

The structure of a tetrameric sponge galectin suggests a basis for glutamate receptor potentiation.
Keywords: galectins, Cinachyrella sp.
Abstract
The galectins are a family of proteins that bind with highest affinity to N-acetyllactosamine disaccharides, which are common constituents of asparagine-linked complex glycans. They play important and diverse physiological roles, particularly in the immune system, and are thought to be critical metastatic agents for many types of cancer cells, including gliomas. A recent bioactivity-based screen of marine sponge (Cinachyrella sp.) extract identified an ancestral member of the galectin family based on its unexpected ability to positively modulate mammalian ionotropic glutamate receptor function. To gain insight into the mechanistic basis of this activity, the 2.1 Å resolution X-ray structure of one member of the family, galectin CchG-1, is reported. While the protomer exhibited structural similarity to mammalian prototype galectin, CchG-1 adopts a novel tetrameric arrangement in which a rigid toroidal-shaped ‘donut’ is stabilized in part by the packing of pairs of vicinal disulfide bonds. Twofold symmetry between binding-site pairs provides a basis for a model for interaction with ionotropic glutamate receptors.
1. Introduction
Lectins are glycan-binding proteins that serve a variety of important functions in most organisms. The family of lectins that selectively interact with β-galactosides, known as galectins (formerly the S-lectins), are small soluble proteins that interact with O- and N-glycans either within the cytoplasm or the extracellular matrix, respectively (Di Lella et al., 2011 ▶). Galectins generally bind with highest affinity to N-acetyllactosamine (LacNAc) disaccharides, a common constituent of the N-linked complex oligosaccharides generated by a series of enzymatic reactions within the Golgi compartment. In mammals, galectins play central roles in diverse processes that include pathogen recognition and cell–cell adhesion. Galectins are also expressed in the mammalian nervous system, where they can have an impact on stem-cell proliferation and differentiation (Ishibashi et al., 2007 ▶), facilitate migration of activated microglia (Doverhag et al., 2010 ▶; Di Lella et al., 2011 ▶) and induce neurodegeneration (Plachta et al., 2007 ▶).
Three structurally distinct subfamilies of galectins have been identified based on similarities in their tertiary structures (Kasai & Hirabayashi, 1996 ▶). ‘Prototype’ galectins are monomeric proteins largely comprised of two antiparallel β-sheets that together form the characteristic β-sandwich fold of a carbohydrate-recognition domain (CRD). Noncovalent dimerization creates the bivalent functional unit of prototype galectins, which include the best-characterized galectin in humans, galectin-1. Galectin-3 represents the sole member of the chimeric galectin subfamily; in addition to a single CRD, this galectin contains a long unstructured N-terminal domain that promotes self-assembly into multimeric complexes. Finally, tandem-repeat galectins typified by galectin-9 contain two CRDs joined by a flexible linker domain. The β-sandwich fold of CRDs is highly conserved, even between galectins that are only weakly homologous in primary amino-acid sequence, but the precise profile of binding specificity for different galactosides varies and higher order oligomerization into functional units can occur to give covalent or noncovalent dimers, tetramers or even aggregated complexes of higher valency. The formation of these oligomers represents a critical parameter of galectin biological activity.
Marine sponges represent an only partially explored reservoir of novel lectins. As part of an effort to identify molecules exhibiting mammalian neuroactivity, a family of novel ‘prototype’ galectins from a Japanese marine sponge, Cinachyrella sp., have been purified and characterized (Ueda et al., 2012 ▶). The proteins, termed CchGs, are distinct at the sequence level from other galectins of marine sponge origin (Atta et al., 1989 ▶; Medeiros et al., 2010 ▶; Ueda et al., 2012 ▶). We have determined the X-ray structure of CchG-1 from three different crystal forms. We find that the CchG galectin associates noncovalently as a toroid-shaped tetramer in an arrangement that has not previously been observed for lectins and that is stabilized in part by twofold-symmetric interaction of a novel vicinal disulfide-bond ‘knuckle’. CchGs were originally isolated from sponge extract based on their potent modulatory action towards mammalian ionotropic glutamate receptors (Ishibashi et al., 2007 ▶; Ueda et al., 2012 ▶). The structure of one such receptor, the GluA2 AMPA receptor, has been determined crystallographically (Sobolevsky et al., 2009 ▶). Based on these respective structures, we propose that CchG cross-links glycans in iGluR dimer pairs, thereby imposing constraints on the conformational changes underlying receptor desensitization.
2. Materials and methods
2.1. Isolation and purification
CchG-1 lectin was purified from a marine sponge of the Cinachyrella sp. collected off of Iriomote island, Okinawa, Japan as described by Ueda et al. (2012 ▶). Briefly, a homogenized aqueous extract was dialyzed against water using a 10 kDa membrane cutoff and the macromolecular fraction was lyophilized, reconstituted in water and purified by anion-exchange chromatography (DE52; GE Healthcare, Tokyo) using 100 mM Tris–HCl pH 8, 0–1.0 M NaCl. Fractions with bioactivity (as measured by both potentiation of iGluR current and hemagglutination) were combined and the lectin was further purified using a lactose-conjugated agarose affinity column (Sigma–Aldrich, St Louis, USA). The lectin was eluted using 0.1 M lactose in TBS (0.15 M NaCl, 50 mM Tris–HCl pH 7.4) and was further purified using reversed-phase HPLC (YMC-Pack Protein-RP; YMC, Kyoto, Japan), yielding a single peak at a retention time of 33 min using a linear gradient elution between water and acetonitrile in the presence of 0.1% TFA. Removal of solvents and lyophilization yielded 3.4 mg of protein. The lectin was remarkably heat-stable, retaining 50% agglutinating activity after 2 h at 368 K (Ueda et al., 2012 ▶).
2.2. Crystallization and data collection
2.2.1. Crystallization
As the protein was purified from one marine sponge, there was a very limited amount available for crystallization experiments. Crystallization was carried out in Corning 3773 96-well sitting-drop plates using an Art Robbins Phoenix Crystallization Robot. Two 96-condition screens were chosen: The Classics II Suite and The PEGs II Suite (Qiagen). Drops were comprised of 1 µl protein in water (10 mg ml−1) and 1 µl screen solution and were set up with a 100 µl volume reservoir of the screen solution. The protein crystallized in several different conditions in both screens and crystals were harvested directly from the drops without additional cryoprotection for diffraction experiments. Three crystals obtained under distinct crystallization conditions were of sufficient quality for the measurement of diffraction data and revealed three different crystal forms of CchG. For data set bal2_C12, the triclinic crystal form, the conditions were 0.2 M CaCl2, 0.1 M Tris pH 8.5, 20% PEG 4000, for data set bal2_G2, the monoclinic crystal form, the conditions were 0.2 M ammonium sulfate, 0.1 M MES pH 6.5, 30% PEG 5000 MME and for data set bal2_B12-1, the orthorhombic crystal form, the conditions were 0.2 M lithium sulfate, 0.1 M Tris pH 8.5, 30% PEG 3000.
2.2.2. Data collection
Crystals were analyzed and each data set was measured at the Life Sciences Collaborative Access Team (LS-CAT) Sector 21 beamline 21-ID-F at the Advanced Photon Source, Argonne National Laboratory, Argonne, Illinois using a MAR 225 CCD detector at a wavelength of 0.97872 Å. Measurement of diffraction data from the monoclinic crystal form at 100 K was straightforward, but both the triclinic and orthorhombic crystals, which were ∼40 µm in size, exhibited diffraction pathologies. In the case of the triclinic form the spots exhibited some streaking and the crystal was sensitive to radiation damage. In the case of the orthorhombic form there was pronounced spot streaking and evidence of splitting throughout the data set. While data were measured over 240° of rotation (yielding high multiplicity for the orthorhombic form), these factors contributed to the relatively high R merge and low completeness of both the P1 and C2221 data sets (Table 1 ▶). The space groups were identified as P1, P21 and C2221, with four, four and two monomers in the asymmetric unit, respectively.
Table 1. Crystallographic statistics.
Values in parentheses are for the high-resolution shell.
| Data set | bal2_G2 | bal2_B12-1 | bal2_C12 |
|---|---|---|---|
| Crystal | |||
| Space group | P21 | C2221 | P1 |
| Unit-cell parameters (Å, °) | a = 41.18, b = 117.14, c = 62.06, α = γ = 90, β = 95.3 | a = 61.00, b = 126.63, c = 80.44, α = β = γ = 90 | a = 38.96, b = 66.51, c = 71.55, α = 117.07, β = 95.39, γ = 99.56 |
| Monomers per asymmetric unit | 4 | 2 | 4 |
| Solvent fraction | 0.47 | 0.49 | 0.51 |
| Data collection | |||
| Resolution (Å) | 30–2.10 (2.16–2.10) | 49–2.98 (3.05–2.98) | 35–2.65 (2.72–2.65) |
| R merge † | 0.080 (0.353) | 0.152 (0.449) | 0.128 (0.528) |
| Completeness (%) | 91.9 (70.9) | 95.6 (74.4) | 85.8 (85.7) |
| Multiplicity | 4.8 (3.3) | 8.7 (5.1) | 2.4 (2.4) |
| 〈I/σ(I)〉 | 17.06 (2.97) | 11.7 (2.1) | 5.8 (1.6) |
| Refinement | |||
| R cryst ‡ | 0.208 (0.274) | 0.193 (0.335) | 0.209 (0.329) |
| R free ‡ | 0.280 (0.361) | 0.288 (0.342) | 0.305 (0.407) |
| No. of protein atoms | 4558 | 2275 | 4572 |
| No. of solvent atoms | 183 | 25 | 87 |
| 〈B iso〉 (Å2) | 32.28 | 54.02 | 27.16 |
| R.m.s.d., bonds (Å) | 0.016 | 0.013 | 0.010 |
| R.m.s.d., angles (°) | 1.956 | 1.761 | 1.509 |
| PDB code | 4agr | 4agg | 4agv |
R
merge = 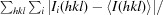
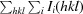 , where 〈I(hkl)〉 is the mean intensity over symmetry equivalents.
, where 〈I(hkl)〉 is the mean intensity over symmetry equivalents.
R
cryst = 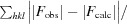
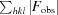 . R
free is R
cryst for 5% of reflections that were omitted from refinement.
. R
free is R
cryst for 5% of reflections that were omitted from refinement.
2.3. Structure determination
2.3.1. Point-group symmetry
The self-rotation functions calculated from the P1 and P21 data sets using the program GLRF (Tong, 2001 ▶) clearly indicated 222 rotational symmetry (Fig. 1 ▶). Although preliminary biochemical studies suggested that the lectin existed as a heterotrimer, this result was only consistent with the existence of a tetrameric oligomerization.
Figure 1.

Self-rotation function. The κ = 180° section of the self-rotation function calculated from data set bal2_C12 (space group P1) demonstrates the 222 symmetry intrinsic to the ‘dimer-of-dimers’ structure of the CchG tetramer. The GLRF software package was used (Tong, 2001 ▶).
2.3.2. Molecular replacement
Once a complete sequence had been determined (see §3.2.3), a BLAST search of the Protein Data Bank identified congerin II (PDB entry 1is5; Shirai et al., 2002 ▶) as having significant sequence identity and its structure was used to construct a model for molecular replacement. Over 98 residues of CchG (18–115), the sequence of 1is5 exhibited 33% identity. (The structure-based alignment after structure determination revealed the sequence identity to be 22% over the complete sequence; Fig. 5.) A model was constructed such that nonconserved residues were pruned to Cβ while leaving conserved residues unchanged (Stein, 2008 ▶). The molecular-replacement model comprised 92 residues (of an expected 147).
Molecular replacement using the P1 data set was carried out with Phaser (McCoy et al., 2007 ▶) and a solution comprising four monomers was identified (TFZ of 5.4, LLG of 77) that largely recapitulated the expected 222 rotational symmetry and converged in REFMAC (Murshudov et al., 2011 ▶) to crystallographic R factors of R cryst = 0.39 and R free = 0.47 at 2.65 Å resolution. The electron-density maps were not interpretable, but a ‘dimer-of-dimers’ arrangement was apparent and one dimer pair was subsequently used as the model for molecular replacement using the P21 data set. A solution of two dimer pairs (TFZ of 11.3, LLG of 133) converged following refinement to crystallographic R factors of R cryst = 0.44 and R free = 0.52 at 2.1 Å resolution.
An initial round of autobuilding in space group P21 carried out using ARP/wARP (Cohen et al., 2008 ▶) yielded a partially complete model (489 residues built, with 395 residues assigned sequence) with crystallographic R factors of R cryst = 0.29 and R free = 0.41. This model provided starting phases for fourfold noncrystallographic symmetry averaging, solvent flattening and histogram matching as implemented in DM (Cowtan & Zhang, 1999 ▶). The resulting electron-density map was of good quality, with NCS averaging correlations of 0.90–0.92 relating each of three monomers to the fourth. This map provided starting phases for a second round of autobuilding using ARP/wARP. The result was an essentially complete model of the CchG tetramer that comprised 566 residues with assigned sequence (of an expected 584 residues) and crystallographic R factors of R cryst = 0.21 and R free = 0.28.
To complete the structure determination, one monomer of the P21 solution was used as a search model for molecular replacement with the P1 and C2221 data sets. A solution comprising four monomers in P1 (TFZ = 23.3, LLG = 596) and a solution comprising two monomers in C2221 (TFZ = 25.3, LLG = 357) were readily obtained and were refined to crystallographic R factors of R cryst = 0.20 and R free = 0.30 and of R cryst = 0.19 and R free = 0.29 for P1 and C2221, respectively. The electron-density map from the P21 solution is shown in Fig. 2 ▶.
Figure 2.

Electron-density map. (a) The final 2F o − F c map from the solution of the P21 data set (over β-strands 10.2.6.7; see Fig. 4 ▶) contoured at 1.2σ. (b) 2F o − F c density at the apex of the CC-loop showing the disulfide bond formed between adjacent residues Cys81/Cys82. The stereo pairs are divergent.
2.3.3. Model of the CchG tetramer
The initial electron-density maps for model rebuilding and correction were generated by tenfold noncrystallographic symmetry averaging over the three different crystal forms as implemented in DMMULTI (Winn et al., 2011 ▶). The NCS averaging correlations over the nine transformations (in three crystals) relative to the first ranged from 0.836 to 0.984. These maps were inspected and the models were rebuilt with Coot (Emsley et al., 2010 ▶) and subsequent refinements for each crystal form were carried out with REFMAC (Murshudov et al., 2011 ▶). Water molecules were placed automatically using ARP (Lamzin et al., 2001 ▶). In each crystal form the structural model includes residues 3–146 of the expected 146-amino-acid sequence. In one monomer of the P21 crystal form the loop 64–67 (which contributes to the ligand-binding site; see below) is disordered. Alternate conformations were modeled in each structure where supported by the electron-density map. Refinement statistics are presented in Table 1 ▶.
The structure determinations in each crystal form revealed a donut-like tetramer exhibiting 222 point symmetry in a dimer-of-dimers arrangement (Fig. 3 ▶ a). The fact that an identical tetrameric relationship occurs in each of the three crystal forms (in the case of C2221 it straddles a crystallographic twofold axis) resolves any ambiguity in assignment of monomers in the crystal-packing context and supports the conclusion that the tetramer is the biochemically relevant species.
Figure 3.


CchG crystal structure. (a) Ribbon representation of the donut-like CchG tetramer in the P1 crystal form. The structures of the P21 and C2221 crystal forms are essentially identical [overlap r.m.s.d.s of the P1 (tetramer) and C2221 (dimer) structures with the P21 tetramer of 0.53 and 0.57 Å, respectively]. The dimensions of the CchG tetramer are ∼72 × 72 × 36 Å; the diameter of the central ‘donut-hole’ is ∼18 Å. (b) A divergent stereo image of the CchG protomer, highlighting the regions discussed in the text. The ribbon representation is colored using a blue–red gradient (N- to C-terminus). (c) The CchG tetramer organization as a dimer of dimers is completely different from those of other tetrameric galectins. Top left, the fungal galectin CGL2 (PDB entry 1ul9; Walser et al., 2004 ▶). Top right, mouse galectin 4 (PDB entry 3i8t; Krejčiříková et al., 2011 ▶). Bottom left, Dioclea rostrata lectin (PDB entry 2zbj; de Oliveira et al., 2008 ▶), a tetrameric plant lectin. Bottom right, ConA (PDB entry 1cjp; Hamodrakas et al., 1997 ▶) oriented to emphasize its pseudo-tetragonal arrangement of binding sites. Images were obtained from the PDB (http://www.pdb.org).
The structures have been submitted to the PDB as the following entries: P21 crystal form, 4agr; P1, 4agv; C2221, 4agg.
3. Results and discussion
3.1. The structure of CchG
3.1.1. Overall fold and comparison of the monomer to other galectin structures
The CchG protein monomer comprises two five-stranded antiparallel β-sheets packed face to face across a hydrophobic core (Fig. 3 ▶ b). This fold is characteristic of galectins and the topology of the β-strands is equivalent to that of galectins such as congerin II (Fig. 4 ▶) and bovine galectin-1. The dimensions of the core β-sheet structure are ∼24 × 23 × 10 Å. The N- and C-terminal strands pack against each other at one end of the β-sandwich, where they both contribute to the dimer interface. At the other end, the two sheets are bridged by a long loop that packs against the edge of the β-sandwich.
Figure 4.

The β-sheet topology of the CchG protomer is similar to those of other galectins. (a) The structure of the congerin II protomer (PDB entry 1is5) is shown as a ribbon diagram with the corresponding topology diagram (PDBSum; Laskowski, 2009 ▶). (b) The topology diagram of the CchG monomer (PDBSum; Laskowski, 2009 ▶). The strand-order relationships revealed by the topology diagrams are indicated beneath the diagrams.
Despite a very low degree of sequence identity of 22%, in the structure-based alignment (Fig. 5 ▶) the CchG protomer can be superposed on congerin II with an r.m.s.d. of 2.41 Å over 127 Cα positions. The most significant deviations between the two structures are localized to bridging loops, particularly residues 78–87, which are N-terminal to strand β7, and residues 129–138, which form a large loop between strands β9 and β10. The strands β4 and β5 and the loop between them that constructs the putative ligand-binding site adopt essentially identical conformations in the two proteins (below).
Figure 5.

(a) Structure-based sequence alignment. The alignment of CchG with congerin II (PDB entry 1is4) was determined using GANGSTA+ (Guerler & Knapp, 2008 ▶). Arrows indicate identical (thick) and similar (thin) residues in the alignment. Note the conserved sequence QNVLVLNS that contributes to the base of the binding site. 97% of the residues align with an r.m.s.d. of 2.65 Å (130 aligned residues). (b) A divergent stereo image of the overlap; the structure of congerin II is shown in light gray and the structure of CchG is shown as a gradient from the N-terminus (blue) to the C-terminus (red). Note particularly the CC-loop that extends at the bottom left of the fold.
3.1.2. The β-sheet dimer interaction and comparison to galectin dimers
Lectins generally assemble as dimers or as dimers of dimers, with the ‘canonical’ dimer-interaction association being via the formation of a continuous β-sheet along one of the two faces of the dimer (Hamelryck, 1999 ▶). The structure of CchG reflects the structure of the canonical dimer, with the addition that both β-sheets of the protomer are continuous across the dimer interface (Fig. 6 ▶). The N-terminal strand mediates the β-sheet that is common to both proteins, and in CchG the C-terminal strand also forms an antiparallel β-sheet interaction with its counterpart across the dimer interface. We term this the ‘β-sheet interface’ of CchG. The buried accessible surface area at the interface is 613 Å2 per protomer (Reynolds et al., 2009 ▶).
Figure 6.

Two interfaces mediate the CchG dimer of dimers. (a) The β-sheet continues across the dimer interface and is unique. The N- and C-terminal strands of two protomers continue the hydrogen-bonding interaction of the β-sheet to form the dimer interface. Divergent stereo images. The view is from the interior of the CchG tetramer (the ‘donut hole’). Residues at the beginning and the end of the C-terminal β-strand of each protomer are labeled. (b) The CC loop mediates the dimer-of-dimers interaction that forms the CchG tetramer. The two adjacent disulfide-bonded residues (Cys80 and Cys81; labeled) pack across the dimer-of-dimers interface and are tinted yellow in this divergent stereo pair. β-Strands β6 and β7 are labeled in each protomer. (c) The location of the two interfaces in the tetramer.
The CchG dimer thus comprises two continuous twisted ten-stranded β-sheets that are packed against each other. When compared with the quaternary structure of the congerin II dimer (for example), the consequence of the formation of a two-β-sheet structure is a rotation of approximately 80° of the second domain relative to the (fixed) first domain. This significantly changes the relationship of the orientations of the two ligand-binding pockets (below) of the dimer when compared with other galectins.
3.1.3. The CC-loop mediates the dimer-of-dimers interaction
The dimer-of-dimers interface is mediated by a novel structure that is stabilized by an unusual disulfide bond between adjacent cysteines (Fig. 2 ▶ b). Such vicinal disulfides are quite rare and they create a tight turn of the polypeptide chain (Carugo et al., 2003 ▶) that, in some proteins, may serve as a redox-regulated conformational switch (Wouters et al., 2010 ▶). Among galectins, the cysteine pair appears to be unique to CchG. It occurs at the apex of the loop prior to strand β6 in the CchG protomer, and two ‘CC-loops’ contributed by adjacent protomers interdigitate ‘knuckle-like’ across the dimer-of-dimers interface (Fig. 6 ▶ b). Two main-chain hydrogen bonds bridge the interface, but otherwise the residues of the interface loops are largely (50%) hydrophobic. The buried surface area is 678 Å2 per protomer.
During the structure determination, we observed some heterogeneity of the electron density at the disulfide bond that was suggestive of its disruption in some fraction of molecules in the crystal. The heterogeneity was not uniform between protomers nor between the three different crystal structures. Disulfide-bond disruption is a well known consequence of X-ray radiation damage (Weik et al., 2000 ▶) and may have arisen that way in this case. However, the observation does suggest the possibility of redox regulation of assembly of the CchG tetramer (Wouters et al., 2010 ▶) and may also help explain the heterogeneity observed during biochemical characterization of the protein (Ueda et al., 2012 ▶).
3.1.4. Comparison of the CchG tetramer with other galectin tetramers
Lectins generally occur as dimers or tetramers and the quaternary structure of lectins has been much discussed (Srinivas et al., 2001 ▶; Hamelryck, 1999 ▶; Brinda et al., 2004 ▶). The quaternary arrangement of a dimer of dimers that creates an almost circular ‘donut hole’ ∼18 Å in diameter at the center of the CchG galectin structure is apparently unique (Figs. 3 ▶ a and 3c ▶). However, given the known diversity of lectin quaternary structures (Hamelryck, 1999 ▶) the ‘donut-hole’ structure is not necessarily significant in itself and its functional importance remains to be determined.
Some lectins are known to undergo solution-dependent quaternary-structure changes (Chatterjee & Mandal, 2003 ▶). The CchG crystal structure itself does not directly reflect the stability or association dynamics of the CchG tetramer. However, the strict noncrystallographic symmetry observed within and between crystal forms (NCS correlations of >0.90; see above) and the superposition r.m.s.d.s ranging from 0.3 to 0.4 Å (protomer–protomer) and from 0.53 to 0.57 Å (tetramer–tetramer) suggest that the CchG tetramer is quite rigid overall. This observation is consistent with the remarkable thermostability of the protein (Ueda et al., 2012 ▶); however, the critical role of the unique CC-loop in maintaining the dimer-of-dimers interface again suggests that the dimer–tetramer equilibrium may be redox-sensitive.
3.2. The carbohydrate-binding sites
3.2.1. Conservation of the binding-site sequence
The most highly conserved sequence motif of CchG that is found in alignment with congerin II comprises nine residues, Glu53–Ser61 (see Fig. 5 ▶), that precede and include strand β5. This strand is the central structural element of the galectin-binding site (as identified in the congerin II structure). Of the eight residues that mediate polar interactions with lactose in the structure of congerin II (PDB entry 1is4; Shirai et al., 2002 ▶), five are identical in the structure of CchG: Arg28, Arg47, Asn60, Trp68 and Glu71 (Fig. 7 ▶ a). Of the remaining residues, CchG residue Glu53 substitutes for Asp54 (in congerin II), with the longer side chain likely compensating for a small shift in the position of the main-chain loop in CchG. Two other substitutions are problematic: Arg51 may substitute functionally for Tyr51 in congerin II, which mediates hydrogen bonding to galactose O1 and O2, and a water-mediated hydrogen bond stabilized by Tyr32 may substitute for His44 in congerin II, which hydrogen bonds galactose O4. The latter deviation is notable, however, because His44 is almost invariant in the sequences of previously characterized galectins (Barondes et al., 1994 ▶; Cooper & Barondes, 1999 ▶) and suggests that CchG binding interactions and specificity may vary compared with other galectins (Shirai et al., 2002 ▶).
Figure 7.

The galectin-binding site. (a) Sequence conservation provides the basis for a binding-site model. The side chains of residues that are conserved between CchG and congerin II are shown in the context of a ribbon diagram of the CchG protomer (left). The superposition of lactose-bound congerin II (PDB entry 1is4; r.m.s.d. of 2.65 Å over 130 residues; Fig. 5 ▶) allows the approximate placement of a model of bound lactose (right; CPK representation) at the putative binding site. Note that some side-chain conformations (e.g. Trp68) are expected to change when ligand is bound. (b) Separation of the binding-site pairs in the tetramer. The four protomers in the surface representation alternate between salmon and teal. The binding pockets are colored yellow and are defined as all atoms within 6 Å of the (modeled) ligand. The pair of sites across the β-sheet interface (left) are separated by 54 Å; the pair of sites across the CC-loop interface are separated by 44 Å.
However, even with the caveat that we do not yet have a full understanding of the structure and function of CchG binding interactions, given hemagglutination studies that establish specificity for lactose (Ueda et al., 2012 ▶) and the substantial conservation of both the primary sequence and the tertiary structure of the galectin-binding pocket (Fig. 7 ▶), it is not unreasonable to propose a working model for substrate interaction with CchG based on the structure of the congerin II–lactose complex structure (PDB entry 1is4). This model forms the basis for considering how the CchG structure determination reported here may illuminate its mode of action in the regulation of receptor function.
3.2.2. Separation of the sites
Two distinct pairs of galectin-binding pockets can be located in the context of the CchG tetramer. The first occurs across the β-sheet interface and mimics somewhat the relationship between sites in other galectin dimers (Fig. 7 ▶ b). The two sites are separated by ∼54 Å (center to center), are on opposite ends of the dimer and are oriented almost perpendicular to each other. The reducing end of the disaccharide bound at each site is directed away from the center. The second pair occurs across the CC-loop interface. The two sites of this pair are separated by ∼44 Å, but are adjacent across the CC-loop interface and are oriented such that the reducing end of the bound disaccharide exits on the same face of the dimer at each site. Thus, the separation, orientation and disposition of ligands bound at the two pairs of sites are distinct.
We note that the creation of a second adjacent pair of ligand-binding sites in the CchG tetramer distinguishes it, perhaps in a functionally interesting way, from other tetrameric lectins such as ConA that adopt a pseudo-tetragonal packing arrangement. The latter assembles as a dimer of dimers by orthogonal packing between the β-sheets of the two dimers such that the four ligand-binding sites are well separated at each apex (Fig. 3 ▶ c). Functional studies of the tetrameric fungal galectin CGL2 have revealed how the organization of sites mediates functional selectivity for particular mammalian blood-group oligosaccharides (Walser et al., 2004 ▶).
3.2.3. Sequence variants
When structure determination was initiated, only an N-terminal sequence determined by Edman degradation was available. Subsequently, the complete CchG nucleotide sequence was determined from cDNA by PCR RACE (Ueda et al., 2012 ▶), which yielded two canonical sequences, termed CchG-1 and CchG-2, that were assigned to different galectin isoforms observed during purification. Surprisingly, subsequent characterization of multiple cDNA and genomic sequencing clones revealed combinatorial variation of the CchG-1 and CchG-2 sequences at 21 distinct amino-acid positions (Ueda et al., 2012 ▶). Galectins serve as components of the innate immune system in sponges and such sequence variation, which has been observed previously (Zhu et al., 2006 ▶), has been thought to perhaps reflect this function (Litman et al., 2007 ▶). However, sequence variation can also arise artifactually from the formation of PCR chimeras when two (or more) closely related underlying sequences are present (Kopczynski et al., 1994 ▶; Cronn et al., 2002 ▶). Neither canonical CchG sequence exactly matched the sequence of the structural model. We therefore used OMIT maps (Bhat, 1988 ▶) to re-evaluate the side-chain electron density at each of the variant positions and were able to unambiguously assign the CchG-1 sequence variant at 13 of 21 positions and the CchG-2 sequence variant at six positions (two positions remained indeterminant owing to isosteric substitution). Although we cannot exclude the presence of minor populations of sequence variants in the crystallized protein, the assignments were consistent across the three different crystal forms and they match the N-terminal 40 residues of isoform CchG-1 obtained by Edman degradation (Ueda et al., 2012 ▶). The source of the discrepancy between the structural and sequence data remains undetermined. The positions of sequence variants when mapped on the crystal structure revealed them to occur in three clusters: one comprising loops adjacent to the β-interface, one distal to the binding pocket adjacent to the CC-loop interface and one which, in the tetrameric molecule, is exposed at the surface of the inner ‘donut-hole’ (Fig. 8 ▶). There is no variation within the putative lactose-binding pocket.
Figure 8.

CchG sequence variation mapped onto its structure. The positions of variation between sequence variants are mapped onto the crystal structure. The binding-site cleft is on the right. The positions of sequence variants cluster at the ‘edges’ at the top right, left and bottom in this perspective. The approximate locations of the structural features including the ‘donut hole’ at the center of the tetramer are indicated to provide context.
3.3. Model for interaction with the kainate receptor
Isolation of CchG was in part guided by a potent bioactivity towards AMPA-type and kainate-type ionotropic glutamate receptors. The desensitization rate was slowed by several-fold and steady-state currents were greatly enhanced (Ueda et al., 2012 ▶). A decade of research into the structural basis of the biophysical properties of glutamate receptors has led to well supported physical models for the conformational changes underlying desensitization. The determination of numerous structures of the N-terminal (ATD) and ligand-binding (LBD) domains of AMPA, kainate and NMDA receptors (Mayer, 2011 ▶), as well as the determination of the complete structure of the AMPA receptor (Sobolevsky et al., 2009 ▶), provided the basis for proposing a speculative model of how the CchG tetramer interacts with a representative CchG-sensitive iGluR, the GluK2 kainate receptor.
N-Glycans that control sensitivity to the plant lectin concanavalin A are located in the linker domain between the ATD and LBD in each of the subunits making up the functional tetrameric receptor (Fay & Bowie, 2006 ▶). A model glycoprotein constructed using the GLYCAM server (Kirschner et al., 2008 ▶) introducing glycans at the ATD/LBD linkers provides the basis for preliminary investigation of how the CchG tetramer may interact with the GluK2 kainate receptor to modulate its function (Fig. 9 ▶). The structure is twofold-symmetric and, given that CchG binding is likely to be bivalent, there are two possible receptor glycan pairs available to mediate binding. However, while it is straightforward to place the CchG tetramer such that one or other of the pairs of galactoside-binding pockets (i.e. those bridging the β-sheet interface or those bridging the CC-loop interface) is ‘appropriately’ spaced to interact with a corresponding pair of carbohydrate substituents of the modelled glycoprotein, it is also clear by inspection that only one of the two unique pairs of ATD/LBD linkers of the receptor tetramer can be selected for binding (Fig. 9 ▶ b). The distance between the alternate pair is too great.
Figure 9.

A model for interaction with the kainate receptor. Three carbohydrate modifications along the linker peptide between the ATD and LBD domains of the kainate receptor GluK2 region of the structure (identified as sites 5, 6 and 7; Fay & Bowie, 2006 ▶) have been shown to mediate the activity of the CchG galectin. (a) Speculative model for the disposition of the CchG tetramer with respect to GluK2, based on the structure of the AMPA receptor (PDB entry 3kg2; Sobolevsky et al., 2009 ▶) and presuming a bivalent binding interaction. The glycan chain has been omitted from the model for clarity. On the left, the pair of sites bridged by the CC-loop interface is oriented towards the linker peptides. On the right, the pair of sites bridged by the β-sheet interface is oriented towards the linkers. The membrane-distal ATD is at the top and LBD chains A and B are labeled. (b) ‘Top’ view of the model. The four ATD subunits A, B, C and D are labeled; the dimer interactions of the ATD are AB and CD, but the dimer interactions of the LBD, which are lateral in this view and obscured, are AD and BC. Note that the four linker peptides of the receptor tetramer are not fourfold equivalent. One pair of adjacent peptides (left and right) are separated by ∼48 Å, closely matching the distances between pairs of CchG binding pockets (Fig. 7 ▶ b). The other pair (top and bottom) are ∼75 Å apart and sterically occluded. (c) Structure of a seven-unit glycan chain (PDB entry 1mco; Guddat et al., 1993 ▶) modeled as extending from the ATD/linker to indicate scale.
The membrane-distal ABD (amino-terminal domain) of the AMPA receptor tetramer comprises AB and CD dimer pairs (the obvious dimer pairs in the top view shown in Fig. 9 ▶ b), whereas the membrane-proximal LBD (ligand-binding domain) comprises AD and BC subunit pairs (Sobolevsky et al., 2009 ▶). The pair of peptide linkers that by simple inspection are available to be cross-linked by bivalent interaction with CchG corresponds to the AD (or BC) subunit pair. Proposed mechanisms for both desensitization and negative allosteric modulation of NMDA receptors (Sobolevsky et al., 2009 ▶) invoke interactions that separate the ‘D1’ subdomains of the membrane-proximal LBD ‘clamshells’, which are communicated as rearrangements of the adjacent ‘D2’ LBD subdomains and subsequently the transmembrane helices to place the pore in a closed-state-like conformation: thus, desensitization by acquisition, while ligand is bound, of a ‘ruptured’ state of the AD pair D1–D1 interface and negative allosteric modulation by separation of ABD subdomain pairs communicated via the ATD/LBD linker to adjacent D1 subdomains of the LBD. The specific ATD/LBD linker peptide pairs cross-linked in the model here could easily be imagined to affect the disposition of the D1 subunits of the adjacent AD LBD pairs. Thus (as in the proposal of Sobolevsky and coworkers), CchG binding could introduce ‘tension of ATD-LBD linkers [that] would, therefore, result in separation of the LBD domains D1’ (Sobolovsky et al., 2009 ▶), and so modulate, specifically, desensitization or negative regulation of kainate receptor gating. Accordingly, the binding of the lectin ConA has been shown to reflect agonist-induced conformational changes in the GluK2 receptor (Fay & Bowie, 2006 ▶).
We note that the arrangement of the ligand-binding sites of the CchG tetramer extends in a plane parallel to the membrane in this model. This has two implications: firstly, that the structure may cross-link extracellular domains of adjacent membrane receptors (in a potentially interesting redox-regulated manner) and, secondly, that given the rigid structure of the CchG tetramer and the planar configuration of its binding-site pairs (e.g. particularly across the CC-loop), the protein could readily function as an allosteric mediator that functions to propagate a particular conformational state from receptor to receptor across the cell surface. Studies to investigate such phenomenona will be of much interest.
Supplementary Material
PDB reference: CchG-1, P21, 4agr
PDB reference: P1, 4agv
PDB reference: C2221, 4agg
Acknowledgments
We acknowledge the assistance of Ludmilla Shuvalova and Bryan Copits. This work used resources of the Northwestern University Structural Biology Facility, which is generously supported by NCI CCSG P30 CA060553 awarded to the Robert H. Lurie Comprehensive Cancer Center, and was supported by the Naito Foundation and a Grant-in-Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science and Technology, Japan (15580183 and 17380125 to RS) and ROI NS44322 from NINDS (to GTS). Use of the Advanced Photon Source was supported by the US Department of Energy, Office of Science, Office of Basic Energy Sciences under Contract No. DE-AC02-06CH11357. Use of LS-CAT Sector 21 was supported by the Michigan Economic Development Corporation and the Michigan Technology Tri-Corridor for the support of this research program (Grant 085P1000817).
References
- Atta, A. M., Barral-Netto, M., Peixinho, S. & Sousa-Atta, M. L. (1989). Braz. J. Med. Biol. Res. 22, 379–385. [PubMed]
- Barondes, S. H., Cooper, D. N., Gitt, M. A. & Leffler, H. (1994). J. Biol. Chem. 269, 20807–20810. [PubMed]
- Bhat, T. N. (1988). J. Appl. Cryst. 21, 279–281.
- Brinda, K. V., Mitra, N., Surolia, A. & Vishveshwara, S. (2004). Protein Sci. 13, 1735–1749. [DOI] [PMC free article] [PubMed]
- Carugo, O., Cemazar, M., Zahariev, S., Hudáky, I., Gáspári, Z., Perczel, A. & Pongor, S. (2003). Protein Eng. 16, 637–639. [DOI] [PubMed]
- Chatterjee, M. & Mandal, D. K. (2003). Biochemistry, 42, 12217–12222. [DOI] [PubMed]
- Cohen, S. X., Ben Jelloul, M., Long, F., Vagin, A., Knipscheer, P., Lebbink, J., Sixma, T. K., Lamzin, V. S., Murshudov, G. N. & Perrakis, A. (2008). Acta Cryst. D64, 49–60. [DOI] [PMC free article] [PubMed]
- Cooper, D. N. & Barondes, S. H. (1999). Glycobiology, 9, 979–984. [DOI] [PubMed]
- Cowtan, K. D. & Zhang, K. Y. J. (1999). Prog. Biophys. Mol. Biol. 72, 245–270. [DOI] [PubMed]
- Cronn, R., Cedroni, M., Haselkorn, T., Grover, C. & Wendel, J. F. (2002). Theor. Appl. Genet. 104, 482–489. [DOI] [PubMed]
- Di Lella, S., Sundblad, V., Cerliani, J. P., Guardia, C. M., Estrin, D. A., Vasta, G. R. & Rabinovich, G. A. (2011). Biochemistry, 50, 7842–7857. [DOI] [PMC free article] [PubMed]
- Doverhag, C., Hedtjärn, M., Poirier, F., Mallard, C., Hagberg, H., Karlsson, A. & Sävman, K. (2010). Neurobiol. Dis. 38, 36–46. [DOI] [PubMed]
- Emsley, P., Lohkamp, B., Scott, W. G. & Cowtan, K. (2010). Acta Cryst. D66, 486–501. [DOI] [PMC free article] [PubMed]
- Fay, A. M. & Bowie, D. (2006). J. Physiol. 572, 201–213. [DOI] [PMC free article] [PubMed]
- Guddat, L. W., Herron, J. N. & Edmundson, A. B. (1993). Proc. Natl Acad. Sci. USA, 90, 4271–4275. [DOI] [PMC free article] [PubMed]
- Guerler, A. & Knapp, E. W. (2008). Protein Sci. 17, 1374–1382. [DOI] [PMC free article] [PubMed]
- Hamelryck, T. W. (1999). PhD thesis. Vrije Universiteit Brussel, Belgium.
- Hamodrakas, S. J., Kanellopoulos, P. N., Pavlou, K. & Tucker, P. A. (1997). J. Struct. Biol. 118, 23–30. [DOI] [PubMed]
- Ishibashi, S., Kuroiwa, T., Sakaguchi, M., Sun, L., Kadoya, T., Okano, H. & Mizusawa, H. (2007). Exp. Neurol. 207, 302–313. [DOI] [PubMed]
- Kasai, K. & Hirabayashi, J. (1996). J. Biochem. 119, 1–8. [DOI] [PubMed]
- Kirschner, K. N., Yongye, A. B., Tschampel, S. M., González-Outeiriño, J., Daniels, C. R., Foley, B. L. & Woods, R. J. (2008). J. Comput. Chem. 29, 622–655. [DOI] [PMC free article] [PubMed]
- Kopczynski, E. D., Bateson, M. M. & Ward, D. M. (1994). Appl. Environ. Microbiol. 60, 746–748. [DOI] [PMC free article] [PubMed]
- Krejčiříková, V., Pachl, P., Fábry, M., Malý, P., Řezáčová, P. & Brynda, J. (2011). Acta Cryst. D67, 204–211. [DOI] [PubMed]
- Lamzin, V., Perrakis, A. & Wilson, K. S. (2001). International Tables for Crystallography, Vol. F, edited by M. G. Rossmann & E. Arnold, pp. 720–722. Dordrecht: Kluwer Academic Publishers.
- Laskowski, R. A. (2009). Nucleic Acids Res. 37, D355–D359. [DOI] [PMC free article] [PubMed]
- Litman, G. W., Dishaw, L. J., Cannon, J. P., Haire, R. N. & Rast, J. P. (2007). Curr. Opin. Immunol. 19, 526–534. [DOI] [PMC free article] [PubMed]
- Mayer, M. L. (2011). Curr. Opin. Neurobiol. 21, 283–290. [DOI] [PMC free article] [PubMed]
- McCoy, A. J., Grosse-Kunstleve, R. W., Adams, P. D., Winn, M. D., Storoni, L. C. & Read, R. J. (2007). J. Appl. Cryst. 40, 658–674. [DOI] [PMC free article] [PubMed]
- Medeiros, D. S., Medeiros, T. L., Ribeiro, J. K., Monteiro, N. K., Migliolo, L., Uchoa, A. F., Vasconcelos, I. M., Oliveira, A. S., de Sales, M. P. & Santos, E. A. (2010). Comp. Biochem. Physiol. B Biochem. Mol. Biol. 155, 211–216. [DOI] [PubMed]
- Murshudov, G. N., Skubák, P., Lebedev, A. A., Pannu, N. S., Steiner, R. A., Nicholls, R. A., Winn, M. D., Long, F. & Vagin, A. A. (2011). Acta Cryst. D67, 355–367. [DOI] [PMC free article] [PubMed]
- Oliveira, T. M. de, Delatorre, P., da Rocha, B. A., de Souza, E. P., Nascimento, K. S., Bezerra, G. A., Moura, T. R., Benevides, R. G., Bezerra, E. H., Moreno, F. B., Freire, V. N., de Azevedo, W. F. & Cavada, B. S. (2008). J. Struct. Biol. 164, 177–182. [DOI] [PubMed]
- Plachta, N., Annaheim, C., Bissière, S., Lin, S., Rüegg, M., Hoving, S., Müller, D., Poirier, F., Bibel, M. & Barde, Y. A. (2007). Nature Neurosci. 10, 712–719. [DOI] [PubMed]
- Reynolds, C., Damerell, D. & Jones, S. (2009). Bioinformatics, 25, 413–414. [DOI] [PubMed]
- Shirai, T., Matsui, Y., Shionyu-Mitsuyama, C., Yamane, T., Kamiya, H., Ishii, C., Ogawa, T. & Muramoto, K. (2002). J. Mol. Biol. 321, 879–889. [DOI] [PubMed]
- Sobolevsky, A. I., Rosconi, M. P. & Gouaux, E. (2009). Nature (London), 462, 745–756. [DOI] [PMC free article] [PubMed]
- Srinivas, V. R., Reddy, G. B., Ahmad, N., Swaminathan, C. P., Mitra, N. & Surolia, A. (2001). Biochim. Biophys. Acta, 1527, 102–111. [DOI] [PubMed]
- Stein, N. (2008). J. Appl. Cryst. 41, 641–643.
- Tong, L. (2001). Acta Cryst. D57, 1383–1389. [DOI] [PubMed]
- Ueda, T., Nakamura, Y., Smith, C., Inoue, A., Ojima, T., Matsumaga, S., Swanson, G. T. & Sakai, R. (2012). Submitted.
- Walser, P. J., Haebel, P. W., Künzler, M., Sargent, D., Kües, U., Aebi, M. & Ban, N. (2004). Structure, 12, 689–702. [DOI] [PubMed]
- Weik, M., Ravelli, R. B., Kryger, G., McSweeney, S., Raves, M. L., Harel, M., Gros, P., Silman, I., Kroon, J. & Sussman, J. L. (2000). Proc. Natl Acad. Sci. USA, 97, 623–628. [DOI] [PMC free article] [PubMed]
- Winn, M. D. et al. (2011). Acta Cryst. D67, 235–242.
- Wouters, M. A., Fan, S. W. & Haworth, N. L. (2010). Antioxid. Redox Signal. 12, 53–91. [DOI] [PubMed]
- Zhu, Y., Ng, P. M. L., Wang, L., Ho, B. & Ding, J. L. (2006). Int. Immunol. 18, 1671–1680. [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PDB reference: CchG-1, P21, 4agr
PDB reference: P1, 4agv
PDB reference: C2221, 4agg