

The crystal structure of the 11.14 kDa orphan ORF 1382 from Archaeoglobus fulgidus (AF1382) has been determined by sulfur SAD phasing using data collected from a moderately diffracting crystal and 1.9 Å synchrotron X-rays.
Keywords: AF1382, orphan ORFs, sulfur SAD, Archaeoglobus fulgidus
Abstract
The crystal structure of the 11.14 kDa orphan ORF 1382 from Archaeoglobus fulgidus (AF1382) has been determined by sulfur SAD phasing using a moderately diffracting crystal and 1.9 Å wavelength synchrotron X-rays. AF1382 was selected as a structural genomics target by the Southeast Collaboratory for Structural Genomics (SECSG) since sequence analyses showed that it did not belong to the Pfam-A database and thus could represent a novel fold. The structure was determined by exploiting longer wavelength X-rays and data redundancy to increase the anomalous signal in the data. AF1382 is a 95-residue protein containing five S atoms associated with four methionine residues and a single cysteine residue that yields a calculated Bijvoet ratio (ΔF anom/F) of 1.39% for 1.9 Å wavelength X-rays. Coupled with an average Bijvoet redundancy of 25 (two 360° data sets), this produced an excellent electron-density map that allowed 69 of the 95 residues to be automatically fitted. The S-SAD model was then manually completed and refined (R = 23.2%, R free = 26.8%) to 2.3 Å resolution (PDB entry 3o3k). High-resolution data were subsequently collected from a better diffracting crystal using 0.97 Å wavelength synchrotron X-rays and the S-SAD model was refined (R = 17.9%, R free = 21.4%) to 1.85 Å resolution (PDB entry 3ov8). AF1382 has a winged-helix–turn–helix structure common to many DNA-binding proteins and most closely resembles the N-terminal domain (residues 1–82) of the Rio2 kinase from A. fulgidus, which has been shown to bind DNA, and a number of MarR-family transcriptional regulators, suggesting a similar DNA-binding function for AF1382. The analysis also points out the advantage gained from carrying out data reduction and structure determination on-site while the crystal is still available for further data collection.
1. Introduction
An ORFan (orphan open reading frame) by definition shares no significant sequence homology to proteins or protein domains in the Pfam-A protein-family classification (Bateman et al., 2004 ▶). These ORFan or Pfam-B proteins are often species-specific and are related to evolutionary and developmental functions. Because of this, these proteins have for the most part been excluded as targets by most PSI-1 structural genomics centers, the focus of which has been on developing a complete picture of protein-fold space based on proteins in the Pfam-A database for molecular modeling and other applications (Norvell & Machalek, 2000 ▶). However, studies have shown that non-Pfam-A proteins represent a significant population (∼20%) of all sequences determined to date (Finn et al., 2008 ▶) and may contain important evolutionary information or have the potential for biomedical or environmental applications (Siew & Fischer, 2004 ▶).
In its search for novel protein folds, the Southeast Collaboratory for Structural Genomics (SECSG; Norvell & Machalek, 2000 ▶; Adams et al., 2003 ▶) thought that there might be value in exploring orphan proteins as possible structural genomics targets. The SECSG analysis (Zhu, 2008 ▶) indicated that approximately 6.5% of the 86 875 protein chains (28 945 structures) in the 30 May 2006 Protein Data Bank (PDB) release (Berman et al., 2000 ▶) were not found in the Pfam-A (v.20.0) database and thus could be classified as ORFans. The SECSG study also showed that for the period spanning 2002–2004 the number of ORFan protein structures deposited in the PDB was less than 10% and the number of novel folds found for ORFan structures was about four times greater than that for other newly deposited protein structures. The SECSG analysis produced a list 1285 ORFan targets, including ORF 1382 (AF1382) from Archaeoglobus fulgidus, which is the focus of this work.
AF1382 is a 95-residue (11.14 kDa) protein containing a single cysteine residue and four methionine residues. When initial crystallization attempts using selenomethionine-labeled protein failed to produce diffraction-quality crystals, the native AF1382 protein was crystallized and sulfur single-wavelength anomalous scattering (S-SAD) phasing was attempted using 1.9 Å wavelength X-rays at SER-CAT (http://www.ser-cat.org). S-SAD phasing was first used to determine the crystal structure of crambin, a small 46-residue plant protein (Hendrickson & Teeter, 1981 ▶). The resolved anomalous scattering (RAS) approach used in this study required both high-resolution (1.5 Å) data and a protein with a high sulfur content. The 46-residue crambin protein contained six cysteine residues (three disulfide bonds) and yielded a Bijvoet ratio (ΔF anom/F) of 1.59% for data collected using Cu Kα X-rays (Hendrickson & Teeter, 1981 ▶).
An alternate approach for resolving the phase ambiguity in SAD data was subsequently proposed by Wang, who showed via simulation that this method could be used to phase a 12 kDa protein using the anomalous scattering signal of a single disulfide using a set of 3 Å resolution error-free data with a Bijvoet ratio of 0.58% for Cu Kα X-rays (Wang, 1985 ▶). In Wang’s iterative single-wavelength anomalous scattering (ISAS) method, the protein envelope and a real/reciprocal-space noise-filtering process are used to identify the true phase from the bimodal phase distribution. Wang’s simulation suggested that the sulfur anomalous scattering signal, even from a protein with low sulfur content, could be used for phasing provided that the data could be measured sufficiently accurately.
By 2000, a combination of synchrotron X-rays, cryogenic methods of stabilizing the crystal in the X-ray beam, CCD-based X-ray area detectors, faster computers and better data-processing software allowed Liu et al. (2000 ▶) to solve the structure of the 22 kDa photoprotein obelin using only the anomalous scattering signal from the eight S atoms found in the native protein and a highly redundant data set recorded with 1.74 Å wavelength synchrotron X-rays (Bijvoet ratio of 1.09%). Since then, there has been growing interest in using the S-SAD approach for macromolecular structure determination (Chayen et al., 2000 ▶; Weiss et al., 2001 ▶; Ramagopal et al., 2003 ▶; Debreczeni et al., 2003 ▶; Olczak et al., 2003 ▶; Mueller-Dieckmann et al., 2004 ▶, 2005 ▶, 2007 ▶). A recent survey showed that there have been more than 50 S-SAD publications and that there are over 65 de novo S-SAD structures in the PDB (Rose et al., 2011 ▶).
Here, we report the procedures used to determine the initial 2.3 Å resolution S-SAD phased structure (determined using two data sets from the same crystal collected at two different times) together with details of the refined 1.85 Å resolution structure obtained using data collected from a further optimized crystal four months later.
2. Materials and methods
2.1. Protein production
The ORF encoding the AF1382 protein was amplified from A. fulgidus DSM4304 genomic DNA by PCR using primers 5′-GAAAACCTGTACTTCCAAGGCGGGTCAGGTATGGTGGAGGACGAAAGAAT-3′ and 5′-GGGGACCACTTTGTACAAGAAAGCTGGGTTCATGTATCATTTTCCATAATATCGA-3′. The PCR product was cloned into the Gateway-based plasmid pDEST-527 containing an N-terminal His6 purification tag followed by a tobacco etch virus (TEV) protease-cleavage site (HHHHHHENLYFQGGSG) and a T7 promoter. The protein was expressed in the Escherichia coli host strain BL21 (DE3) (Stratagene, USA). Cells were allowed to grow in 1 l LB medium for 3 h post induction with IPTG (1 mM). The cells were harvested by centrifugation (5000 rev min−1) for 10 min and resuspended in 25 ml lysis buffer consisting of 20 mM sodium phosphate pH 7.6, 1.2 M NaCl. Protein purification was carried out using a three-step (nickel affinity, ion exchange and size exclusion) chromatography process at 277 K. For the Ni-affinity chromatography buffer A [20 mM sodium phosphate pH 7.6, 500 mM NaCl, 5%(v/v) glycerol, 20 mM imidazole] was used for loading, while buffer B [20 mM sodium phosphate pH 7.6, 500 mM NaCl, 5%(v/v) glycerol, 300 mM imidazole] was used to elute the His6-tagged protein. The peak fractions from the Ni-affinity column (HisTrap FF column; GE Healthcare, USA) were collected and dialyzed against dialysis buffer [25 mM HEPES pH 7.6, 50 mM NaCl, 5 mM EDTA, 1 mM DTT, 5%(v/v) glycerol]. The His6 purification tag was then removed by TEV protease cleavage (Haspel et al., 2001 ▶) followed by overnight dialysis. The protein sample was loaded onto a HiTrap Q-Sepharose column (GE Healthcare, USA) and eluted with a salt gradient from 50 mM to 1 M NaCl. The peak fractions that contained the AF1382 protein were collected, concentrated to 3 ml and loaded onto a Superdex 200 26/60 size-exclusion column (GE Healthcare, USA) which had been equilibrated with 25 mM HEPES buffer pH 7.6, 100 mM NaCl, 5 mM EDTA, 1 mM DTT, 5%(v/v) glycerol. After size exclusion, the purity of the AF1382 protein fractions was assayed by SDS–PAGE and they were pooled and concentrated to 2.8 mg ml−1 for crystallization trials.
2.2. Crystallization
Initial crystallization screening trials were set up using the modified microbatch-under-oil method (Chayen et al., 1990 ▶) against the 384-condition SECSG screen (Shah et al., 2005 ▶) employing 48-well Nunc plates set up using a Douglas Instruments Oryx 6 crystallization robot (Douglas Instruments). Each well of the plate was first filled with a 70:30 silicone:paraffin oil mixture. A 1 µl drop consisting of equal volumes of protein solution (2.8 mg ml−1; see above) and precipitant cocktail was then placed in the bottom of the well. A 2 mm layer of the oil mixture described above was then used to seal the plate and to allow the slow evaporation of water from the well to mimic the vapor-diffusion process. The plates were then incubated at 291 K for 30 d. Periodically, each well in the plate was visually inspected for the presence of crystals.
Crystals from two different setups were used in the analysis. The crystal used for S-SAD phasing was obtained from drops consisting of 0.1 M sodium citrate/citric acid pH 5.5, 0.1 M sodium chloride, 0.1 M lithium sulfate, 30%(v/v) polyethylene glycol 400. The crystal used for high-resolution data collection was obtained by continued crystal screening/optimization. The optimized high-resolution crystal was grown in drops consisting of 0.2 M ammonium sulfate pH 4.6, 0.1 M sodium acetate, 30%(w/v) polyethylene glycol monomethyl ether 2000.
2.3. Data collection and processing
For data collection, crystals were harvested and mounted using the loop-mounting method (Teng, 1990 ▶) without cryoprotectant. The size of the CryoLoop (Hampton Research) was matched to the size of the crystal to reduce excess mother liquor around the crystal. Once harvested, the crystal was flash-cooled to 100 K and shipped to SER-CAT, Advanced Photon Source, Argonne National Laboratory and transferred from the liquid-nitrogen-filled storage dewar using a locally modified ALS/Berkeley-style crystal-mounting robot (Cork et al., 2006 ▶) for data collection.
Diffraction data were collected at cryogenic temperatures (100 K) on beamline 22-ID using a 100 µm beam and a MAR 300 CCD detector. The S-SAD data used for phasing were collected using 1.9 Å wavelength (6.25 keV) X-rays to increase the sulfur anomalous scattering signal (Δf′′ is 0.8746 e− for 1.9 Å wavelength X-rays). Although some have suggested X-ray wavelengths greater than 2 Å as optimal for the S-SAD experiment (Weiss et al., 2004 ▶; Mueller-Dieckmann et al., 2004 ▶), the wavelength of 1.9 Å used reflects the beam-stability issues that were associated (in 2007) with SER-CAT 22-ID at wavelengths greater than 2 Å. A helium beam path was used to minimize air absorption.
Data were collected according to the SECSG S-SAD data-collection protocol (Rose et al., 2004 ▶; Li et al., 2008 ▶). A 360° data set (D1) was collected using 1° oscillation steps, a crystal-to-detector distance of 125 mm and a 3 s exposure time (using a highly attenuated X-ray beam). After data collection, the crystal was recovered and returned to the storage dewar. The D1 data set was indexed, integrated and scaled (anomalous and absorption flags set) using HKL-2000 (Otwinowski & Minor, 1997 ▶). Automated structure determination using the SGXPro crystallographer’s workbench (Fu et al., 2005 ▶) was then attempted. When SGXPro failed to produce an interpretable electron-density map, the AF1382 crystal was remounted, manually centered and a second 360° data set (D2) was collected as described above but using a 2 s exposure time and an attenuated X-ray beam. The D2 data set was processed using HKL-2000 as described above. Attempts to solve the structure using the D2 data set and SGXPro were again unsuccessful. At this point, the D1 and D2 data sets were merged and scaled using SCALEPACK with the anomalous and absorption flags set. Using the merged D1D2 data set, SGXPro was able to solve the structure (see below). As part of the analysis, we also calculated values for R meas (the redundancy-independent merging R factor of Diederichs & Karplus, 1997 ▶) and R p.i.m. (the precision-indicating merging R factor of Weiss & Hilgenfeld, 1997 ▶). These calculations were made using SCALEPACK scaled data with the ‘NO MERGE ORIGINAL INDEX’ flag set.
The high-resolution D3 data set was collected several months later from a better diffracting crystal using 0.9729 Å wavelength (12.75 keV) X-rays to reduce absorption effects. A single 360° data set was collected using 1° oscillation steps, a crystal-to-detector distance of 230 mm and an exposure time of 1 s. These data were processed by HKL-2000 but with the anomalous and absorption correction flags unchecked. The data-collection and processing statistics for the four data sets used in the analysis are summarized in Table 1 ▶.
Table 1. Data-collection and refinement statistics.
Values in parentheses are for the highest resolution shell.
| D1 data set | D2 data set | D1D2 data set | D3 data set | |
|---|---|---|---|---|
| Data collection | ||||
| X-ray source | APS 22-ID | APS 22-ID | APS 22-ID | |
| Wavelength (Å) | 1.9 | 1.9 | 0.9724 | |
| Crystal-to-detector distance (mm) | 125 | 125 | 230 | |
| Exposure time (s) | 3 | 2 | 1 | |
| Space group | P42 | P42 | P42 | P42 |
| Unit-cell parameters (Å) | a = b = 53.54, c = 41.25 | a = b = 53.52, c = 41.24 | a = b = 53.54, c = 41.25 | a = b = 53.03, c = 40.97 |
| Resolution range (Å) | 50.00–2.30 (2.38–2.30) | 50.00–2.30 (2.38–2.30) | 50.00–2.30 (2.38–2.30) | 50.00–1.85 (1.92–1.85) |
| Completeness (%) | 95.9 (59.3) | 99.8 (98.5) | 99.9 (99.2) | 99.9 (99.8) |
| Redundancy | 12.0 (3.9) | 13.5 (9.7) | 25 (13.4) | 13.3 (12.5) |
| R merge † (%) | 4.1 (32.3) | 4.1 (21.3) | 4.5 (25.4) | 5.1 (23.3) |
| 〈I/σ(I)〉 | 72.4 (3.1) | 94.1 (14.5) | 124.7 (17.38) | 60.66 (12.96) |
| Mosaicity (°) | 0.48–0.65 | 0.50–0.65 | 0.56 | |
| Unique reflections | 5347 | 5325 | 5325 | 9859 |
| Rmeas ‡ (%) | 4.4 (36.2) | 4.2 (22.2) | 4.8 (28.4) | |
| R p.i.m. § (%) | 1.2 (16.1) | 1.1 (6.7) | 0.9 (7.5) | |
| SHELXD CCall/CCweak | 40.8/11.2 | 29.75/13.49 | 47.93/29.15 | |
| RTSI (see text) | 43.33 | 37.63 | 68.00 | |
| Traceable map | No | No | Yes | |
| Refinement | ||||
| Resolution limits (Å) | 37.86–2.30 | 37.50–1.85 | ||
| Reflections used | 5325 | 9761 | ||
| No. of protein atoms refined | 740 | 748 | ||
| No. of water molecules | 32 | 76 | ||
| R work/R free ¶ (%) | 23.2/26.8 | 17.90/21.40 | ||
| R.m.s.d. bond lengths (Å) | 0.009 | 0.008 | ||
| R.m.s.d. bond angles (°) | 1.300 | 0.998 | ||
| Mean B value (Å2) | 50.0 | 28.7 | ||
| MolProbity all-atom clash score | 20.42 | 12.35 | ||
| Ramachandran favored (%) | 87/88 | 88/89 | ||
| PDB entry | 3o3k †† | 3ov8 | ||
R
merge = 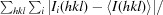
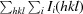 , where I(hkl) is the observed intensity of reflections.
, where I(hkl) is the observed intensity of reflections.
R meas is the redundancy-independent merging R factor of Diederichs & Karplus (1997 ▶).
R p.i.m. is the precision-indicating merging R factor of Weiss & Hilgenfeld (1997 ▶).
R
work = 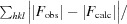
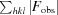 , where F
calc and F
obs are the calculated and observed structure-factor amplitudes, respectively. R
free is the same as R
work but calculated using an ∼5% test set of reflections that were excluded from refinement.
, where F
calc and F
obs are the calculated and observed structure-factor amplitudes, respectively. R
free is the same as R
work but calculated using an ∼5% test set of reflections that were excluded from refinement.
The accompanying 2.3 Å resolution S-SAD data set has been deposited in the PDB.
2.4. Phasing and refinement
Initial phases were generated using SGXPro (Fu et al., 2005 ▶). Within the SGXPro environment, SHELXD (Usón & Sheldrick, 1999 ▶) was used to determine the anomalous scattering substructure, ISAS (Wang, 1985 ▶) was used to determine the initial protein phases and handedness of the anomalous substructure, RESOLVE (Terwilliger, 2003 ▶) was used for phase improvement and auto-fitting, and Coot (Emsley & Cowtan, 2004 ▶) was used for visualization and model building.
When neither the D1 data set nor the D2 data set produced an interpretable electron-density map, the individual low-energy data sets were merged together and scaled using SCALEPACK. Using the merged D1D2 data set, SHELXD gave four of the five sulfur positions present in the protein. The four sulfur positions and the merged D1D2 data set were input into the ISAS suite. ISAS was able to determine the handedness of the sulfur coordinate set, to generate a set of initial protein phases and to extend these phases to 2.3 Å resolution. The 2.3 Å resolution ISAS phases were then refined using RESOLVE and a polyalanine model was automatically built into the electron-density map.
The RESOLVE model was then completed (the chain was extended and side chains were added) manually using Coot (Emsley & Cowtan, 2004 ▶) and refined using CNS v.1.3 (Brunger, 2007 ▶; Brünger et al., 1998 ▶) against the 2.3 Å resolution D1D2 data set. Solvent, modeled as water, was added according to the following criteria: the presence of a positive peak above 3.2σ in the F o − F c difference electron-density map with good hydrogen-bonding geometry to either the protein or other solvent atoms.
For the high-resolution refinement, the 2.3 Å resolution S-SAD phased structure was refined against the 1.85 Å resolution D3 data set using PHENIX (v.1.6.4_486). After several rounds of refinement (positional and atomic displacement parameters), a TLS model (four TLS groups) was calculated and included in the refinement. Validation of the model was carried out using MolProbity (Davis et al., 2007 ▶) and PROCHECK (Laskowski et al., 1993 ▶). Refinement statistics are presented in Table 1 ▶.
3. Results and discussion
AF1382 was selected as an SECSG target since sequence analyses (Altschul et al., 1990 ▶) at the time indicated that it was a protein of unknown function that belonged to Pfam-B family 114426 (Finn et al., 2008 ▶). Selenomethionine-labeled protein was then produced and crystallized using the SECSG high-throughput gene-to-structure pipeline (Liu et al., 2005 ▶; Wang et al., 2005 ▶). However, the crystals of the selenomethionine protein were small and did not diffract well. The focus was then shifted to crystallizing the native protein as a possible target for S-SAD phasing.
3.1. Data collection
The crystals of AF1382 belonged to space group P42 (identified during structure determination), with unit-cell parameters a = b = 53.54, c = 41.25 Å. There is one molecule in the crystallographic asymmetric unit, giving a Matthews coefficient V M of 2.70 Å3 Da−1 and a calculated solvent content of 54.40% (Matthews, 1968 ▶).
As mentioned above, when the D1 data set failed to produce an interpretable electron-density map, the initial crystal was recovered from the dewar 3 h later, centered in the X-ray beam and a second 360° data set (D2) was collected using a shorter 2 s exposure time since there was some concern about the possibility of radiation damage. However, after data reduction, analysis of the scaling statistics did not indicate any appreciable crystal decay in either data set. In addition, the increased anomalous scattering signal (see below) observed for the D1D2 data set coupled with the successful S-SAD phasing and structure refinement would tend to suggest that radiation damage in the D2 data set was not problematic.
The statistics [number of reflections, completeness, redundancy and 〈I/σ(I)〉] for the D2 data set were unexpectedly a little better than those observed for the D1 data set (see Table 1 ▶). This is interesting because one would expect that the D1 data set would have the better statistics. The improved statistics for the D2 data set may be a consequence of a combination of several factors. (i) Since the crystal was remounted several hours later for the D2 data collection, there is a possibility that the X-ray beam was less attenuated or better optimized, increasing the total flux on the sample. (ii) Although analysis of the scale factors for both data sets did not suggest that the crystal was misaligned, the crystal may have been centered on a better diffracting crystal domain for the D2 data set since the size of the crystal used was twice the beam diameter. (iii) The mosaicity for the D2 data set is only about 1.8% lower than that observed for the D1 data set. This would suggest that crystal annealing is not a major contributor since this process generally produces a more dramatic decrease in crystal mosaicity (Harp et al., 1998 ▶).
The D2 data set alone again failed to give an interpretable electron-density map and the two data sets were then merged and scaled together. The R merge for the D1D2 data set is only slightly higher than that observed for either single data set and is still in the range (R merge < 6%) expected for synchrotron X-ray diffraction data. The 〈I/σ(I)〉 for the D1D2 data set was 124.7, which is higher than that observed for either single data set. The increased 〈I/σ(I)〉 for the merged data set is expected since the merged D1D2 data set essentially doubled the redundancy of the data and this should increase the signal-to-noise level in the data.
It should also be pointed out that a variety of HKL-2000 integration-box sizes were explored during the integration process for the D1 and D2 data sets. This strategy is based on our experience, which shows that choosing the proper spot size for integration can significantly have an impact on the strength of the sulfur anomalous signal for data collected from moderately diffracting crystals such as those of AF1382. In this process each D1, D2 data-set pair was integrated using spot sizes of 0.5, 0.7, 0.9 and 1.1 mm. The integrated intensities were then scaled and automated structure determination was attempted using SGXPro. The results of this analysis showed that only the merged D1D2 data set produced using data processed with an integration spot size of 0.9 mm yielded a structure (see below).
3.2. Phasing, model building and refinement for the 2.3 Å resolution S-SAD structure
Only the merged D1D2 data set produced a SHELXD solution that yielded an interpretable RESOLVE electron-density map. The D1D2 SHELXD solution (CCall = 47.93, CCweak = 29.15) found four of the five AF1382 sulfur positions corresponding to Met19, Met59, Met91 and Cys62 in the refined structure. Residue Met1, which is located at the flexible N-terminus of the protein, was not observed in either the 2.3 or the 1.85 Å resolution crystal structures. In contrast, the D1 and D2 data sets gave SHELXD solutions with CCall/CCweak values of 40.80/11.20 and 29.75/13.49 for the D1 and D2 data sets, respectively.
The ISAS program suite was used to confirm the correct hand of the anomalous scattering substructure, to generate an initial set of protein phases to 3.5 Å resolution and to extend the phase set to 2.3 Å resolution. Phase refinement and auto-fitting (using a 95-residue polyalanine model) was carried out using RESOLVE running within the SGXPro workflow. The D1D2 data set gave an initial SOLVE FOM of 0.36 and produced a final RESOLVE FOM of 0.68. RESOLVE autotracing yielded 76 (80%) of the 95 residues present in the asymmetric unit, corresponding to a RESOLVE tracing success index (RTSI) of 68.00.
Here, we have defined RTSI as
A similar analysis using the D1 and D2 data sets produced solutions with RTSIs of 43.33 and 37.63, respectively.
The quality of the electron-density map generated from the D1D2 data set was excellent (see Fig. 1 ▶ a) and the AF1382 structure was quickly completed manually using Coot. The initial AF1382 model was then refined using CNS followed by manual adjustment. Solvent was added when the free R factor refined to below 30% using the criteria described above. At this point 32 solvent atoms (modeled as waters) were added. The model was then refined to convergence, yielding an R factor of 23.2% and a free R factor of 26.8%.
Figure 1.

S-SAD phasing of AF1382. (a) A stereoview of a section (residues 43–59) of the S-SAD-phased 2F o − F c experimental electron-density map contoured at 1σ generated by CCP4mg (McNicholas et al., 2011 ▶). The coordinates for residues 43–59 of the 2.3 Å resolution refined S-SAD model are also shown. The density in red corresponds to the anomalous electron density for SD of residue Met59 in the Bijvoet difference Fourier map using phases generated from the corresponding RESOLVE-fitted polyalanine model. (b) A plot of peak heights for the top ten peaks observed in the Bijvoet difference Fourier maps for the D1 (green), D2 (blue) and D1D2 (red) data sets. The maps were produced using phases generated from the corresponding RESOLVE model from SGXPro. Peaks are labeled corresponding to the residue contributing the S atom at that position. Note that the SD atom of Met91 is disordered. (c) A comparison of the anomalous scattering signal-to-noise index R as as a function of resolution for the D1, D2 and D1D2 data sets. (d) A comparison of the SHELXC anomalous scattering signal 〈d′′/sig〉 as a function of resolution for the D1, D2 and D1D2 data sets.
The refined 2.3 Å resolution model contained residues 4–93 (740 protein atoms) and 32 solvent molecules. Residues 1–3 and 94–95 were not observed in the electron-density maps. These residues are most probably disordered. Validation of the final model using PROCHECK and MolProbity indicated that the model had excellent stereochemistry. The r.m.s. deviations in bond lengths and angles for the refined model were 0.009 Å and 1.30°, respectively. The PROCHECK Ramachandran plot showed that all residues (excluding glycine) lie within the additionally allowed regions of the Ramachandran plot, with 96.4% of the residues lying in the most favored regions. We also explored refining the high-resolution structure (see below) against the 2.3 Å resolution D1D2 data set; however, little improvement in the number of missing residues, the stereochemistry of the structure or in the R factor and free R factor were observed.
Optimizing the signal-to-noise (S/N) level in the data is the key to successful SAD structure determination and is critical in the case of sulfur SAD. This study exploits two common approaches for increasing the anomalous scattering signal in SAD data: the use of soft or longer wavelength X-rays for data collection and the collection of highly redundant data (Wu et al., 2000 ▶; Liu et al., 2000 ▶; Weiss et al., 2001 ▶; Dauter & Adamiak, 2001 ▶; Ramagopal et al., 2003 ▶; Sarma & Karplus, 2006 ▶; Cianci et al., 2008 ▶).
The S-SAD data used for phasing were collected using 1.9 Å wavelength X-rays to increase the sulfur anomalous scattering signal. The Bijvoet ratio for the protein (95 residues and five S atoms) at this wavelength is 1.39%, which is slightly above the average Bijvoet ratio of 1.37% observed for proteins for which structures have been determined by de novo S-SAD (Rose et al., in preparation).
Increasing data redundancy is also a common method of increasing the S/N in the data. Thus, when the D1 data set failed to give a SHELXD solution, the crystal was quickly remounted and a second data set (D2) was collected, which when scaled together with the first data set doubled the redundancy of the D1D2 data set, leading to the S-SAD-phased structure. Analysis of peak heights in the anomalous difference Fourier maps (Fig. 1 ▶ b) calculated using the D1, D2 and D1D2 data sets and their corresponding experimental phase sets from RESOLVE clearly showed a markedly improved anomalous signal for the merged data set. This trend was also supported by the RTSI values for the corresponding data sets.
Fu et al. (2004 ▶) have proposed the R as index as a means of estimating the anomalous scattering signal-to-noise level in a data set, with an R as value at 3 Å resolution of greater than 1.5 being a good indicator for S-SAD phasing success. As shown in Fig. 1 ▶(c), the 3 Å resolution R as value of 1.36 for the D1D2 data set is higher than those of either the D1 (1.12) or the D2 (1.18) data sets. The fact that the successfully phased D1D2 data set had a 3 Å resolution R as value of 1.36 suggests that the 1.5 R as minimum needed for phasing success should be revised. A similar trend was observed in the plot of the SHELXC anomalous scattering indicator 〈d′′/sig〉 versus resolution shown in Fig. 1 ▶(d) (Sheldrick, 2010 ▶). Again, the D1D2 data set has a significantly improved anomalous signal. It should be noted that although increasing the data redundancy is an easy means of increasing the anomalous S/N in the data in theory, one must be extremely careful of radiation damage since it can introduce significant noise into the data set (Weiss et al., 2005 ▶). This is especially true for S-SAD since prolonged exposure of the crystal to X-rays has been found to preferentially damage S atoms owing to the significantly higher absorption cross-section of sulfur (compared with other atoms commonly found in a proteins), as manifested by the reduction of disulfide bonds leading to the disruption of the anomalous substructure (Chen et al., 2000 ▶; Weik et al., 2000 ▶).
3.3. Phasing, model building, refinement and analysis of the 1.85 Å high-resolution structure
The final 2.3 Å resolution S-SAD phased model was then refined against the high-resolution D3 data set (99% completeness) to 1.85 Å resolution. After several rounds of refinement, 76 solvent molecules (modeled as water), one acetate and two chloride ions were identified in the electron-density map and included in refinement. The refinement was continued to convergence and gave an R factor of 17.9% and a free R factor of 21.4%. The refined high-resolution model had good stereochemistry, with r.m.s. deviations from ideality of 0.008 Å for bond lengths and 0.998° for bond angles. PROCHECK analysis showed that all residues (excluding glycine) lay within the additionally allowed regions of the Ramachandran plot, with 97.6% of the residues lying in the most favored regions.
A comparison of the 2.3 and 1.85 Å resolution structures showed that the two models could be superimposed (Holm & Sander, 1998 ▶) to give an average r.m.s. deviation (on Cα atoms) of 0.3 Å, with most of the structural differences being observed in the N- and C-terminal residues of the protein.
The refined AF1382 structure (PDB entry 3ov8) is a mixed α+β structure containing five α-helices and a three-stranded (β1↑β3↓β2↑) antiparallel β-sheet (Fig. 2 ▶ a) with a solvent-accessible surface of 6124 Å2 (Krissinel & Henrick, 2007 ▶). The polypeptide chain (Fig. 2 ▶ b) begins with a short α-helix, αA (residues 4–10), which transitions immediately to helix αB (residues 11–27). Residues 31–32 make up the short β1 strand, which is followed by helix αC (residues 33–40). A short loop connects helix αC to helix αD (residues 43–57), which is followed by strand β2 (residues 59–65). Residues 65–68 form a type II′ β-turn that connects strand β2 to strand β3 (residues 68–73). Strand β3 is immediately followed by helix αE (residues 74–93). The chain then terminates at residue 94. A notable feature of this mostly α-structure is the β-hairpin (hairpin class 2:2 IIP) formed by residues in strands β2 (60–65) and β3 (68–73). The refined crystal structure also contained three bound ions: a single acetate ion (ACT96) and two chloride ions (Cl97 and Cl98). ACT96 is located at the head of helix αB and is hydrogen bonded to the main-chain NH atoms of Glu12 (2.85 Å) and Lys13 (3.16 Å) via hydrogen bonding (2.24 Å) to HOH114, which in turn is hydrogen bonded (3.38 Å) to the main-chain O atom of Phe10. Cl97 lies at the head of helix αE and makes a hydrogen bond (3.21 Å) to the main-chain N atom of Glu75, while Cl98 is hydrogen bonded (2.94 Å) to Asp30 OD1.
Figure 2.

The refined high-resolution AF1382 structure. (a) A ribbon representation of the refined AF1382 model colored blue to red based on residue number, with secondary-structural elements labeled. (b) The corresponding topology diagram generated by PDBsum (Laskowski, 2009 ▶). (c) The putative AF1382 dimer generated by PISA (Krissinel & Henrick, 2007 ▶). The model is colored blue to red based on residue number and is viewed down the crystallographic twofold axis. Figs. 2, 3 and 4 were generated using Chimera (Pettersen et al., 2004 ▶) unless specified otherwise.
A search of the SUPERFAMILY database (Gough et al., 2001 ▶) indicates that AF1382 structure belongs to the winged-helix DNA-binding domain superfamily characterized by a winged-helix–turn–helix (wHTH) motif (Aravind et al., 2005 ▶). In the AF1382 wHTH motif helices αC and αD comprise the HTH DNA-binding motif, while strands β2 and β3 form the wing. As the second helix in the HTH motif, helix αD is assumed to be the DNA-recognition helix.
An analysis of the AF1382 structure using PISA (Protein Interfaces, Surfaces and Assemblies; Krissinel & Henrick, 2007 ▶) predicts that AF1382 is most likely to form a dimer in solution. The formation of a dimer buries 1146 Å2 of the total monomer surface area (the next largest interface buries 99 Å2). The dimer (Fig. 2 ▶ c) is stabilized by hydrophobic interactions between helices αE and αE* (where * denotes a symmetry-related monomer), three pairs of hydrogen bonds (Lys11 NZ⋯Lys7* O, 2.77 Å; Lys11 N⋯Leu9* O, 2.77 Å; Ser86 N⋯Gln83 OE1*, 2.75 Å) and one pair of salt bridges (Lys76 NZ–Asp94* OD1, 3.01 Å). However, the PISA complex significance score (CSS) was only 0.451. This may suggest that the dimer interface observed in the crystal structure may be a crystal-packing artifact.
A search for proteins with a similar structure to AF1382 using DALI (Holm & Sander, 1998 ▶) revealed that the wHTH AF1382 fold was very common, giving over 900 hits with a Z score greater than 2. The top ten nonredundant DALI hits are listed in Table 2 ▶. The top hit, PDB entry 1zar (Fig. 3 ▶ a), corresponds to the N-terminal wHTH DNA-binding domain of Rio2 kinase (LaRonde-LeBlanc & Wlodawer, 2004 ▶). Rio2 has been shown to be important for late 18S rRNA processing (LaRonde-LeBlanc & Wlodawer, 2005 ▶; Granneman et al., 2010 ▶). The other hits all show sequence similarity to HTH MarR-type DNA-binding domains (Magrane, 2011 ▶). The functions of three of these proteins (NNB1585, MepR and SarZ) have been experimentally verified. NNB1585 from Neisseria meningitides (PDB entry 3g3z) is a MarR-family transcriptional regulator that has been shown to act as an auto-repressor (Nichols et al., 2009 ▶), MepR from Staphylococcus aureus (PDB entry 3eco) is a transcription regulator that represses the multidrug efflux system (Kumaraswami et al., 2009 ▶), and SarZ from S. aureus (PDB entry 3hrm) belongs to the OhrR–MgrA class of proteins that play key roles in bacterial virulence and oxidative resistance (Poor et al., 2009 ▶).
Table 2. Top ten DALI hits.
| PDB code | Chain | Z | R.m.s.d. (Å) | Length aligned | No. of residues | Identity (%) | PDB description |
|---|---|---|---|---|---|---|---|
| 3ov8 | A | 21.2 | 0.0 | 91 | 91 | 100 | Protein AF_1382 |
| 1zar | C | 12.8 | 2.5 | 82 | 267 | 13 | Rio2 kinase† |
| 3eco | A | 12.7 | 1.6 | 89 | 135 | 21 | MepR‡ |
| 2obp | A | 12.7 | 1.8 | 78 | 81 | 17 | Putative DNA-binding protein |
| 3s2w | A | 12.7 | 2.1 | 87 | 146 | 17 | MarR-family transcriptional regulator |
| 3e6m | C | 12.6 | 1.8 | 87 | 146 | 14 | MarR-family transcriptional regulator |
| 3oop | A | 12.5 | 1.6 | 86 | 139 | 17 | Lin 2960 protein |
| 3nrv | C | 12.5 | 2.2 | 87 | 142 | 22 | Putative MarR-family transcriptional regulator |
| 3g3z | B | 12.5 | 2.0 | 87 | 142 | 16 | MarR-family transcriptional regulator§ |
| 3cdh | B | 12.2 | 1.7 | 86 | 136 | 15 | MarR-family transcriptional regulator |
| 3hrm | B | 12.2 | 2.1 | 87 | 134 | 20 | HTH-type transcriptional regulator SarZ¶ |
Figure 3.

Superposition of AF1382 with DALI hits. (a) Superposition (Meng et al., 2006 ▶) of the AF1382 structure (white) onto the structure of the Rio2 kinase (PDB entry 1zar) from A. fulgidus (magenta), which was the top hit in the DALI search. (b) Superposition (Chimera) of residues 11–94 of the AF1382 structure (white) with DALI hits 1zar (magenta), 3eco (cyan), 2obp (green), 3oop (blue), 3nrv (light blue), 3g3z (yellow) and 3hrm (red). The orientation is similar to that in Fig. 2 ▶(a).
The remaining DALI hits are annotated as structural genomics targets whose function has not been experimentally verified. These include putative MarR-family transcriptional regulators from Methanosarcina mazei Go1 (PDB entry 3s2w; Midwest Center for Structural Genomics, unpublished work), Silicibacter pomeroyi DSS (PDB entry 3e6m; Midwest Center for Structural Genomics, unpublished work) and Acinetobacter sp. ADP1 (PDB entry 3nrv; Midwest Center for Structural Genomics, unpublished work), the putative DNA-binding protein YP_298295.1 from Ralstonia eutropha (PDB entry 2obp; Joint Center for Structural Genomics, unpublished work) and a protein of unknown function from Listeria innocua strain Clip11262 (PDB entry 3oop; Midwest Center for Structural Genomics, unpublished work). Fig. 3 ▶(b) shows a superposition of the AF1382 structure (residues 11–94) onto the structures 1zar, 3eco, 2obp, 3oop, 3nrv, 3g3z and 3hrm.
3.4. Putative interactions with DNA
The winged-helix motif is associated with many DNA-binding proteins, in which the third helix of the wHTH motif generally serves as the DNA-recognition helix. The recognition helix inserts into the major groove of the DNA, while the wing generally interacts with bases in the minor groove. A display of the surface electrostatic potential (McNicholas et al., 2011 ▶) of AF1382 is shown in Fig. 4 ▶(a). Residues in both the wing and the N-terminus of AF1382 are observed to carry a positive charge, which should promote DNA interaction. A tentative model of the AF1382 protein–DNA complex is shown in Fig. 4 ▶(b). The top panel shows the PISA-predicted AF1382 dimer and the bottom panel shows the model of the AF1382–DNA complex in a similar orientation (bottom panel). The model of the protein–DNA complex was generated using the Chimera MatchMaker tool (Meng et al., 2006 ▶) to superimpose the AF1382 monomer onto each of the Bacillus subtilis replication terminator protein (RTP) monomers observed in the structure of the complex between RTP and a 21-base-pair double-stranded DNA fragment (Wilce et al., 2001 ▶; PDB entry 1f4k). An initial DALI superposition of the two structures gave an r.m.s. deviation (on 84 Cα atoms) of 2.5 Å, which is similar to the top DALI hit (PDB entry 1zar) in Table 2 ▶. However, unlike DALI, the Chimera MatchMaker tool uses residue similarity, secondary structure and gap penalties to aid in superposition. This approach has been shown to be very useful for the superposition of more distantly related proteins such as AF1382 and RTP, which show only 10% sequence identity (Meng et al., 2006 ▶). The MatchMaker alignment gave an r.m.s. deviation of 1.9 Å (based on 43 Cα pairs) and shows that AF1382 residues Leu9*, Lys11*, Tyr32, His44, Val47, Gln83*, Lys85 and Asp89 in AF1382 are conserved in RTP (the residues marked with an asterisk are involved in dimer formation). The alignment also shows that with some minor repositioning of helices αB, αC and αE a significant improvement in the superposition can be achieved. The DNA-recognition helix αD and the β2–β3 wing showed the best overlap between the two structures. A MatchMaker superposition using only the 43-residue AF1382 winged-HTH motif (residues 31–74) and the RTP structure gave an r.m.s. deviation of 1.6 Å (based on 30 Cα pairs), with helix αD and the β2–β3 wing again having the best overlap.
Figure 4.

Putative AF1382–DNA interactions. (a) An electrostatic potential surface representation of the AF1382 dimer predicted by PISA (blue, positive; red, negative) generated by CCP4mg. (b) A ribbon diagram showing a possible mode of protein–DNA interaction. Top, a view of the PISA-predicted AF1382 dimer oriented similarly to that found in the model AF1382–DNA complex shown below. The DNA-recognition helices are highlighted in orange. Bottom, a putative model of the AF1382–DNA complex based on the crystal structure (PDB entry 1f4k) of RTP complexed with DNA (Wilce et al., 2001 ▶). The model was generated by superimposing (Meng et al., 2006 ▶) two AF1382 monomers onto the two RTP monomers found in the RTP–DNA complex.
The RTP-based model correctly places the recognition helix (αD) in the major grove of the DNA, with the positively charged β-strands of the wing and the N-terminus interacting with the ribose-phosphate backbone of the DNA. Compared with the PISA-predicted AF1382 dimer (Fig. 4 ▶ b, upper panel), generation of the DNA-bound AF1382 dimer would require some rotation and translation of the two AF1382 monomers to correctly orient the αD helices for DNA binding.
4. Conclusions
The structure of the ORFan protein AF1382 from A. fulgidus has been determined by S-SAD and refined to 1.85 Å resolution. The AF1382 structure is consistent in terms of surface electrostatics and fold with the structures of other wHTH proteins, which would support its annotation as a putative DNA-binding protein. However, it is difficult to predict based on structure alone if and how AF1382 binds to DNA, since the functional studies required lie outside the scope of the SECSG structural genomics project.
The structure was determined by S-SAD from one crystal, which diffracted to 2.3 Å resolution. A recent analysis of the PDB (65 confirmed de novo S-SAD structures) showed that the average data resolution for successful de novo S-SAD structure determination was 1.81 Å (Rose et al., 2011 ▶). Two common methods (the use of longer wavelength X-rays and the collection of a highly redundant data set) were employed to enhance the anomalous scattering S/N in the SAD data. The use of longer wavelength X-rays enhances the strength of the sulfur anomalous scattering signal (as reflected by the wavelength-dependent Δf′′). However, beam stability at longer wavelengths and absorption effects can introduce error (noise) into the experiment, preventing S-SAD phasing success. Thus, one must be able to identify and address these problems.
In the study reported here, two 360° data sets were merged together to increase data redundancy and thus improve the anomalous scattering signal in the resulting data set. However, highly redundant data sets can suffer from radiation damage. Thus, one must carefully monitor the data collection to identify and mitigate radiation damage if present. Finally, the data were collected without the aid of a data-collection strategy, which would ensure that the unique data are collected first. Since each data set covered a complete rotation of the crystal in the X-ray beam and analysis of the resulting data did not show signs of radiation damage, the lack of a data-collection strategy in this case had little effect on the structure determination. However, in cases where beam time is limited or radiation damage is suspected, the use of data-collection strategy programs such as BEST (Bourenkov & Popov, 2006 ▶) to design and optimize the experiment is highly recommended.
The structure determination also greatly benefited from a highly automated synchrotron beamline (SER-CAT 22-ID) and rapid on-site structure solution. The analysis also points out the advantage gained from carrying out data reduction and structure determination on-site while the crystal is still available for further data collection. The study used the SGXPro structure-determination pipeline to quickly judge phasing success. However, other high-throughput pipelines such as Auto-Rickshaw (Panjikar et al., 2009 ▶), HKL-3000 (Minor et al., 2006 ▶) and PHENIX (Adams et al., 2002 ▶) are also available for rapid SAD structure determination.
Finally, it should be noted that this study was part of an ongoing PSI-1 structural genomics project in 2007 that was focused on developing methods and technology for high-throughput structure determination to search for novel protein folds. Thus, once the structure had been determined, its fold classified and the coordinates and structure factors deposited in the PDB, the focus shifted to the next target in the pipeline. Given this limitation, further analyses such as functional studies lies outside of the scope of the SECSG project, which was aimed at solving protein structures with least possible effort.
Atomic coordinates and structure factors for the AF1382 structure have been deposited in the Protein Data Bank under accession codes 3o3k (the 2.3 Å resolution AF1382 structure fitted and refined against sulfur SAD data) and 3ov8 (the refined 1.85 Å high-resolution structure).
Supplementary Material
PDB reference: 2.3 Å resolution AF1382 structure fitted and refined against sulfur SAD data, 3o3k
PDB reference: the refined 1.85 Å high-resolution AF1382 structure, 3ov8
Acknowledgments
This work was supported in part by funds from the National Institutes of Health (GM62407), The Georgia Research Alliance and the University of Georgia Research Foundation. Data were collected on the Southeast Regional Collaborative Access Team (SER-CAT) 22-ID beamline at the Advanced Photon Source, Argonne National Laboratory. Supporting institutions may be found at http://www.ser-cat.org/members.html. Use of the Advanced Photon Source was supported by the US Department of Energy, Office of Science, Office of Basic Energy Sciences under Contract No. W-31-109-Eng-38.
References
- Adams, M. W. W., Dailey, H. A., DeLucas, L. J., Luo, M., Prestegard, J. H., Rose, J. P. & Wang, B.-C. (2003). Acc. Chem. Res. 36, 191–198. [DOI] [PubMed]
- Adams, P. D., Grosse-Kunstleve, R. W., Hung, L.-W., Ioerger, T. R., McCoy, A. J., Moriarty, N. W., Read, R. J., Sacchettini, J. C., Sauter, N. K. & Terwilliger, T. C. (2002). Acta Cryst. D58, 1948–1954. [DOI] [PubMed]
- Altschul, S. F., Gish, W., Miller, W., Myers, E. W. & Lipman, D. J. (1990). J. Mol. Biol. 215, 403–410. [DOI] [PubMed]
- Aravind, L., Anantharaman, V., Balaji, S., Babu, M. M. & Iyer, L. M. (2005). FEMS Microbiol. Rev. 29, 231–262. [DOI] [PubMed]
- Bateman, A., Coin, L., Durbin, R., Finn, R. D., Hollich, V., Griffiths-Jones, S., Khanna, A., Marshall, M., Moxon, S., Sonnhammer, E. L., Studholme, D. J., Yeats, C. & Eddy, S. R. (2004). Nucleic Acids Res. 32, D138–D141. [DOI] [PMC free article] [PubMed]
- Berman, H. M., Westbrook, J., Feng, Z., Gilliland, G., Bhat, T. N., Weissig, H., Shindyalov, I. N. & Bourne, P. E. (2000). Nucleic Acids Res. 28, 235–242. [DOI] [PMC free article] [PubMed]
- Bourenkov, G. P. & Popov, A. N. (2006). Acta Cryst. D62, 58–64. [DOI] [PubMed]
- Brunger, A. T. (2007). Nature Protoc. 2, 2728–2733. [DOI] [PubMed]
- Brünger, A. T., Adams, P. D., Clore, G. M., DeLano, W. L., Gros, P., Grosse-Kunstleve, R. W., Jiang, J.-S., Kuszewski, J., Nilges, M., Pannu, N. S., Read, R. J., Rice, L. M., Simonson, T. & Warren, G. L. (1998). Acta Cryst. D54, 905–921. [DOI] [PubMed]
- Chayen, N. E., Cianci, M., Olczak, A., Raftery, J., Rizkallah, P. J., Zagalsky, P. F. & Helliwell, J. R. (2000). Acta Cryst. D56, 1064–1066. [DOI] [PubMed]
- Chayen, N. E., Shaw Stewart, P. D., Maeder, D. L. & Blow, D. M. (1990). J. Appl. Cryst. 23, 297–302.
- Chen, C.-J., Wang, B.-C., Liu, Z.-J., Rose, J. P., Rosenbaum, G., Ma, K., Schiffer, M., Pokkulur, P. R., Joachimiak, A., Zhang, R., Howard, A., Chrzas, J. & Robins, A. H. (2000). Am. Crystallogr. Assoc. Annu. Meet., p. 76, Abstract 10.01.05.
- Cianci, M., Helliwell, J. R. & Suzuki, A. (2008). Acta Cryst. D64, 1196–1209. [DOI] [PubMed]
- Cork, C., O’Neill, J., Taylor, J. & Earnest, T. (2006). Acta Cryst. D62, 852–858. [DOI] [PubMed]
- Dauter, Z. & Adamiak, D. A. (2001). Acta Cryst. D57, 990–995. [DOI] [PubMed]
- Davis, I. W., Leaver-Fay, A., Chen, V. B., Block, J. N., Kapral, G. J., Wang, X., Murray, L. W., Arendall, W. B. III, Snoeyink, J., Richardson, J. S. & Richardson, D. C. (2007). Nucleic Acids Res. 35, W375–W383. [DOI] [PMC free article] [PubMed]
- Debreczeni, J. É., Bunkóczi, G., Girmann, B. & Sheldrick, G. M. (2003). Acta Cryst. D59, 393–395. [DOI] [PubMed]
- Diederichs, K. & Karplus, P. A. (1997). Nature Struct. Biol. 4, 269–275. [DOI] [PubMed]
- Emsley, P. & Cowtan, K. (2004). Acta Cryst. D60, 2126–2132. [DOI] [PubMed]
- Finn, R. D., Tate, J., Mistry, J., Coggill, P. C., Sammut, S. J., Hotz, H. R., Ceric, G., Forslund, K., Eddy, S. R., Sonnhammer, E. L. & Bateman, A. (2008). Nucleic Acids Res. 36, D281–D288. [DOI] [PMC free article] [PubMed]
- Fu, Z.-Q., Rose, J. P. & Wang, B.-C. (2004). Acta Cryst. D60, 499–506. [DOI] [PubMed]
- Fu, Z.-Q., Rose, J. & Wang, B.-C. (2005). Acta Cryst. D61, 951–959. [DOI] [PubMed]
- Gough, J., Karplus, K., Hughey, R. & Chothia, C. (2001). J. Mol. Biol. 313, 903–919. [DOI] [PubMed]
- Granneman, S., Petfalski, E., Swiatkowska, A. & Tollervey, D. (2010). EMBO J. 29, 2026–2036. [DOI] [PMC free article] [PubMed]
- Harp, J. M., Timm, D. E. & Bunick, G. J. (1998). Acta Cryst. D54, 622–628. [DOI] [PubMed]
- Haspel, J., Blanco, C., Jacob, J. & Grumet, M. (2001). Biotechniques, 30, 60–66. [DOI] [PubMed]
- Hendrickson, W. A. & Teeter, M. M. (1981). Nature (London), 290, 107–113. [DOI] [PMC free article] [PubMed]
- Holm, L. & Sander, C. (1998). Nucleic Acids Res. 26, 316–319. [DOI] [PMC free article] [PubMed]
- Krissinel, E. & Henrick, K. (2007). J. Mol. Biol. 372, 774–797. [DOI] [PubMed]
- Kumaraswami, M., Schuman, J. T., Seo, S. M., Kaatz, G. W. & Brennan, R. G. (2009). Nucleic Acids Res. 37, 1211–1224. [DOI] [PMC free article] [PubMed]
- LaRonde-LeBlanc, N. & Wlodawer, A. (2004). Structure, 12, 1585–1594. [DOI] [PubMed]
- LaRonde-LeBlanc, N. & Wlodawer, A. (2005). Biochim. Biophys. Acta, 1754, 14–24. [DOI] [PubMed]
- Laskowski, R. A. (2009). Nucleic Acids Res. 37, D355–D359. [DOI] [PMC free article] [PubMed]
- Laskowski, R. A., MacArthur, M. W., Moss, D. S. & Thornton, J. M. (1993). J. Appl. Cryst. 26, 283–291.
- Li, Y. et al. (2008). Proteins, 71, 2109–2113. [DOI] [PubMed]
- Liu, Z.-J. et al. (2005). Acta Cryst. D61, 679–684.
- Liu, Z.-J., Vysotski, E. S., Chen, C. J., Rose, J. P., Lee, J. & Wang, B.-C. (2000). Protein Sci. 9, 2085–2093. [DOI] [PMC free article] [PubMed]
- Magrane, M. (2011). Database, 2011, bar009. [DOI] [PMC free article] [PubMed]
- Matthews, B. W. (1968). J. Mol. Biol. 33, 491–497. [DOI] [PubMed]
- McNicholas, S., Potterton, E., Wilson, K. S. & Noble, M. E. M. (2011). Acta Cryst. D67, 386–394. [DOI] [PMC free article] [PubMed]
- Meng, E. C., Pettersen, E. F., Couch, G. S., Huang, C. C. & Ferrin, T. E. (2006). BMC Bioinformatics, 7, 339. [DOI] [PMC free article] [PubMed]
- Minor, W., Cymborowski, M., Otwinowski, Z. & Chruszcz, M. (2006). Acta Cryst. D62, 859–866. [DOI] [PubMed]
- Mueller-Dieckmann, C., Panjikar, S., Schmidt, A., Mueller, S., Kuper, J., Geerlof, A., Wilmanns, M., Singh, R. K., Tucker, P. A. & Weiss, M. S. (2007). Acta Cryst. D63, 366–380. [DOI] [PubMed]
- Mueller-Dieckmann, C., Panjikar, S., Tucker, P. A. & Weiss, M. S. (2005). Acta Cryst. D61, 1263–1272. [DOI] [PubMed]
- Mueller-Dieckmann, C., Polentarutti, M., Djinovic Carugo, K., Panjikar, S., Tucker, P. A. & Weiss, M. S. (2004). Acta Cryst. D60, 28–38. [DOI] [PubMed]
- Nichols, C. E., Sainsbury, S., Ren, J., Walter, T. S., Verma, A., Stammers, D. K., Saunders, N. J. & Owens, R. J. (2009). Acta Cryst. F65, 204–209. [DOI] [PMC free article] [PubMed]
- Norvell, J. C. & Machalek, A. Z. (2000). Nature Struct. Biol. 7, 931. [DOI] [PubMed]
- Olczak, A., Cianci, M., Hao, Q., Rizkallah, P. J., Raftery, J. & Helliwell, J. R. (2003). Acta Cryst. A59, 327–334. [DOI] [PubMed]
- Otwinowski, Z. & Minor, W. (1997). Methods Enzymol. 276, 307–326. [DOI] [PubMed]
- Panjikar, S., Parthasarathy, V., Lamzin, V. S., Weiss, M. S. & Tucker, P. A. (2009). Acta Cryst. D65, 1089–1097. [DOI] [PMC free article] [PubMed]
- Pettersen, E. F., Goddard, T. D., Huang, C. C., Couch, G. S., Greenblatt, D. M., Meng, E. C. & Ferrin, T. E. (2004). J. Comput. Chem. 25, 1605–1612. [DOI] [PubMed]
- Poor, C. B., Chen, P. R., Duguid, E., Rice, P. A. & He, C. (2009). J. Biol. Chem. 284, 23517–23524. [DOI] [PMC free article] [PubMed]
- Ramagopal, U. A., Dauter, M. & Dauter, Z. (2003). Acta Cryst. D59, 1020–1027. [DOI] [PubMed]
- Rose, J. P., Liu, Z- J., Temple, W., Chen, L., Lee, D., Newton, M. G. & Wang, B.-C. (2004). Rigaku J. 21, 1–9.
- Rose, J. P., Rasheed, Y., Zhang, H., Weiss, M. S. & Wang, B.-C. (2011). Am. Crystallogr. Assoc. Annu. Meet., Abstract P02.01.
- Sarma, G. N. & Karplus, P. A. (2006). Acta Cryst. D62, 707–716. [DOI] [PubMed]
- Shah, A. K., Liu, Z.-J., Stewart, P. D., Schubot, F. D., Rose, J. P., Newton, M. G. & Wang, B.-C. (2005). Acta Cryst. D61, 123–129. [DOI] [PubMed]
- Sheldrick, G. M. (2010). Acta Cryst. D66, 479–485. [DOI] [PMC free article] [PubMed]
- Siew, N. & Fischer, D. (2004). J. Mol. Biol. 342, 369–373. [DOI] [PubMed]
- Teng, T.-Y. (1990). J. Appl. Cryst. 23, 387–391.
- Terwilliger, T. C. (2003). Methods Enzymol. 374, 22–37. [DOI] [PubMed]
- Usón, I. & Sheldrick, G. M. (1999). Curr. Opin. Struct. Biol. 9, 643–648. [DOI] [PubMed]
- Wang, B.-C. (1985). Methods Enzymol. 115, 90–112. [DOI] [PubMed]
- Wang, B.-C. et al. (2005). J. Struct. Funct. Genomics, 6, 233–243. [DOI] [PubMed]
- Weik, M., Ravelli, R. B., Kryger, G., McSweeney, S., Raves, M. L., Harel, M., Gros, P., Silman, I., Kroon, J. & Sussman, J. L. (2000). Proc. Natl Acad. Sci. USA, 97, 623–628. [DOI] [PMC free article] [PubMed]
- Weiss, M. S. & Hilgenfeld, R. (1997). J. Appl. Cryst. 30, 203–205.
- Weiss, M. S., Mander, G., Hedderich, R., Diederichs, K., Ermler, U. & Warkentin, E. (2004). Acta Cryst. D60, 686–695. [DOI] [PubMed]
- Weiss, M. S., Panjikar, S., Mueller-Dieckmann, C. & Tucker, P. A. (2005). J. Synchrotron Rad. 12, 304–309. [DOI] [PubMed]
- Weiss, M. S., Sicker, T., Djinovic-Carugo, K. & Hilgenfeld, R. (2001). Acta Cryst. D57, 689–695. [DOI] [PubMed]
- Wilce, J. A., Vivian, J. P., Hastings, A. F., Otting, G., Folmer, R. H., Duggin, I. G., Wake, R. G. & Wilce, M. C. (2001). Nature Struct. Biol. 8, 206–210. [DOI] [PubMed]
- Wu, C.-K., Dailey, T. A., Dailey, H. A., Francavilla, A., Starzl, T. E., Wang, B.-C. & Rose, J. P. (2000). Protein Pept. Lett. 7, 25–32.
- Zhu, J. (2008). Dissertation. University of Georgia, Athens, Georgia, USA.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PDB reference: 2.3 Å resolution AF1382 structure fitted and refined against sulfur SAD data, 3o3k
PDB reference: the refined 1.85 Å high-resolution AF1382 structure, 3ov8