

Protecting groups play major roles in carbohydrate chemistry serving, in addition to blocking hydroxyl groups, as modulators of reactivity and as stereodirecting groups in glycosylation.[1] Cyclic protecting groups are prominent in the latter respect and are of current interest.[2] For example, it has been demonstrated that a 2N,3O-oxazoldinone group both strongly favors α- over β-glycosides in the glucosaminopyranosides, i.e, enhances the anomeric effect, and facilitates anomerization.[3] In the sialic acids, a 4O,5N-oxazolidinone, both with and without an additional N-acetyl group, favors the formation of α-O-,[4] C-,[5] and S-sialosides,[6] and presents the best solution currently available to the challenging problem of α-sialoside formation. The mechanism by which the oxazolidinone group directs stereoselective sialidation is therefore of considerable interest and prompted the efforts described herein.
We first investigated the influence of the oxazolidinone ring on the anomeric effect. Studies on the mutarotation of sialosyl hemiacetals were thwarted by the discovery that the oxazolidinone protected systems exist very significantly in the open chain keto form, reflecting the strain imposed on the cyclic form by the presence of the trans-fused oxazolidinone moiety. The equilibration of actual glycosides under acidic conditions,[7] was ruled out owing to the lability of the N-acetyloxazolidinone system. We turned to the persistent radical effect (PRE) for an alternative means of equilibration at the anomeric center avoiding any ring-opened species.[8] Anomeric radical reactions are well-known under kinetic conditions when they give rise to axially quenched products,[9] with the notable exception of the sialic acids when poor selectivity is found.[10] Conversely, the equilibration of anomeric stereochemistry via radical intermediates is rare[11] and has not been applied explicitly to the study of the anomeric effect.
Attempted synthesis of sialosyl glycosides of the persistent radical 2,2,6,6-tetramethylpiperidine N-oxide (TEMPO) were fruitless and we focused on radical approaches to the desired glycosides. The known S-sialosyl xanthate 1[12] was photolyzed in dichloroethane at room temperature in the presence of TEMPO (20 equiv) leading to a 69% yield of the desired TEMPO glycosides 2 as a separable 1:2 α,β-anomeric mixture (Scheme 1). Resubjecting this mixture to the photolysis conditions did not result in a change in this ratio indicating its kinetic origin. Attempted equilibration of the xanthate itself, by photolysis alone, gave only the glycal 3 in a process reminiscent of the photolytic elimination of thionobenzoate esters (Scheme 1).[13]
Scheme 1.
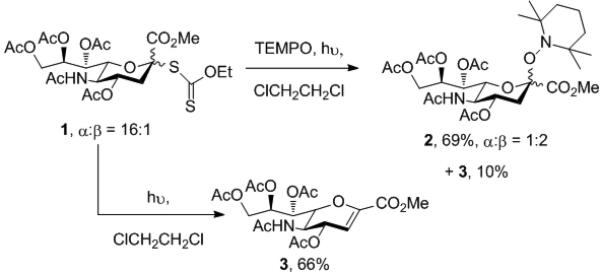
Preparation of the TEMPO sialoside 2
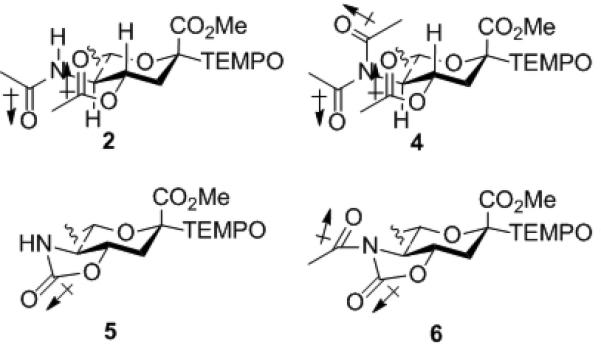
A series of standard manipulations of 2 yielded the variously protected TEMPO sialosides 4-6 as described in the Supporting Information. Dilute solutions (0.05-0.1 M) of the various TEMPO sialosides were heated under nitrogen at 90 °C in ClCD2CD2Cl with periodic monitoring by NMR spectroscopy until equilibrium was attained (Scheme 3).[14] In this process equilibration takes place by homolytic scission of the anomeric-TEMPO C-O bond to give the captodatively stabilized anomeric radical and the persistent radical TEMPO. Because of the PRE radical combination takes place to give the hetero- rather than any homodimers, enabling scrambling at the anomeric center. Because of spin delocalization onto the ester group the anomeric radical is considered to be best represented as a planar sp2 hybridized carbon, rather than as the rapidly inverting sp3 hydribized species found for simple anomeric radicals. In reality the exact nature of the intermediate anomeric radical is of no consequence in this thermodynamically controlled process.
Scheme 3.
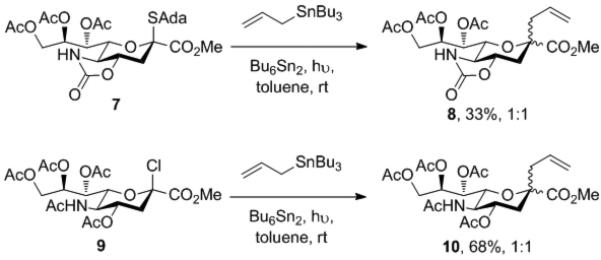
Lack of influence of the oxazolidinone on the kinetic selectivity of the 2-sialyl radical
From the equilibrium ratios obtained (Table 1) it is apparent that the protecting group array at O4 and N5 influences the magnitude of the anomeric effect.
Table 1.
Anomeric equilibrium ratios for TEMPO sialosides[a]
| Entry | Compound | α:β ratio |
|---|---|---|
| 1 |
|
3.1±0.2:1 |
| 2 |
|
3.9±0.2:1 |
| 3 |
|
7.0±0.2:1 |
| 4 |
|
6.2±0.2:1 |
Equilibrium ratios were the same within experimental error irrespective of the anomer used as substrate.
In all cases studied, the α-glycoside in which the bulky TEMPO adopts the equatorial position is preferred (Table 1). This is presumably for steric reasons as in the simple methyl glycoside of N-acetylneuraminic acid methyl ester the β-isomer, with the axial glycosidic bond, is reportedly very highly favoured[7] and avoids 1,3-diaxial interactions with the bulky carboxylate and H's 4 and 6. While steric reasons obviously underlie the overall preference for the α-isomers in the TEMPO sialosides, they do not explain the changes in the anomeric ratios observed in the series of compounds presented in Table 1 according to the nature of the protecting group employed. We suggest that they correlate best with the electron-withdrawing ability of the 4O,5N-protecting group array as manifest in its dipole moment with respect to acyclic equivalents (Fig 1).[15]
Figure 1.
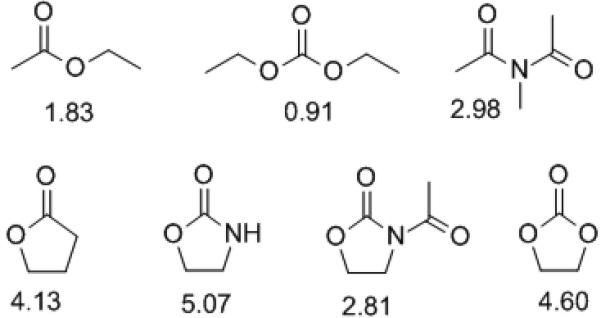
Dipole moments (Debye units) of open chain and cyclic carbonyl functionalities in non-polar solvents
Among the protecting systems employed the greatest stabilization of the α-anomer is obtained with the simple 4O,5N-oxazolidinone which has the highest dipole moment (Table 1, entry 3, and Fig 1). The second highest selectivity in favour of the α-anomer is obtained with the N-acetyloxazoldinone protecting system consistent with this protecting array also having a high dipole moment (Table 1, entry 4 and Fig 1). In both the oxazolidinone 5 and N-acetyloxazolidinone 6 systems the overall dipole moment of the heterocyclic system is aligned parallel with the pyranose C4-O4 and C5-N5 bonds, thereby enhancing their inherent electron-withdrawing ability (Fig 2). This contrasts with the situation in the simple amide 2[7], where the carbonyl dipole is aligned syn-coplanar with the C5-N5 bond (the result of the trans-amide conformation and the approximately 180° H5-C5-N5-NH torsion angle with 3JNH,H5 = 10 Hz). In the N-acetyl system 6 the dipole moment of the heterocyclic system is moderated by the presence of the N-acetyl group, which preferentially adopts an antiperiplanar conformation with respect to the oxazolidinone carbonyl bond as is known for imides in general and as is seen in X-ray crystallographic analyses of derivatives of 6,[4c] thereby moderating the influence of the oxazolidinone.
Figure 2.
Orientations of the key O4 and N5 protecting group carbonyl dipole moments in the TEMPO sialosides (Side chains omitted for clarity)
To probe the influence of the oxazolidinone ring on the kinetic selectivity of the 2-sialyl radical, the thioglycoside 7[4d] was photolyzed at room temperature in toluene in the presence of hexabutyldistannane and allyl tributylstannane,[16] leading to a 33% yield of a 1:1 mixture of the known[5] C-glycosides 8 (Scheme 3). This lack of selectivity mirrors that observed on reaction of the sialyl chloride 9 with allyl tributylstannane under similar conditions (Scheme 3).[10] Clearly, the oxazolidinone ring has no significant influence on the face selectivity of the 2-sialyl radical under kinetic conditions, i.e, the presence of the oxazolidinone does not bias the pyranose ring toward reaction on either one of its two faces.
As there is no reason to consider the putative 2-sialyl oxocarbenium ion 11 to adopt a different conformation to that of the 2-sialyl radical 12 in the presence of the conformationally locking oxazolidinone ring (Fig 3), and as the reactions of glycosyl oxocarbenium ions are exothermic with early transition states,[17] we deduce that reactions of the oxocarbenium ion 11 would show a similar lack of face selectivity to those of the radical 12.
Figure 3.
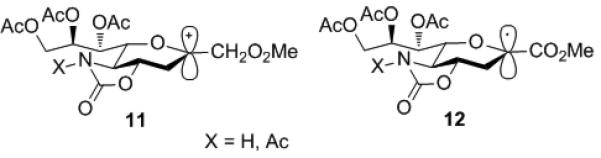
Conformationally related 2-sialyl oxocarbenium ion 11 and radical 12
Indeed, it is apparent from consideration of the only reasonable conformations available to oxonium ion 11, which approximate the 4H5 half chair conformer, that significant selectivity in its reactions with nucleophiles is unlikely. Thus, neither face of 11 appears to be significantly more shielded than the other. Moreover, attack of a nucleophile on the α-face of 11 provides the α-product 13 in the 4S2 twist boat conformation, whereas attack on the β-face affords the β-product 14 directly in the 2C5 chair (Scheme 4).
Scheme 4.
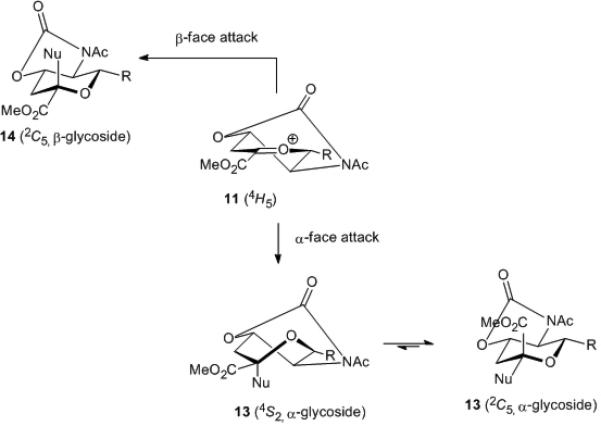
Conformational analysis of nucleophilic attack on the putative glycosyl oxocarbenium ion 11
To probe the influence of protecting roups on the energetics of sialyl oxocarbenium ions we turned to mass spectrometry and an investigation of the threshold energy required for the fragmentation of a suitable anomeric derivative as a function of protecting group. The use of threshold fragmentation energies has been previously identified by Denekmap and Sandlers as a useful means of assessing the influence of protecting groups on the stability of glycosyl oxocarbenium ions, albeit not in the sialic acid series.[18]
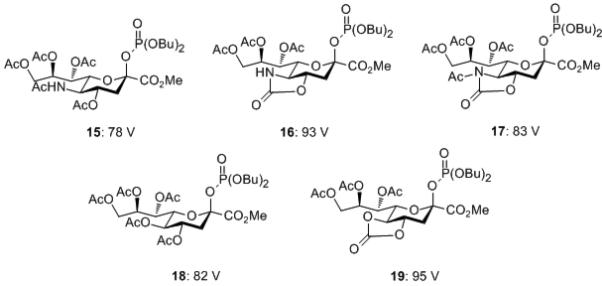
With the thioglycosides the loss of an acetoxy group was the primary fragmentation pathway; therefore, we turned to the glycosyl phosphates introduced by the Wong group,[4f] and 15-17 were synthesized (see Supporting Information) following the Wong methodology. We also synthesized the 2-keto-3-deoxy-D-glycero-D-galactonulosonic acid (KDN) phosphates 18 and 19 (see Supporting Information) as we have previously demonstrated[4j] that the 4,5-O-cyclic carbonate in the KDN series serves the same stereodirecting purpose as the oxazolidinone in neuraminic acid. All sialyl phosphates were isolated as single β-anomers, with the exception of 18 which was obtained as a mixture highly enriched in the β-isomer, and their anomeric stereochemistry was confirmed by the the 3JH3ax-CO2 coupling constants.[19]
Methanolic solutions of phosphates 15-19 were examined by ESI mass spectrometry. At the standard cone voltage of 40 volts, each substance exhibited a clean sodiated molecular ion and the absence of any fragment ions from loss of the phosphate group. The cone voltage was then increased incrementaly until the onset of fragmentation leading to the values reported in Figure 4. In each case the fragment ion observed resulted from the overall loss of dibutyl phosphoric acid from the molecular ion, and was typically accompanied by peaks arising from the overall replacement of dibutyl phosphate by methanol or water, namely “glycosylation“ or “hydrolysis“ by the infusion solvent.
Figure 4.
Onset cone voltages for fragmentation of sialyl phosphates
We interpret the observed fragmentations in terms of expulsion of the dibutyl phosphate anion to give the corresponding sialyl oxocarbenium ion followed by deprotonation to afford the observed fragment ion (Scheme 5, path a). Alternatively, a one-step pericyclic syn-elimination (McLafferty rearrangement) leads directly to the fragment ion (Scheme 5, path b). As such a concerted mechanism is likely to be asynchronous, and to involve a transition state with significant positive charge on the anomeric carbon, the main difference between Scheme 7 paths a and b for the purposes of this experiment is simply one of the differing extents of positive charge build-up on the anomeric carbon. As the appearance of the fragment ion is coincident with that of the “glycosylation“ and “hydrolysis“ products, which necessarily arise from the oxocarbenium ion, we consider path a (Scheme 5) to be the more likely of the two alternatives.
Scheme 5.
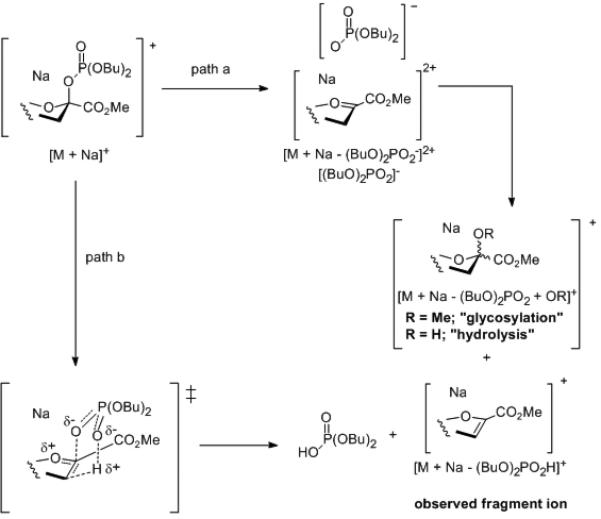
Mass spectral fragmentation and adduct formation
A greater cone voltage is required to fragment a molecular ion carrying a cyclic protecting group spanning positions 4 and 5, than one lacking the cyclic protection (Fig 4). Furthermore, the simple oxazolidinone-protected system requires a higher cone voltage for fragmentation than the corresponding N-acetyl oxazolidinone. We interpret these observations in terms of the oxazolidinone and carbonate groups exerting a powerful electron-withdrawing effect that retards the build-up of positive charge at the anomeric center, necessitating the higher cone voltages for the onset of fragmentation. Consistent with the explanation advanced for the influence of the oxazolidinone on the anomeric effect, the powerful electron withdrawing effect of the oxazolidinone and of the cyclic carbonate is seen as a function of their large dipole moment (Fig 1) that is aligned with the carbonyl group in the mean plane of the pyranose ring and which reinforces the inherent electron-withdrawing effect of the C4-O4 and C5-N5/O5 bonds (Fig 5). In contrast, the carbonyl dipole in the O4 ester group and the O5 ester/N5 amide of the systems lacking cyclic protection are roughly orthogonal to the dipoles of the C4-O4 and C5-O5/N5 bonds and do not reinforce their electron-withdrawing effect to the same extent. Again consistent with the explanation advanced for influence on the anomeric effect, the antiperiplanar orientation of the N-acetyl and oxazoldinone dipoles in 17 moderates its electron-withdrawing influence relative to the simple oxazolidinone thereby lowering the mimimum cone voltage required for fragmentation.
Figure 5.
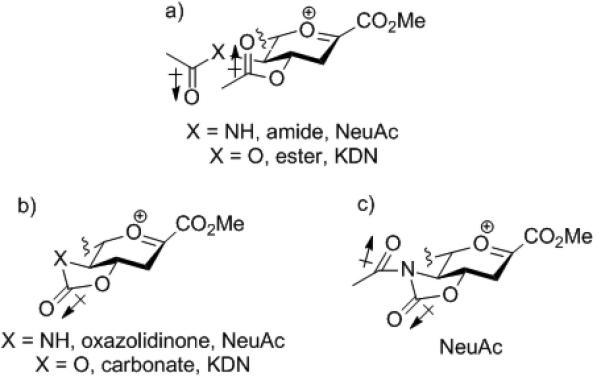
Protecting group dipoles in relations to the oxocarbenium ion: a) acyclic; b) oxazolidinone or carbonate; and c) N-acetyl oxazolidinone protection. Side chains are omitted for clarity.
We conclude that the oxazolidinone and/or carbonate groups are powerfully electron-withdrawing protecting groups when trans-fused to pyranose ring systems by virtue of their strong dipole moments in the mean plane of the ring system. This powerful effect has two consequences. First, equatorial glycosides are stabilized with respect to monocyclic systems by the diminished propensity of the pyranosyl ring oxygen to donate electron density into the σ* orbital of an axial glycosidic bond.[20] Second, the oxazolidinone and carbonate groups exert their highly effective stereodirecting influence by destabilizing the glycosyl oxocarbenium ions and, thus, by suppressing unselective dissociative reaction pathways. In doing so they favor pathways closer to the associative end of the SN1–SN2 mechanistic continuum[21] for glycosylation, i.e., mechanisms involving SN2-like pathways, possibly with so-called exploded transition states with their relatively long partial bonds, or pathways taking place through functionally equivalent contact or tight ion pairs. Possible intermediates in such mechanisms include β-glycosyl nitrilium ions,[22] β-glycosyl triflates, glycosyl sulfoxonium ions,[23] and spirocyclic intermediates arising from participation of the anomeric ester group related to the spirocyclic α-lactones sometimes discussed[24] in relation to the mechanism of sialidase enzymes and Kajihara's proposed spirocyclic imidate.[25] The exact nature of these intermediates will depend on the reaction conditions and the promotion system employed. Although SN2 reactions at tertiary centers are usually considered to be strongly disfavoured,[26] there is considerable stereochemical[27] and kinetic[28] precedent for such mechanisms at tertiary centers adjacent to carboxylate esters.
Experimental Section
Equilibration
A solution of the TEMPO sialoside (0.05-0.1 M) in deuterated dichloroethane in an NMR tube was degassed, filled with nitrogen and heated at 90 °C. With periodic monitoring by 1H NMR spectroscopy, heating was continued until equilibrium was reached. Mass Spectral Fragmentation. A methanolic solution of the phosphate (~ 10 nM) was infused in to the ESIMS source at room temperature and the cone voltage gradually increased from 40 V through the fragmentation threshold.
Supplementary Material
Scheme 2.
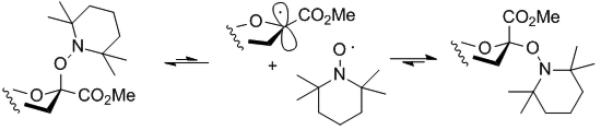
Equilibration via the persistent radical effect
Acknowledgments
We thank the NIH (GM 62160) for support of this work.
Footnotes
Dedicated to Professor W. B. Motherwell, FRS
Supporting information for this article is available on the WWW under http://www.angewandte.org or from the author
References
- 1.a Fraser-Reid B, Lopez C. Top. Curr. Chem. 2011;301:1–30. doi: 10.1007/128_2010_105. [DOI] [PubMed] [Google Scholar]; b Gomez AM. Top. Curr. Chem. 2011;301:31–68. doi: 10.1007/128_2010_112. [DOI] [PubMed] [Google Scholar]; c Shiao TC, Roy R. Top. Curr. Chem. 2011;301:69–108. doi: 10.1007/128_2010_108. [DOI] [PubMed] [Google Scholar]; d Kim KS, Suk D-H. Top. Curr. Chem. 2011;301:109–140. doi: 10.1007/128_2010_107. [DOI] [PubMed] [Google Scholar]; e Aubry S, Sasaki K, Sharma I, Crich D. Top. Curr. Chem. 2011;301:141–188. doi: 10.1007/128_2010_102. [DOI] [PubMed] [Google Scholar]; f Premathilake HD, Demchenko AV. Top. Curr. Chem. 2011;301:189–222. doi: 10.1007/128_2010_106. [DOI] [PMC free article] [PubMed] [Google Scholar]; g Wu C-Y, Wong C-H. Top. Curr. Chem. 2011;301:223–252. doi: 10.1007/128_2010_109. [DOI] [PubMed] [Google Scholar]; h Codée JDC, Christina AE, Walvoort MTC, Overkleeft HS, van der Marel GA. Top. Curr. Chem. 2011;301:253–290. doi: 10.1007/128_2010_111. [DOI] [PubMed] [Google Scholar]
- 2.a Codée JDC, Ali A, Overkleeft HS, van der Marel GA. CR Chimie. 2011;14:178–193. [Google Scholar]; b Litjens REJN, van den Bos LJ, Codée JDC, Overkleeft HS, van der Marel G. Carbohydr. Res. 2007;342:419–429. doi: 10.1016/j.carres.2006.09.002. [DOI] [PubMed] [Google Scholar]
- 3.a Crich D, Vinod AU. J. Org. Chem. 2005;70:1291–1296. doi: 10.1021/jo0482559. [DOI] [PubMed] [Google Scholar]; b Manabe S, Ishii K, Ito Y. J. Am. Chem. Soc. 2006;128:10666–10667. doi: 10.1021/ja062531e. [DOI] [PubMed] [Google Scholar]; c Kerns RJ, Zha C, Benakli K, Liang Y-Z. Tetrahedron Lett. 2003;44:8069–8072. [Google Scholar]; d Olsson JDM, Eriksson L, Lahmann M, Oscarson S. J. Org. Chem. 2008;73:7181–7188. doi: 10.1021/jo800971s. [DOI] [PubMed] [Google Scholar]; e Satoh H, Manabe S, Ito Y, Luthi HP, Laino T, Hutter J. J. Am. Chem. Soc. 2011;133:5610–5619. doi: 10.1021/ja201024a. [DOI] [PubMed] [Google Scholar]
- 4.a Tanaka H, Nishiura Y, Takahashi T. J. Am. Chem. Soc. 2006;128:7124–7125. doi: 10.1021/ja0613613. [DOI] [PubMed] [Google Scholar]; b Farris MD, De Meo C. Tetrahedron Lett. 2007;48:1225–1227. [Google Scholar]; c Crich D, Li W. J. Org. Chem. 2007;72:2387–2391. doi: 10.1021/jo062431r. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Crich D, Li W. J. Org. Chem. 2007;72:7794–7797. doi: 10.1021/jo7012912. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Crich D, Wu B. Org. Lett. 2008;10:4033–4035. doi: 10.1021/ol801548k. [DOI] [PMC free article] [PubMed] [Google Scholar]; f Hsu C-H, Chu K-C, Lin Y-S, Han J-L, Peng Y-S, Ren C-T, Wu C-Y, Wong C-H. Chem. Eur. J. 2010;16:1754–1760. doi: 10.1002/chem.200903035. [DOI] [PubMed] [Google Scholar]; g Chu K-C, Ren C-T, Lu C-P, Hsu C-H, Sun T-H, Han J-L, Pal B, Chao T-A, Lin Y-F, Wu S-H, Wong C-H, Wu C-Y. Angew. Chem. Int. Ed. 2011;50:9391–9395. doi: 10.1002/anie.201101794. [DOI] [PubMed] [Google Scholar]; h Harris BN, Patel PP, Gobble CP, Stark MJ, De Meo C. Eur. J. Org. Chem. 2011:4023–4027. [Google Scholar]; i Wang Y-J, Jia J, Gu Z-Y, Liang F-F, Li R-C, Huang M-H, Xu C-S, Zhang J-X, Men Y, Xing G-W. Carbohydr. Res. 2011;346:1271–1276. doi: 10.1016/j.carres.2011.04.029. [DOI] [PubMed] [Google Scholar]; j Crich D, Navuluri C. Angew. Chem. Int. Ed. 2010;49:3049–3052. doi: 10.1002/anie.200907178. [DOI] [PMC free article] [PubMed] [Google Scholar]; k Gu Z.-y., Zhang J.-x., Guo-wen Xing G.-x. Chem. Asian J. 2012;7:1524–1528. doi: 10.1002/asia.201200172. [DOI] [PubMed] [Google Scholar]
- 5.Noel A, Delpech B, Crich D. Org. Lett. 2012;14:1342–1345. doi: 10.1021/ol300255q. [DOI] [PubMed] [Google Scholar]
- 6.Noel A, Delpech B, Crich D. Org. Lett. 2012;14:4138–4141. doi: 10.1021/ol301779e. [DOI] [PubMed] [Google Scholar]
- 7.a Yu PK, Ledeen R. J. Biol. Chem. 1969;244:1306–1313. [PubMed] [Google Scholar]; b Kuhn R, Lutz P, Macdonald DL. Chem. Ber. 1966;99:611–617. doi: 10.1002/cber.19660990235. [DOI] [PubMed] [Google Scholar]
- 8.a Fischer H. Chem. Rev. 2001;101:3581–3610. doi: 10.1021/cr990124y. [DOI] [PubMed] [Google Scholar]; b Studer A. Chem. Soc. Rev. 2004;33:267–273. doi: 10.1039/b307652k. [DOI] [PubMed] [Google Scholar]; c Tebben L, Studer A. Angew. Chem. Int. Ed. 2011;50:5034–5068. doi: 10.1002/anie.201002547. [DOI] [PubMed] [Google Scholar]
- 9.a Adlington RM, Baldwin JE, Basak A, Kozyrod RP. J. Chem. Soc,, Chem. Commun. 1983:944–945. [Google Scholar]; b Giese B, Dupuis J. Angew. Chem. Int. Ed. 1983;22:622–623. [Google Scholar]; c Baumberger F, Vasella A. Helv. Chim. Acta. 1983;66:2210–2222. [Google Scholar]; d Pearce AJ, Mallet J-M, Sinay P. In: Radicals in Organic Synthesis. Renaud P, Sibi M, editors. Vol. 2. Wiley-VCH; Weinheim: 2001. pp. 538–577. [Google Scholar]
- 10.Nagy JO, Bednarski MD. Tetrahedron Lett. 1991;32:3953–3956. [Google Scholar]
- 11.a Akhlaq MS, Schuchmann HP, Von Sonntag C. Int. J. Rad. Biol. 1987;5:91–102. doi: 10.1080/09553008714550531. [DOI] [PubMed] [Google Scholar]; b Dang H, Roberts BP. Tetrahedron Lett. 1999;40:4271–4274. [Google Scholar]; c Yamago S, Miyazoe H, Yoshida J.-i. Tetrahedron Lett. 1999;40:2339–2342. [Google Scholar]; d Cai Y, Roberts BP. J. Chem. Soc., Chem Comm. 1998:1145–1146. [Google Scholar]; e Yu GX, Tyler DR, Branchaud BP. J. Org. Chem. 2001;66:5687–5691. doi: 10.1021/jo005595v. [DOI] [PubMed] [Google Scholar]; f Escoubet S, Gastaldi S, Vanthuyne N, Gil G, Siri D, Bertrand MP. J. Org. Chem. 2002;71:7288–7292. doi: 10.1021/jo061033l. [DOI] [PubMed] [Google Scholar]
- 12.Martichonok V, Whitesides GM. J. Org. Chem. 1996;61:1702–1706. doi: 10.1021/jo951711w. [DOI] [PubMed] [Google Scholar]
- 13.a Achmatowicz S, Barton DHR, Magnus PD, Poulton GA, West PJ. J. Chem. Soc., D. 1971:1014–1015. doi: 10.1039/p19730001567. [DOI] [PubMed] [Google Scholar]; b Barton DHR, Bolton M, Magnus PD, West PJ, Porter G, Wirz J. J. Chem. Soc., Chem. Commun. 1972:632–633. [Google Scholar]
- 14.No decomposition was observed under these conditions
- 15.a McClellan AL. Tables of Experimental Dipole Moments. Freeman; San Francisco: 1963. [Google Scholar]; b Lee CM, Kumler WD. J. Am. Chem. Soc. 1961;83:4596–4600. [Google Scholar]; c Lee CM, Kumler WD. J. Am. Chem. Soc. 1962;84:571–578. [Google Scholar]
- 16.Keck GE, Enholm EJ, Yates JB, Wiley MR. Tetrahedron. 1985;41:4079–4094. [Google Scholar]
- 17.Cumpstey I. Org. Biomol. Chem. 2012;10:2503–2508. doi: 10.1039/c2ob06696c. [DOI] [PubMed] [Google Scholar]
- 18.a Denekamp C, Sandlers Y. J. Mass Spectrom. 2005;40:1055–1063. doi: 10.1002/jms.880. [DOI] [PubMed] [Google Scholar]; b Denekamp C, Sandlers Y. J. Mass Spectrom. 2005;40:765–771. doi: 10.1002/jms.848. [DOI] [PubMed] [Google Scholar]
- 19.a Hori H, Nakajima T, Nishida Y, Ohrui H, Meguro H. Tetrahedron Lett. 1988;29:6317–6320. [Google Scholar]; b Prytulla S, Lauterwein J, Klessinger M, Thiem J. Carbohydr. Res. 1991;215:345–349. [Google Scholar]
- 20.Alternatively, it may be considered that this thermodynamic effect arises from minimization of repulsion between the anomeric C-O dipole and the strong dipole of the oxazolidinone or carbonate ring
- 21.a Peters KS. Chem. Rev. 2007;107:859–873. doi: 10.1021/cr068021k. [DOI] [PubMed] [Google Scholar]; b Bohé L, Crich D. CR Chimie. 2011;14:3–16. [Google Scholar]
- 22.Murase T, Kameyama A, Kartha KPR, Ishida H, Kiso M, Hasegawa A. J. Carbohydr. Chem. 1989;8:265–283. [Google Scholar]
- 23.a Crich D, Li W. Org. Lett. 2006;8:959–962. doi: 10.1021/ol060030s. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Haberman JM, Gin DY. Org. Lett. 2003;5:2539–2541. doi: 10.1021/ol034815z. [DOI] [PubMed] [Google Scholar]
- 24.Horenstein NA. Adv. Phys. Org. Chem. 2006;41:275–314. [Google Scholar]
- 25.Okamoto R, Souma S, Kajihara Y. J. Org. Chem. 2008;73:3460–3466. doi: 10.1021/jo702609p. [DOI] [PubMed] [Google Scholar]
- 26.Ingold CK. Structure and Mechanism in Organic Chemistry. 2 ed. Cornell Univ Press; Ithaca: 1969. [Google Scholar]
- 27.Green JE, Bender DM, Jackson S, O'Donnell MJ, McCarthy JR. Org. Lett. 2009;11:807–810. doi: 10.1021/ol802325h. [DOI] [PubMed] [Google Scholar]
- 28.Peng C-H, Kong J, Seeliger F, Matyjaszewski K. Macromolecules. 2011;44:7546–7557. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.