and, on the other hand, are not transparent to wavelengths longer than ~1100 nm owing to the inevitable presence of water. Thus, the optical window (also called the near-infrared region) permitting a deep light penetration in the body is limited to the range of 650–900 nm (König, 2000 ▶).
Up to now, a number of far-red FPs approaching the 650 nm barrier have been generated from various fluorescent and chromoproteins by the directed evolution approach (Table 1 ▶). The first generation of recombinant far-red FPs included HcRed (Gurskaya et al., 2001 ▶), mPlum (Wang et al., 2004 ▶) and AQ143 (Shkrob et al., 2005 ▶) engineered from Heteractis crispa chromoprotein hcCP (Lukyanov et al., 2000 ▶), a monomeric DsRed-like protein mRFP1.2 (Campbell et al., 2002 ▶) and Actinia equina chromoprotein aeCP597 (Shkrob et al., 2005 ▶) (Table 1 ▶). These proteins were successfully demonstrated to be useful for numerous imaging applications (Chudakov et al., 2005 ▶; Hoffman, 2005 ▶; Hoffman & Yang, 2005 ▶; Shcherbo et al., 2007 ▶); however, all of them have relatively low brightness [about tenfold lower than that of the enhanced green fluorescent protein (EGFP) (Yang et al., 1996 ▶)], limiting their suitability for deep-tissue imaging.
Table 1. Evolution of far-red fluorescent proteins.
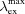 /
/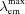 , excitation and emission maxima. E
mol, extinction coefficient. ΦF, quantum yield. B, brightness (E
mol × ΦF)/1000.
, excitation and emission maxima. E
mol, extinction coefficient. ΦF, quantum yield. B, brightness (E
mol × ΦF)/1000. 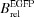 , brightness relative to EGFP [the product of ΦF and E
mol compared to the brightness of EGFP (53 000 M
−1 cm−1 × 0.60) (Patterson et al., 1997 ▶)].
, brightness relative to EGFP [the product of ΦF and E
mol compared to the brightness of EGFP (53 000 M
−1 cm−1 × 0.60) (Patterson et al., 1997 ▶)].
| FP variant |
/ (nm) |
E mol (M −1 cm−1) | ΦF | B |
|
Reference |
|---|---|---|---|---|---|---|
| EGFP† | 489/509 | 53000 | 0.60 | 31.8 | 1.00 | Patterson et al. (1997 ▶) |
| WT progenitors | ||||||
| DsRed | 558/583 | 75000 | 0.70 | 52.5 | 1.65 | Yarbrough et al. (2001 ▶) |
| eqFP578 | 552/578 | 102000 | 0.54 | 55.1 | 1.73 | Merzlyak et al. (2007 ▶) |
| eqFP611 | 559/611 | 78000 | 0.45 | 35.1 | 1.10 | Wiedenmann et al. (2002 ▶) |
| First generation | ||||||
| HcRed | 594/649 | 70000 | 0.05 | 3.5 | 0.11 | Gurskaya et al. (2001 ▶) |
| mPlum | 590/649 | 41000 | 0.10 | 4.1 | 0.13 | Wang et al. (2004 ▶) |
| AQ143 | 595/655 | 90000 | 0.04 | 3.6 | 0.11 | Shkrob et al. (2005 ▶) |
| Second generation | ||||||
| Katushka | 588/635 | 65000 | 0.34 | 22.1 | 0.69 | Shcherbo et al. (2007 ▶) |
| mKate | 588/635 | 45000 | 0.33 | 14.9 | 0.49 | Shcherbo et al. (2007 ▶) |
| RFP637 | 587/637 | 72000 | 0.23 | 16.5 | 0.52 | Kredel et al. (2008 ▶) |
| RFP639 | 588/639 | 69000 | 0.18 | 12.4 | 0.39 | Kredel et al. (2008 ▶) |
| Third generation | ||||||
| mKate2 | 588/633 | 62500 | 0.40 | 25.0 | 0.79 | Shcherbo et al. (2009 ▶) |
| Neptune | 600/650 | 72000 | 0.18 | 13.0 | 0.41 | Lin et al. (2009 ▶) |
| mNeptune | 600/650 | 67000 | 0.20 | 13.4 | 0.42 | Lin et al. (2009 ▶) |
| E2-Crimson | 611/646 | 126000 | 0.23 | 29.0 | 0.91 | Strack et al. (2009 ▶) |
| TagRFP657 | 611/657 | 34000 | 0.10 | 3.4 | 0.11 | Morozova et al. (2010 ▶) |
| eqFP650 | 592/650 | 65000 | 0.24 | 15.6 | 0.49 | Shcherbo et al. (2010 ▶) |
| eqFP670 | 605/670 | 70000 | 0.06 | 4.2 | 0.13 | Shcherbo et al. (2010 ▶) |
The values for EGFP are given for comparison.
The second generation of far-red FPs was substantially brighter [with a brightness only half of that of EGFP (Yang et al., 1996 ▶)], but their emission maxima at best reached 640 nm (Table 1 ▶). Typical members of this group are the dimeric far-red FP Katushka (Shcherbo et al., 2007 ▶), its monomeric version mKate (Shcherbo et al., 2007 ▶) and RFP639 (Kredel et al., 2008 ▶). Both Katushka and mKate were generated from Entacmaea quadricolor red fluorescent protein eqFP578 (Merzlyak et al., 2007 ▶), whereas RFP639 was derived from eqFP578-related protein eqFP611 (Wiedenmann et al., 2002 ▶). The major drawback of the second generation of far-red FPs is their hypsochromically (blue) shifted excitation band, impairing their efficient excitation in living tissues.
The third generation of far-red FPs aptly combines the advantages of both generations of the previously created far-red FPs. Their excitation and emission maxima demonstrate bathochromic (red) shift as compared to the first generation, whereas their brightness is usually higher than that of the second generation (Table 1 ▶). Among these proteins are four successors of mKate: mKate2 (Shcherbo et al., 2009 ▶), Neptune (Lin et al., 2009 ▶), mNeptune (Lin et al., 2009 ▶) and TagRFP657 (Morozova et al., 2010 ▶); one derivative of DsRed-Express2, E2-Crimson (Strack et al., 2009 ▶); as well as two Katushka variants, eqFP650 and eqFP670 (Shcherbo et al., 2010 ▶). The structural properties of the latter two FPs are described here.
Crystallographic data available for wild-type red FPs and their far-red successors indicate that all these proteins have chemically identical chromophores. Therefore, a bathochromic shift of up to 90 nm of their excitation and emission bands is caused solely by variation of the environment nearest to the chromophore. The most common reasons for red shift of the excitation maxima in red FPs are: trans-to-cis isomerization of the chromophore; chromophore deprotonation; the appearance of additional π–π stacking interactions between the chromophore and the protein matrix; and the formation of new hydrogen bonds. For example, eqFP578 (Pletneva et al., 2011 ▶) and eqFP611 (Petersen et al., 2003 ▶) contain the trans form of the chromophore, whereas their far-red successors Katushka (Pletneva et al., 2011 ▶) and RFP639 (Nienhaus et al., 2008 ▶) contain the cis form. In both cases, trans–cis isomerization of the chromophore provided by variation of its closest environment resulted in over 25 nm bathochromic shifts of the emission maxima.
The Ser197Tyr substitution in E2-Crimson, which presumably introduced additional π–π stacking interactions between Tyr197 and the chromophore, was reported to be one of the key mutations responsible for red shift of the fluorescence bands in this protein (Strack et al., 2009 ▶). The crystal structure of Neptune revealed the presence of a new water-mediated hydrogen bond between the acylimine oxygen of the chromophore and the hydroxyl of Ser28, corresponding to a new way of red shifting the emission maxima of FPs (Lin et al., 2009 ▶). Other ways of red shifting the emission bands of FPs are also possible.
To explore further the reasons for bathochromic shift in FPs we have examined the crystal structures of two recently generated far-red fluorescent proteins, eqFP650 and eqFP670. These structures were determined at 1.8 and 1.6 Å resolution, respectively, providing detailed views of the environment of the chromophores. With the help of structural data and the results of mutagenesis we discuss the cause of the 15 nm bathochromic shift observed for eqFP650 and the 35 nm red shift observed for eqFP670.
2. Materials and methods
2.1. Protein expression, purification and spectroscopic characterization
For protein expression, PCR-amplified BamHI/HindIII fragments encoding target proteins with N-terminal His-tags have been cloned into pQE30 vector (Qiagen) and transformed into E. coli XL1 Blue strain (Invitrogen Corp.). Bacterial cultures were grown overnight at 310 K and additionally incubated for 12 h at 298 K to achieve complete protein maturation. No IPTG (isopropyl β-d-1-thiogalactopyranoside) induction was necessary since the promoter leakage was sufficient for effective expression. The cells were pelleted by centrifugation, re-suspended in phosphate buffer saline (PBS) and lysed by sonication. The proteins were purified by immobilized metal affinity chromatography (IMAC) using TALON resin (Clontech Laboratories, Inc.) followed by size exclusion chromatography (SEC) using Superdex 200 (16/60) column (Amersham Biosciences). For IMAC, supernatants clarified by centrifugation were loaded on the column pre-washed with 10–20 column volumes of PBS and the proteins were eluted with PBS + 100 mM EDTA solution. For SEC, protein fractions recovered from IMAC were transferred to 20 mM Tris–HCl, 200 mM NaCl, 5 mM EDTA buffer pH 8.0, concentrated to 2 ml, and passed through the column using the same buffer as eluent.
Absorbance and fluorescence spectra were measured using a Cary 500 spectrophotometer (Varian Inc.) and a FluoroMax-4 spectrofluorometer (Horiba Jobin Yvon Inc.), respectively.
2.2. Crystallization
For crystallization, the proteins were transferred to a buffer containing 10 mM Tris–HCl, 100 mM NaCl, 2.5 mM EDTA pH 8.0 and were concentrated to 25 mg ml−1. An initial search for crystallization conditions was carried out using a Mosquito Robotic Crystallization System (TTP LabTech Ltd). To ensure the proper fluorescent state of the proteins, only neutral pH hits were used for further optimization. Large-scale crystallization was set up by the hanging-drop method at room temperature. The best crystals of both eqFP650 and eqFP670 were obtained from 2.5 M sodium acetate, 0.1 M bis-Tris propane pH 7.0.
2.3. Diffraction data collection and processing
X-ray diffraction data were collected at the Advanced Photon Source on SER-CAT 22-ID beamline (Argonne National Laboratory, Argonne, IL, USA). Diffraction intensities were registered on a MAR 300 CCD detector (Rayonix). Prior to data collection the crystals were incubated in a cryoprotecting solution consisting of 20% glycerol and 80% of well solution for 1 min and were flash-frozen in a 100 K nitrogen stream. The cryogenic temperature was maintained by a CryoJetXL cooling device (Oxford Cryosystems). Diffraction images were indexed, integrated and scaled with the HKL2000 software (Otwinowski & Minor, 1997 ▶). Data processing statistics are given in Table 2 ▶.
Table 2. Data collection statistics.
Values in parentheses are for the outermost resolution shells.
| Protein | eqFP650 | eqFP670 |
|---|---|---|
| Space group | P6122 | P6122 |
| Unit-cell parameters (Å) | a,b = 104.5, c = 214.6 | a,b = 104.6, c = 214.9 |
| Temperature (K) | 100 | 100 |
| Wavelength (Å) | 1.00 | 1.00 |
| Resolution (Å) | 30.0–1.8 (1.86–1.80) | 30.0–1.6 (1.66 –1.60) |
| Total reflections | 941107 | 1889671 |
| Unique reflections | 64734 | 92328 |
| Completeness (%) | 99.9 (100.0) | 100.0 (100.0) |
| 〈I/σ(I)〉 | 45.2 (5.5) | 52.2 (4.4) |
| R merge | 0.056 (0.524) | 0.059 (0.515) |
| Multiplicity | 14.5 (14.7) | 20.5 (12.4) |
2.4. Structure solution and refinement
The structures of eqFP650 and eqFP670 were solved by the molecular replacement method with MOLREP (Vagin & Teplyakov, 1997 ▶) using as a search model a single monomer of Katushka (PDB ID: 3pj5; Pletneva et al., 2011 ▶), excluding its chromophore. Structure refinement was performed with REFMAC (Murshudov et al., 1997 ▶), COOT (Emsley & Cowtan, 2004 ▶) and PHENIX.REFINE (Adams et al., 2002 ▶). Manual structure rebuilding and addition of ordered solvent molecules were done using COOT. Structure validation was performed with COOT and PROCHECK (Laskowski et al., 1993 ▶), and the refinement statistics are given in Table 3 ▶. The coordinates and structure factors were deposited in the Protein Data Bank under accession codes 4edo and 4eds for eqFP650 and eqFP670, respectively.
Table 3. Refinement statistics.
| Protein | eqFP650 | eqFP670 |
|---|---|---|
| No. of protein atoms | 3671 | 3951 |
| No. of solvent atoms | 280 | 360 |
| Resolution range (Å) | 30.0–1.8 | 30.0–1.6 |
| R work | 0.189 | 0.179 |
| R free | 0.211 | 0.197 |
| R.m.s. deviation bond lengths (Å) | 0.014 | 0.013 |
| R.m.s. deviation angles (°) | 1.64 | 1.58 |
| R.m.s. deviation chirality (Å3) | 0.12 | 0.11 |
| R.m.s. deviation planarity (Å) | 0.007 | 0.007 |
| R.m.s. deviation dihedral (°) | 17.1 | 17.6 |
| Mean B factors (Å2) for protein atoms | ||
| Overall | 32.8 | 24.0 |
| Main chain | 30.8 | 22.4 |
| Side chain | 34.7 | 25.6 |
| Ramachandran statistics (%) (for non-Gly/Pro residues) | ||
| Most favorable | 92.7 | 92.4 |
| Additional allowed | 7.3 | 7.6 |
3. Results
Both eqFP650 and eqFP670 have been engineered from Katushka (Shcherbo et al., 2010 ▶). eqFP650 differs from its progenitor by two amino-acid substitutions, Asp21Gly and Met41Ala (Fig. 1 ▶). Such a seemingly minor difference results, however, in a 15 nm bathochromic shift of the emission band of eqFP650 (Fig. 2 ▶). eqFP670 differs from Katushka by five amino-acid substitutions: Met11Thr, Asp21Gly, Met41Cys, Ser143Asn and Ser158Asn (Fig. 1 ▶). These five replacements result in a 35 nm bathochromic shift, as compared to Katushka (Fig. 2 ▶). Note that the bathochromic shift of eqFP670 is accompanied by a substantial loss of brightness as compared to both Katushka and eqFP650 (Table 1 ▶). In the structure of Katushka (Pletneva et al., 2011 ▶) residues 11, 41, 143 and 158 belong to the immediate environment of the chromophore whereas residue 21 resides on the surface of the protein (Fig. 1 ▶).
Figure 1.

Sequence alignment and overall structures of the far-red fluorescent protein Katushka and its successors, eqFP650 and eqFP670. Amino-acid differences are marked in blue, the chromophore is marked in red.
Figure 2.

Excitation, emission and absorption spectra of eqFP650 and eqFP670. (a) Absorption spectra of eqFP650 (red) and eqFP670 (blue). (b) Excitation (solid) and emission (dashed) spectra of eqFP650 (red) and eqFP670 (blue).
3.1. Crystal structures of eqFP650 and eqFP670
The asymmetric unit of the crystals contains two monomers oriented at an angle of about 60° with respect to each other, measured between the principal axes of their β-barrels. Crystallographic symmetry operations complete full tetramers. The final electron density is generally well defined in both structures with the exception of the N-terminal His-tags, as well as residues 1–2 and 224–233 of the target proteins. Multiple conformations of side chains are observed for the total of 22 (9.4%) and 56 (24.0%) residues in eqFP650 and eqFP670, respectively. The final atomic models of both eqFP650 and eqFP670 are of high quality, as indicated by low deviations of bonds, angles, dihedrals and planes (Table 3 ▶) from their library values. Superposition of the Cα atoms in eqFP650 and eqFP670 results in a root-mean-square deviation of only 0.13 Å (well within the refinement error limits), indicating close similarity of the structures.
3.2. Chromophores of eqFP650 and eqFP670 and their environment
The electron-density maps show that the structure of the chromophore in both eqFP650 and eqFP670 is typical for DsRed-like proteins (Fig. 3 ▶). Both moieties possess a cis conformation and exhibit a noticeable but not severe deviation from coplanarity between the imidazolinone and p-hydroxyphenyl rings, with κ2 angles (i.e. rotation around the Cβ2—Cγ2 bond) being around −18 and −13° for eqFP650 and eqFP670, respectively.
Figure 3.

The chromophores of (a) eqFP650 and (b) eqFP670 in 2F o − F c electron-density maps contoured at the 1.0σ level.
The slightly larger distortion from planarity of the eqFP650 chromophore is presumably caused by Arg197 adopting two alternative conformations (Fig. 4 ▶ b). In its major conformation [occupancy 0.6 (A)] Arg197 is stabilized by multiple hydrogen bonds with the side chains of Glu145, Lys67 and Glu215 (2.9, 3.1 and 3.1 Å, respectively). The guanidinium group of Arg197 lies parallel to the chromophore (about 4 Å below the chromophore methylidene bond), causing partial stacking which further stabilizes this conformation. The minor conformation of Arg197 [occupancy 0.4 (B)] is stabilized by hydrogen bonds with the hydroxyls of Ser143 and Ser158 (3.1 and 2.4 Å, respectively). The residue protrudes towards the p-hydroxyphenyl ring of the chromophore, which lies about 3.3 Å away from it and slightly pushes it out of plane (Fig. 4 ▶ b). In eqFP670, Arg197 adopts only a single conformation which corresponds to the major conformation of Arg197 in eqFP650 (Fig. 4 ▶ c). Apparently, bulkier Asn158 makes it impossible for Arg197 to acquire the minor conformation observed in eqFP650.
Figure 4.

Immediate chromophore environment of (a) Katushka, (b) eqFP650 and (c) eqFP670. Water molecules and hydrogen bonds measured in Å are shown as red spheres and dashed red lines, respectively.
As was mentioned above, eqFP650 differs from its progenitor Katushka by two amino-acid substitutions, Asp21Gly and Met41Ala (Fig. 1 ▶). Residue 21 is located in a β-turn region at the surface of the protein and its side chain faces an exterior of the protein. Residue 41, in turn, belongs to the nearest chromophore environment and occupies a hydrophobic cavity near the methionine group of the chromophore. As compared to Katushka, the presence of Gly21 in eqFP650 results in an increase of the flexibility of the protein scaffold, whereas the presence of Ala41, with its side chain much shorter than that of Met, allows the chromophore greater freedom which may optimally accommodate itself inside the β-barrel. Ala41 also creates additional space near the chromophore that is filled by a water molecule. The presence of this water molecule enables hydrogen bonding between the carbonyl of Phe62 (part of the chromophore conjugated system) and the hydroxyl of Ser28 (Fig. 4 ▶ b). A combination of these two substitutions is seen as a cause of the bathochromic shift of the eqFP650 emission band.
eqFP670 differs from eqFP650 by the four following substitutions: Met11Thr, Ala41Cys, Ser143Asn and Ser158Asn (Fig. 1 ▶). Residue 11, located 6–7 Å away from the chromophore, fills the internal cavity of the β-barrel adjacent to the acylimine group of the chromophore (Figs. 4 ▶ b and 4 ▶ c). Thr11 in eqFP670 increases the hydrophilicity of the area and makes available more space in the vicinity of the acylimine group of the chromophore, compared to Met11 in eqFP650. In eqFP670, this space is filled by three water molecules which form an extensive hydrogen-bonded network involving the main-chain carbonyls of Phe62 and Thr40, as well as the side chains of the surrounding residues Ser28, Thr11 and Gln39 (Fig. 4 ▶ c).
Cys41 does not seem to introduce any noticeable changes in the structure of eqFP670 as compared to eqFP650. The side chain of Cys41 re-adjusts to occupy nearly the same volume as Ala41 in eqFP650, with its sulfhydryl group turned away from the water-filled pocket described above.
Ser143Asn and Ser158Asn seem to be the two most important substitutions in eqFP670. Both residues are located at the side of the p-hydroxyphenyl ring and lie in about the same plane as the chromophore (Fig. 4 ▶ c). Both Asn143 and Asn158 adopt two alternative conformations – a major conformation (A, occupancy 0.7 and 0.6 for Asn143 and Asn158, respectively) and a minor one (B, occupancy 0.3 and 0.4 for Asn143 and Asn158, respectively). In its major conformation, the side-chain amide group of Asn143 forms two strong hydrogen bonds: one with the hydroxyl group of the p-hydroxyphenyl ring of the chromophore (2.4 Å) and another with the side chain of Asn158 in its major conformation (2.6 Å). Transition of Asn143 to the minor conformation terminates its hydrogen bond with the chromophore and is accompanied by transition of Asn158 to its minor conformation. Asn158 forms hydrogen bonds with two water molecules in each of its conformations; these bonds are part of an extensive hydrogen-bonded network surrounding the chromophore at its p-hydroxyphenyl site.
3.3. Site-directed mutagenesis of the residues at positions 11, 143 and 158 in Katushka, eqFP650 and eqFP670
To examine the influence of the residues proximal to the p-hydroxyphenyl and acylimine sites of the chromophore on the bathochromic shift in eqFP650 and eqFP670, we have generated eight mutants and characterized their absorbance and fluorescence (Table 4 ▶).
Table 4. Spectroscopic properties of Katushka, eqFP650 and eqFP670 mutants.
ΦF, quantum yield. E
mol, extinction coefficient. B, brightness (E
mol × ΦF). , brightness relative to EGFP [product of ΦF and E
mol compared to the brightness of EGFP (53 000 M
−1 cm−1 × 0.60) (Patterson et al., 1997 ▶)]. 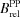 , brightness relative to parental protein. /, excitation and emission maxima.
, brightness relative to parental protein. /, excitation and emission maxima.
| Mutant | ΦF | E mol | B |
(%) |
(%) |
(nm) |
(nm) |
|---|---|---|---|---|---|---|---|
| EGFP† | 0.60 | 53000 | 31800 | 100 | 489 | 509 | |
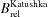 (%) (%) |
|||||||
| Katushka | 0.34 | 65000 | 22100 | 69 | 100 | 588 | 635 |
| Katushka/S143N | 0.02 | 33500 | 500 | 2 | 2 | 565 | 604 |
| Katushka/S158N | 0.29 | 75500 | 21600 | 68 | 98 | 588 | 635 |
| Katushka/S143N/S158N | 0.10 | 60500 | 5900 | 19 | 27 | 605 | 652 |
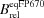 (%) (%) |
|||||||
| eqFP670 | 0.06 | 70000 | 3900 | 12 | 100 | 605 | 670 |
| eqFP670/N143S‡ | 0.17 | 77000 | 13500 | 42 | 346 | 580 | 650 |
| eqFP670/N158S | 0.02 | 46500 | 1000 | 3 | 24 | 570 | 620 |
| eqFP670/N143S/N158S‡ | 0.24 | 48000 | 11800 | 37 | 301 | 588 | 647 |
| eqFP670/T11M | 0.06 | 56500 | 3200 | 10 | 82 | 605 | 670 |
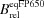 (%) (%) |
|||||||
| eqFP650 | 0.24 | 65000 | 15600 | 49 | 100 | 592 | 650 |
| eqFP650/M11T‡ | 0.24 | 69000 | 16600 | 52 | 107 | 588 | 648 |
The values for EGFP are given for comparison.
For mutants eqFP670/N143S, eqFP670/N143S/N158S and eqFP650/M11T the extinction coefficient was corrected for an additional peak at 380 nm (E mol = 70000) upon NaOH denaturation.
First, we substituted Ser143 and Ser158 residues in the parental Katushka by asparagines, producing three corresponding mutants: Katushka/S143N, Katushka/S158N and Katushka/S143N/S158N. Katushka, a progenitor of eqFP650 and eqFP670, has neither a direct nor water-mediated hydrogen-bond connection between the carbonyl of Phe62 (the acylimine site of the chromophore) and the protein matrix that has been demonstrated to produce a bathochromic shift of the emission/excitation bands in mPlum (Shu et al., 2009 ▶), mNeptune (Lin et al., 2009 ▶) and eqFP650. Therefore, mutants generated from Katushka should impartially reveal the role of Asn143 and Asn158 in the optical properties of eqFP670. Substitution Katushka/S143N resulted in a very dim mutant with blue-shifted excitation/emission bands. Katushka/S158N has spectroscopic properties similar to those of Katushka, while the double mutant Katushka/S143N/S158N has both excitation and emission bands red shifted and demonstrates a 3.7-fold lower brightness than Katushka itself.
In a similar manner, for eqFP670 with both 143 and 158 positions occupied by asparagines, we replaced them by serines (one at a time and two together) to examine the influence of three water molecules at the acylimine site of the chromophore on its fluorescence (Table 4 ▶). All three of these mutants demonstrated a 20–50 nm hypsochromic shift of their excitation and emission bands. eqFP670/N143S and eqFP670/N143S/N158S were found to be 3.5- and 3.0-fold brighter than original eqFP670, respectively, whereas eqFP670/N158S was 4.2-fold dimmer.
The last group of mutants was generated in order to clarify the influence of Met11/Thr11 on the spectroscopic properties of eqFP650 and eqFP670. It seems that the size and the ability of residue 11 to form hydrogen bonds define the number of water molecules present at the acylimine site of the chromophore. Therefore, it was assumed that substitutions eqFP650/M11T and eqFP670/T11M should generate eqFP650 and eqFP670 variants with three and one water molecules, respectively. Both eqFP650/M11T and eqFP670/T11M retained the excitation and emission bands of their respective progenitors but have shown variation of brightness as compared to their parent FPs: 7% increase for eqFP650/M11T and 18% decrease for eqFP670/T11M.
4. Discussion
Red and far-red fluorescent proteins engineered from the same wild-type precursors have chemically identical chromophores; thus their sometimes dramatically different spectral properties must be predominantly caused by the variations in the immediate environment of the chromophore. The key factors responsible for the red shift of the emission bands of the FPs that have been identified so far are trans-to-cis isomerization of the chromophore, its deprotonation, as well as participation in stacking interactions and in new hydrogen bonds between the chromophore and its immediate surroundings. These modifications mostly affect the distribution of electronic density over the chromophore. The more electron density is accumulated around the chromophore, the lower is its excitation energy, and the more red shifted the excitation and emission bands are.
4.1. Structural basis for the red shift in eqFP650
The high-resolution structure of eqFP650 (592/650 nm) reveals the presence of a water molecule occupying the cavity adjacent to the acylimine oxygen of the chromophore (Fig. 4 ▶ b). This is not the case for Katushka (588/635 nm), the progenitor of eqFP650 (Fig. 4 ▶ a). The presence of this water molecule enables the formation of a hydrogen bond between the acylimine oxygen of the chromophore and the hydroxyl of Ser28 (Fig. 4 ▶ b). A similar water-mediated hydrogen bond was reported for another far-red FP, Neptune, in which Met41 was substituted by glycine, resulting in a shift of its emission band maximum from 635 to 650 nm (Lin et al., 2009 ▶). In that case it was suggested that donation of the hydrogen bond to the terminal carbonyl oxygen of the chromophore could stabilize its π-electron system in the excited state (Lin et al., 2009 ▶). Similarly, the cavity resulting from the Met41Ala substitution in eqFP650 enables the appearance of a water molecule at the acylimine site of the chromophore. This water molecule provides hydrogen bonding between the chromophore and the protein scaffold, presumably causing a 15 nm red shift of the emission band. The importance of the hydrogen bond between the acylimine oxygen of the chromophore and the protein scaffold for bathochromic shift was also demonstrated for the far-red FP mPlum, in which the acylimine oxygen forms a direct hydrogen bond with Glu16 (Shu et al., 2009 ▶).
4.2. Structural basis for the red shift in eqFP670
Four substitutions that comprise the difference between eqFP650 and eqFP670 affect two sites of the chromophore, its acylimine group and its phenolic group. Substitutions Met11Thr and Ala41Cys result in the formation of a larger cavity, close to the acylimine group of the chromophore in eqFP670, than in eqFP650. This cavity is filled with three water molecules arranged in a triangle and hydrogen bonding the acylimine oxygen of the chromophore with the side chains of Thr11, Ser28, Gln39, and the main-chain carbonyl of Thr40 (Fig. 4 ▶ c). The appearance of three water molecules at this chromophore site increases its interactions with the protein scaffold.
Dramatic changes that take place at the p-hydroxyphenyl site of the chromophore in eqFP670 are caused apparently by the substitutions Ser143Asn and Ser158Asn. Both asparagines are capable of adopting two alternative conformations and their side chains are positioned roughly in the same plane as the chromophore. In its major conformation, the amide nitrogen of Asn143 forms an unusually short hydrogen bond with the hydroxyl oxygen of the p-hydroxyphenyl group of the chromophore (Fig. 4 ▶ c). Moreover, the major conformation of Asn143 is stabilized by a hydrogen bond between the amide O atom and the side chain of Asn158. With the help of an extensive network of hydrogen bonds (Fig. 4 ▶ c), this hydrogen bond connects the p-hydroxyphenyl site of the chromophore with the protein scaffold. Thus, in the major conformation, residues Asn143 and Asn158 increase the number of hydrogen bonds between the chromophore and the protein matrix, compared to Ser143 and Ser158.
Both Asn143 and Asn158 assume their minor conformations simultaneously. Even in their minor conformations, the side chains of Asn143 and Asn158 maintain hydrogen bonds between themselves and between Asn158 and the protein scaffold. However, the hydrogen bond between Asn143 and the p-hydroxyphenyl ring of the chromophore is broken, disabling interactions of the latter with the protein matrix (Fig. 4 ▶ c). The respective occupancies of 0.7 and 0.3 for the major and minor conformations of Asn143 are in good agreement with a threefold decrease of the brightness of eqFP670 as compared to eqFP650, where the p-hydroxyphenyl group of the chromophore always retains a hydrogen bond with Ser143. Both Asn143 and Asn158 are important for the fluorescent properties of eqFP670 and, as was shown by mutagenesis, replacement of either of them with serines (and presumably with any other amino acid) results in variants with blue-shifted fluorescence bands.
In eqFP670, because of the steric restrictions imposed by the side chain of Asn158, Arg197 adopts a single conformation parallel to the chromophore moiety. This is not the case for eqFP650 in which a smaller Ser158 permits Arg197 to adopt two conformations: one similar to that observed in eqFP670 and another protruding towards the p-hydroxyphenyl ring of the chromophore. As a result, the chromophore of eqFP650 is less planar than that of eqFP670. Therefore, a pair Asn143 and Asn158 provides not only stronger hydrogen-bond connection of the chromophore with the protein matrix than Ser143 and Ser158 but also favors the planarity of the chromophore.
4.3. The influence of the residues at positions 11, 143 and 158 on the spectroscopic properties of Katushka, eqFP650 and eqFP670
To examine the influence of serine and asparagine at positions 143 and 158 on the spectral properties of Katushka and its successors we have created six mutants probing all possible serine–asparagine combinations at these sites (Table 4 ▶). The generated mutants provided four possible combinations: Ser143+Ser158, Asn143+Ser158, Ser143+Asn158 and Asn143+Asn158.
Ser143+Ser158 (eqFP670/N143S/N158S) gives a hypsochromic shift of absorbance/fluorescence bands and increases the fluorescence brightness. The spectral properties of eqFP670/N143S/N158S are close to those of eqFP650, suggesting that in eqFP670 a combination of Asn143 and Asn158 causes an ∼20 nm bathochromic shift of the excitation/emission bands and a decrease in brightness.
Asn143+Ser158 (Katushka/S143N and eqFP670/N158S) gives extremely dim fluorescent proteins that are the most blue shifted among the generated mutants. A combination of Asn143 and Ser158 was earlier observed for eqFP578 (Pletneva et al., 2011 ▶), eqFP611 (Petersen et al., 2003 ▶) and TagRFP (Subach et al., 2010 ▶), and was reported to cause a trans conformation of the chromophore, which is presumably the case for Katushka/S143N and eqFP670/N158S.
Ser143+Asn158 (Katushka/S158N and eqFP670/N143S) gives no clear correlation between the structure and the optical properties of the mutants, suggesting the importance of cooperative interactions of the numerous residues surrounding the chromophore.
The Asn143+Asn158 (Katushka/S143N/S158N) combination produced a variant with significantly red-shifted excitation/emission bands at the expense of the quantum yield and brightness of the chromophore, compared to a more common Ser143+Ser158 combination. Asn143+Asn158 forms a much more extensive hydrogen-bonded network than Ser143+Ser158, which provides greater polarization of the chromophore tyrosine site favoring red shift of its emission, and simultaneously increases the number of possible non-radiative transitions of the excited chromophore, reducing fluorescence brightness.
Two other mutants, eqFP650/M11T and eqFP670/T11M, allow us to examine the influence of the water-mediated network of hydrogen bonds at the acylimine site of the chromophore on its spectral properties. Based on structural data, it was assumed that the mutants with threonine and methionine at position 11 have three and one water molecules at the acylimine site, respectively. Table 4 ▶ clearly shows that both replacements did not affect the position of emission/excitation bands but that Thr11 increases the brightness of the chromophore fluorescence (eqFP650/M11T versus eqFP650), whereas its replacement with Met11 decreases it (eqFP670/T11M versus eqFP670) (Table 4 ▶).
Recently Chica et al. (2010 ▶) applied a structure-based rational design approach to generate fluorescent proteins with longer emission wavelengths. They tested the following three hypotheses. (i) Stabilization of the excited state of the chromophore by creating hydrogen bond(s) between its acylimine oxygen and protein matrix. (ii) Destabilization of the ground state of the chromophore by surrounding its charged phenolate group with hydrophobic residues. (iii) Stabilization of polarization of the excited state of the chromophore by creating π-stacking interactions between the chromophore phenolate and Tyr197. The crystal structures of mRojoA and mRouge have brilliantly confirmed hypotheses (i) and (iii), whereas the validity of hypothesis (ii) remained unclear. Our observations presented here are also in good agreement with hypotheses (i) and (iii). Both eqFP650 and eqFP670 have acylimine oxygen linked to the protein matrix via water-mediated hydrogen bonds [hypothesis (i)]; in both eqFP650 and eqFP670, Arg197 is involved in π-stacking interactions with the chromophore [hypothesis (iii)].
5. Conclusions
Considerable knowledge about the influence of the nearest chromophore environment on the spectral properties of red fluorescent proteins acquired from the extensive studies of this field presents credible guidelines for the rational development of new far-red biomarkers. eqFP650 is the third generation of far-red FPs generated from Katushka; it differs from its progenitor by two amino-acid substitutions (Asp21Gly and Met41Ala) and by the presence of one water molecule which hydrogen bonds the carbonyl of Phe62 (part of the chromophore conjugated system) with the hydroxyl of Ser28 (Fig. 4 ▶ b). This seemingly minor change results in a 15 nm red shift.
The second far-red fluorescent protein described here, eqFP670, has three water molecules in the pocket close to the Phe62 carbonyl. These water molecules create an extensive network of hydrogen bonds connecting the chromophore π-electron system with the residues of the protein scaffold (Fig. 4 ▶ c). Additional water molecules at the acylimine site of the chromophore increase the brightness of the protein. The combination of Asn143 and Asn158 residues in their major conformations provides a stronger hydrogen-bond connection of the chromophore p-hydroxyphenyl ring with the protein matrix and also favors planarity of the chromophore causing an additional 20 nm red shift of its excitation/emission as compared to eqFP650.
Supplementary Material
PDB reference: eqFP650, 4edo
PDB reference: eqFP670, 4eds
Acknowledgments
Diffraction data were collected at the SER-CAT ID22 beamline at the Advanced Photon Source, Argonne National Laboratory. Use of the Advanced Photon Source was supported by the US Department of Energy, Office of Science, Office of Basic Energy Sciences, under contract No. W-31-109-Eng-38. This project has been funded in part with Federal funds from the Frederick National Laboratory for Cancer Research, National Institutes of Health (NIH) contract No. HHSN261200800001E, and the Intramural Research Program of the NIH, Frederick National Laboratory, Center for Cancer Research, by grants from the President of the Russian Federation (No. MK-539.2011.4), the Russian Foundation for Basic Research (No. 11-04-00241), the Russian Ministry of Education and Science (Nos. 16.740.11.0367 and 16.512.11.2139), the Molecular and Cell Biology program of the Russian Academy of Sciences and Measures to Attract Leading Scientists to Russian Educational Institutions (No. 11.G34.31.0017). The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products or organizations imply endorsement by the US Government.
References
- Adams, P. D., Grosse-Kunstleve, R. W., Hung, L.-W., Ioerger, T. R., McCoy, A. J., Moriarty, N. W., Read, R. J., Sacchettini, J. C., Sauter, N. K. & Terwilliger, T. C. (2002). Acta Cryst. D58, 1948–1954. [DOI] [PubMed]
- Campbell, R. E., Tour, O., Palmer, A. E., Steinbach, P. A., Baird, G. S., Zacharias, D. A. & Tsien, R. Y. (2002). Proc. Natl Acad. Sci. USA, 99, 7877–7882. [DOI] [PMC free article] [PubMed]
- Chica, R. A., Moore, M. M., Allen, B. D. & Mayo, S. L. (2010). Proc. Natl Acad. Sci. USA, 107, 20257–20262. [DOI] [PMC free article] [PubMed]
- Chudakov, D. M., Lukyanov, S. & Lukyanov, K. A. (2005). Trends Biotechnol. 23, 605–613. [DOI] [PubMed]
- Emsley, P. & Cowtan, K. (2004). Acta Cryst. D60, 2126–2132. [DOI] [PubMed]
- Gurskaya, N. G., Fradkov, A. F., Terskikh, A., Matz, M. V., Labas, Y. A., Martynov, V. I., Yanushevich, Y. G., Lukyanov, K. A. & Lukyanov, S. A. (2001). FEBS Lett. 507, 16–20. [DOI] [PubMed]
- Hoffman, R. M. (2005). J. Biomed. Opt. 10, 41202. [DOI] [PubMed]
- Hoffman, R. M. & Yang, M. (2005). Nat. Biotechnol. 23, 790. [DOI] [PubMed]
- König, K. (2000). J. Microsc. 200, 83–104. [DOI] [PubMed]
- Kredel, S., Nienhaus, K., Oswald, F., Wolff, M., Ivanchenko, S., Cymer, F., Jeromin, A., Michel, F. J., Spindler, K. D., Heilker, R., Nienhaus, G. U. & Wiedenmann, J. (2008). Chem. Biol. 15, 224–233. [DOI] [PubMed]
- Laskowski, R. A., MacArthur, M. W., Moss, D. S. & Thornton, J. M. (1993). J. Appl. Cryst. 26, 283–291.
- Lin, M. Z., McKeown, M. R., Ng, H. L., Aguilera, T. A., Shaner, N. C., Campbell, R. E., Adams, S. R., Gross, L. A., Ma, W., Alber, T. & Tsien, R. Y. (2009). Chem. Biol. 16, 1169–1179. [DOI] [PMC free article] [PubMed]
- Lukyanov, K. A., Fradkov, A. F., Gurskaya, N. G., Matz, M. V., Labas, Y. A., Savitsky, A. P., Markelov, M. L., Zaraisky, A. G., Zhao, X., Fang, Y., Tan, W. & Lukyanov, S. A. (2000). J. Biol. Chem. 275, 25879–25882. [DOI] [PubMed]
- Merzlyak, E. M., Goedhart, J., Shcherbo, D., Bulina, M. E., Shcheglov, A. S., Fradkov, A. F., Gaintzeva, A., Lukyanov, K. A., Lukyanov, S., Gadella, T. W. & Chudakov, D. M. (2007). Nat. Methods, 4, 555–557. [DOI] [PubMed]
- Morozova, K. S., Piatkevich, K. D., Gould, T. J., Zhang, J., Bewersdorf, J. & Verkhusha, V. V. (2010). Biophys. J. 99, L13–L15. [DOI] [PMC free article] [PubMed]
- Murshudov, G. N., Vagin, A. A. & Dodson, E. J. (1997). Acta Cryst. D53, 240–255. [DOI] [PubMed]
- Nienhaus, K., Nar, H., Heilker, R., Wiedenmann, J. & Nienhaus, G. U. (2008). J. Am. Chem. Soc. 130, 12578–12579. [DOI] [PubMed]
- Otwinowski, Z. & Minor, W. (1997). Methods Enzymol. 276, 307–326. [DOI] [PubMed]
- Patterson, G. H., Knobel, S. M., Sharif, W. D., Kain, S. R. & Piston, D. W. (1997). Biophys. J. 73, 2782–2790. [DOI] [PMC free article] [PubMed]
- Petersen, J., Wilmann, P. G., Beddoe, T., Oakley, A. J., Devenish, R. J., Prescott, M. & Rossjohn, J. (2003). J. Biol. Chem. 278, 44626–44631. [DOI] [PubMed]
- Pletneva, N. V., Pletnev, V. Z., Shemiakina, I. I., Chudakov, D. M., Artemyev, I., Wlodawer, A., Dauter, Z. & Pletnev, S. (2011). Protein Sci. 20, 1265–1274. [DOI] [PMC free article] [PubMed]
- Shcherbo, D., Merzlyak, E. M., Chepurnykh, T. V., Fradkov, A. F., Ermakova, G. V., Solovieva, E. A., Lukyanov, K. A., Bogdanova, E. A., Zaraisky, A. G., Lukyanov, S. & Chudakov, D. M. (2007). Nat. Methods, 4, 741–746. [DOI] [PubMed]
- Shcherbo, D., Murphy, C. S., Ermakova, G. V., Solovieva, E. A., Chepurnykh, T. V., Shcheglov, A. S., Verkhusha, V. V., Pletnev, V. Z., Hazelwood, K. L., Roche, P. M., Lukyanov, S., Zaraisky, A. G., Davidson, M. W. & Chudakov, D. M. (2009). Biochem. J. 418, 567–574. [DOI] [PMC free article] [PubMed]
- Shcherbo, D., Shemiakina, I. I., Ryabova, A. V., Luker, K. E., Schmidt, B. T., Souslova, E. A., Gorodnicheva, T. V., Strukova, L., Shidlovskiy, K. M., Britanova, O. V., Zaraisky, A. G., Lukyanov, K. A., Loschenov, V. B., Luker, G. D. & Chudakov, D. M. (2010). Nat. Methods, 7, 827–829. [DOI] [PMC free article] [PubMed]
- Shkrob, M. A., Yanushevich, Y. G., Chudakov, D. M., Gurskaya, N. G., Labas, Y. A., Poponov, S. Y., Mudrik, N. N., Lukyanov, S. & Lukyanov, K. A. (2005). Biochem. J. 392, 649–654. [DOI] [PMC free article] [PubMed]
- Shu, X., Wang, L., Colip, L., Kallio, K. & Remington, S. J. (2009). Protein Sci. 18, 460–466. [DOI] [PMC free article] [PubMed]
- Strack, R. L., Hein, B., Bhattacharyya, D., Hell, S. W., Keenan, R. J. & Glick, B. S. (2009). Biochemistry, 48, 8279–8281. [DOI] [PMC free article] [PubMed]
- Subach, O. M., Malashkevich, V. N., Zencheck, W. D., Morozova, K. S., Piatkevich, K. D., Almo, S. C. & Verkhusha, V. V. (2010). Chem. Biol. 17, 333–341. [DOI] [PMC free article] [PubMed]
- Vagin, A. & Teplyakov, A. (1997). J. Appl. Cryst. 30, 1022–1025.
- Wang, L., Jackson, W. C., Steinbach, P. A. & Tsien, R. Y. (2004). Proc. Natl Acad. Sci. USA, 101, 16745–16749. [DOI] [PMC free article] [PubMed]
- Wiedenmann, J., Schenk, A., Röcker, C., Girod, A., Spindler, K. D. & Nienhaus, G. U. (2002). Proc. Natl Acad. Sci. USA, 99, 11646–11651. [DOI] [PMC free article] [PubMed]
- Yang, T. T., Cheng, L. & Kain, S. R. (1996). Nucleic Acids Res. 24, 4592–4593. [DOI] [PMC free article] [PubMed]
- Yarbrough, D., Wachter, R. M., Kallio, K., Matz, M. V. & Remington, S. J. (2001). Proc. Natl Acad. Sci. USA, 98, 462–467. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PDB reference: eqFP650, 4edo
PDB reference: eqFP670, 4eds