

Abstract
Nitrite is currently recognized as a biomarker of the state of nitric oxide metabolism. Therefore, assessing nitrite levels in various organs and compartments is an important issue. As nitrite levels in most organs and tissues are low (in high nanomolar or low micromolar range) several new sensitive methods for quantifying nitrite in various biological samples have been developed. Chemiluminescence, combined with tri-iodide reducing solution, is currently considered the most sensitive method, allowing quantification in the low nanomolar range of nitrite concentrations. Here, we present an overview of chemiluminescence-based determination of nitrite in blood and blood compartments – red blood cells and plasma. We also explain how to preserve the original physiological nitrite concentration in nitrite-hostile environments, such as an excess of hemoglobin in blood.
Keywords: Nitrite, hemoglobin, red blood cell, chemiluminescence
1. Introduction
Recently, nitrite has emerged as a central component of the nitric oxide (NO) cycle – it is both a NO precursor and a product of NO oxidation. This dual role not only makes it a prime target for therapies to correct NO deficiency but also allows insights into NO metabolism and its changes in health and disease. It is thought that simple measurement of nitrite levels over time under different conditions and in different organs/biocompartments can give a rough estimation of NO pathway activity and efficiency. Attempts have been made to use nitrite as a biomarker of some health conditions.
The most common clinical measurements of nitrite are those in blood and urine, due to easy access to both fluids during clinical examination of a patient. Here we will describe measurement of nitrite in blood and its compartments (red blood cells (RBC) and plasma) using the gas phase chemiluminescence method with tri-iodide reducing solution and a nitrite-preserving “stop” solution, based on original published protocols (1–3). Major steps in necessary pretreatment of blood and sample processing are outlined in Fig. 4.1. Final nitrite concentration determination procedures are outlined in Fig. 4.2.
Fig. 4.1.
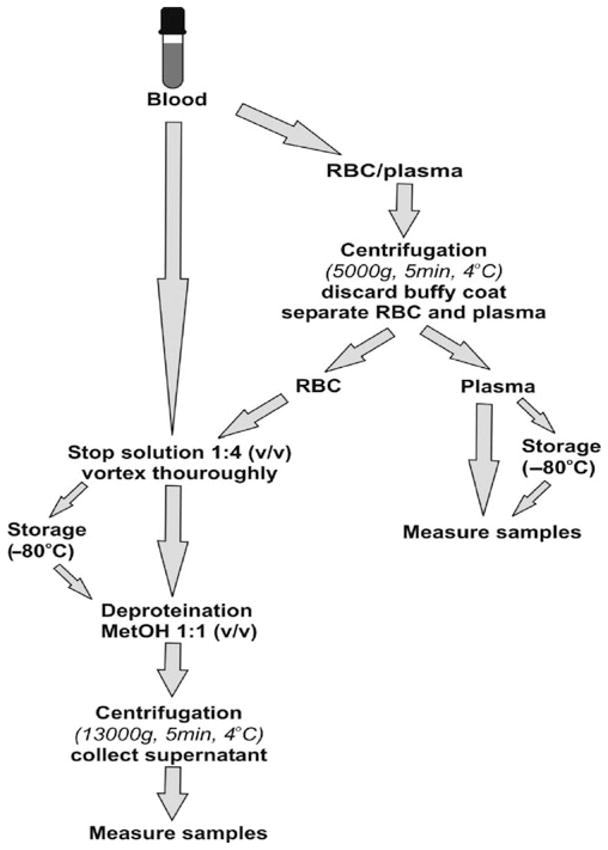
Outline guide to blood processing for nitrite determination in whole blood, red blood cells (RBC), and plasma.
Fig. 4.2.

Outline guide to sample measurements and data processing for nitrite determination by chemiluminescence method (CL). Samples prepared as shown in Fig. 4.1.
2. Materials
2.1. Nitrite-Preserving “Stop” Solution
Nitrite rapidly reacts with oxy- and deoxy hemoglobin (Hb) in blood and is destroyed in both reactions (see Note 1). Standard pretreatment of blood and most biological samples, to prevent nitrite destruction, consists of oxidizing the heme to form metHb or heme(FeIII)-protein. This pretreatment is not necessary if the sample does not contain Hb or other heme(FeII)-proteins and it is usually skipped for plasma nitrite measurement as shown in Fig. 4.1. The roles of the individual reagents in the stop solution are explained in Note 2.
2.1.1. Reagents
K3Fe(CN)6 (potassium ferricyanide, MW 329.24),
NEM (N-ethylmaleimide, MW 125.12) – light sensitive, store at 4°C,
Nonidet P-40 (octyl phenoxylpolyethoxylethanol),
H2O – we recommend using either molecular biology grade water or fresh distilled water filtered using Milli-Q (Millipore, Billerica, MA) water purification system with conductivity of 18 MΩ cm (see Note 3).
2.1.2. Preparation
Prepare the solution containing 890.9 mM potassium ferricyanide and 118.13 mM NEM. This solution needs to be vortexed vigorously for several minutes until all NEM and ferricyanide crystals are dissolved and the color is clear yellow. Add NP-40 in a 1:9 ratio (v/v, NP-40/solution), and gently mix ingredients together by inverting the test tube several times. At this stage, avoid unnecessary mixing, as this will cause excessive foaming (see Note 4 and 5).
2.2. Sample Collection and Pretreatment
Blood is collected using a 20 G needle for venous blood and an 18 G needle for arterial blood to prevent hemolysis. 5 IU heparin/mL blood is used to prevent coagulation. Note 6 emphasizes the crucial points in sample collection and processing, and Fig. 4.1 provides a general overview of the procedures involved.
2.3. Chemiluminescence
2.3.1. Chemiluminescence Principle, Reagents, and Reactions
Only free NO gas is detected by chemiluminescence. In order to detect NO metabolites, they have to be converted into free NO prior to quantification. Several reducing/oxidizing solutions, depending on the nature of the NO metabolite, are used for this purpose (see the following paragraph for details about nitrite-reducing solutions). For a simplified presentation of the chemiluminescence setup see Fig. 4.3 or the more detailed descriptions in (4, 5). Briefly, free NO gas is purged from the reaction vessel by an inert carrier gas (He, N2, Ar) into the chemiluminescence analyzer. In the analyzer reaction chamber, ozone (O3) is combined with NO to form nitrogen dioxide (NO2) in its activated state (see reaction 5 in Note 7). Upon deactivation, NO2* emits a photon (see reaction 6 in Note 7) in the infrared region that is detected by photomultiplier equipped with a long pass filter, detecting only emission above 600 nm. The intensity of emitted light is proportional to the concentration of NO in the reaction chamber, and using proper calibration curves, it can be related to the amount of NO in the original sample (see Note 7).
Fig. 4.3.

Chemiluminescence apparatus setup. Reaction vessel is filed with I3 solution with He carrier gas gently bubbling though. Sample is injected using Hamilton syringe through septum into I3 solution where NOx components are reduced to NO gas and carried into NO analyzer. Cold trap, NaOH-filled trap, and filter protect analyzer against humidity and acid vapors. In reaction chamber (RC) NO gas is combined with O3 (generated in O3 generator) from O2 either from O2 tank or from room air. Chemiluminescence signal from NO2* is detected by photomultiplier tube (PMT) and further amplified and processed. Data acquisition and analysis are carried out on PC. Vacuum pump created low pressure in the reaction chamber (RC) and evacuates toxic NO2 gas after chemiluminescence measurement through charcoal filter (CF).
All NO metabolites and adducts, such as nitrite, nitrate, R-nitrosothiols (R-SNO), R-nitrosoamines (R-NNO), or metal-NO, must be converted into free NO gas in order to quantify their original amounts via chemiluminescence. A reducing solution is used for nitrite, nitrate, R-SNO, and R-NNO. Here we will focus on the most popular – the I3-based reducing solution (see Note 8). I3-based reducing mixture is not specific for nitrite, and all NOx metabolites, except nitrate, will be reduced to NO, contributing to the signal. Details about how to account for this “false” nitrite signal using acid sulfanilamide (AS) treatment are in Note 9.
Reagents for I3 solution: KI (potassium iodide, MW 166.0) – light sensitive, I2 (crystalline iodide, MW 253.81) – light sensitive, glacial acetic acid, H2O.
Prepare 301.2 mM KI together with 137.8 mM I2 solution in water.
Mix this solution with acetic acid in a 2:7 ratio. Thorough mixing requires ~ 20–30 min on the magnetic stirring plate. The resulting solution has a brown color. For further information about storage and preparation see Note 10.
A slightly different, simpler version of reducing solution is described in Note 11.
A recently proposed ascorbic acid/acetic acid reducing solution which has been shown to be specific for nitrite is described in Note 12. The simplicity and specificity for nitrite could soon make this new method very popular in clinical practice.
2.3.2. Chemiluminescence –Instrumentation
Currently, there are two commercially available NO analyzers: Sievers NO analyzer (NOA, model 280i, GE Analytical Instruments, Boulder, CO, USA) and CLD 88Y (EcoPhysics, Duernten, Switzerland). The Sievers NOA requires an O2 tank attached to the analyzer as a source of ozone (O3), while the CLD 88Y derives O3 from O2 in normal atmospheric conditions. Inert gas (He, N2, or Ar) from a source tank with a constant flow rate of 100–150 mL/min is used as the NO carrier in both models. Sensitivity of both analyzers for NO detection is comparable and is in the low nM/high picomolar range of nitrite. According to manufacturers, it is possible to detect upward from 1 pM of NO gas in the reaction chamber. However, the sensitivity of each individual unit should be determined using calibration either with a known amount of NO gas or using NO donors.
3. Methods
3.1. Nitrite-Preserving “Stop” Solution
3.1.1
Stop solution is added to blood sample in a ratio 1:4 (v/v, stop solution/sample). Sample is then vortexed vigorously for ~10 s to allow complete lysis of cells and thorough mixing. Samples containing Hb will become brown, which indicates the presence of oxidized heme in the form of metHb.
3.1.2
Stop solution will cause lysis of cells; so if it is desirable to measure nitrite content in cells and supernatant separately, intact cells must be separated from supernatant prior to addition of stop solution (Fig. 4.1).
3.2. Sample Collection and Pretreatment
3.2.1. Whole Blood Treatment
Collected blood samples are aliquoted into 1.5–2 mL Eppen-dorf tubes containing stop solution in a 1:4 ratio (v/v, stop solution/sample). For preparation of nitrite preserving stop solution, see previous Section 2.1.
After vigorous vortexing, samples can be frozen on dry ice and stored at −80°C for later analysis (stable up to several months). If processed immediately, the deproteination step follows.
3.2.2. RBC Treatment
Whole blood samples are centrifuged at 5,000×g for 5 min to separate RBC from plasma. Collected RBCs are aliquoted into 1.5–2 mL tubes containing stop solution in a 1:4 ratio (v/v, stop solution/RBC).
After vigorous vortexing, samples are either frozen on dry ice and stored at −80°C for delayed processing or measured immediately after deproteination (see Section 3.2.4).
3.2.3. Plasma Treatment
Whole blood samples are centrifuged at 5,000×g for 5 min to separate RBC from plasma. Collected supernatant – plasma – is aliquoted into 1.5–2 mL tubes. Stop solution is not needed if plasma is clear and without hemolysis. If significant hemolysis is observed, stop solution must be used as described above for RBC treatment.
Aliquots can be either frozen on dry ice and stored at −80°C for delayed processing or measured immediately. Deproteination is not needed for plasma.
3.2.4. Deproteination Step
Whole blood and RBC aliquots must be deproteinated before injection into the chemiluminescence reaction vessel to avoid excessive foaming. If processing previously frozen samples, they should slowly thaw on ice. Proteins are precipitated by adding cold methanol to samples in a 1:1 ratio (v/v) and mixture vortexing. Proteins are then removed from mixture by centrifugation of the mixture at 13,000×g for 5 min at 4°C. Supernatant is carefully transferred to a new 1.5 mL tube and used for nitrite analysis (see Note 13 and 14).
It is necessary to keep small aliquots (~1 mL) of all solutions used in your sample treatments––stop solution, methanol, water, or any additional treatment that was used. All these solutions might contain nitrite contamination, which will contribute to the nitrite concentration measured in samples. Nitrite content in all these solutions is determined at the same time as nitrite in samples and, if necessary, corrections for nitrite contamination from these external sources can be made.
3.3. Chemiluminescence–Data Acquisition
There is a slight difference in initial set up of the CLD 88Y and Sievers NOA instruments, but data collection and processing are similar. CLD 88Y is slightly more user-friendly than Sievers NOA.
3.3.1. Procedure for CLD 88Y from EcoPhysics
Turn on the main switch located on the rear panel.
Turn on the computer attached to the instrument via the USB cable and start the CLD Excel macro.
The computer should detect the connected instrument and start running CLD macro, displaying a signal from the photomultiplier in millivolts as a function of time (see Note 15).
Check that input gas line connector located on the rear panel is tightly screwed on and that there is a dry HPLC filter between the bubbler (filled with 12 mL of 1 M sodium hydroxide (NaOH) to completely cover glass frit) and instrument. Any water residues left in the line or filter will result in an unstable baseline. Do not connect the line between bubbler and instrument at this point.
Wait for the CLD 88Y to automatically turn on the vacuum pump to evacuate the instrument’s internal reaction chamber. This will show on the computer screen as a large decrease in signal. Wait until an appropriate vacuum in the chamber is achieved and the baseline is stable – the complete process takes ~1–1.5 h.
Connect the carrier gas input line from the gas tank to the appropriate port of the reaction vessel. Start the carrier gas flow prior to pouring I3 solution into the glass vessel. Carrier gas should flow at a rate that allows the addition of 7–9 mL of I3 solution into the reaction vessel without letting the liquid go below the glass frit. Adjust the gas flow rate so that I3 solution is gently bubbling but not over-flowing beyond the cap or into the cooling part of reaction vessel.
Add 50–100 μL of Antifoam B into I3 solution in the reaction vessel (see Note 16).
Connect the NaOH-filled bubbler to the input gas line with HPLC filter. The second port of the bubbler should be still open with gentle bubbling observed, as the CLD 88Y creates a slight negative pressure to aspirate the gas from the line. If bubbling is missing, check all connections, including those on the HPLC filter, for tightness and all lines for some mechanical obstruction or humidity.
Carefully connect the remaining gas output line from the reaction vessel to the bubbler, watching for any possible I3 solution overflow that can be prevented by adjusting carrier gas flow. Once the carrier gas flow is adjusted, it has to remain the same for the duration of the experiment. If it is necessary to adjust the flow during the experiment, a new standard curve needs to be acquired at this flow rate. A detailed diagram of the lines and reaction vessel connections is given in Fig. 4.3.
About ~30 min after all lines are connected the baseline should be flat and relative noise should not exceed 0.1–0.3 mV. Possible reasons for a high/unstable baseline and ideas for troubleshooting are in Note 17.
Stop the acquisition and run a new CLD Excel macro file. After acquiring a few minutes of a steady baseline, proceed with nitrite standards and sample measurements.
3.3.2. Procedure for Sievers NOA Setup Is Described Separately in Note 18
While waiting for the NO analyzer to warm up and stabilize, wash all Hamilton syringes you plan to use thoroughly. See Note 19 for precautions and useful tips regarding syringes. Prepare 1 μM sodium nitrite standard solution (NaNO2, MW 68.99) for the calibration curve. After the NO analyzer is ready for measurements, acquire the chemiluminescence signal from your samples and nitrite standards as follows:
Using a well-washed Hamilton syringe, inject 50 μL of 1μM nitrite solution into I3 reaction mixture. Wait between injections until the chemiluminescence signal peak drops back to baseline and baseline is stable for at least 2 min to achieve good separation of peaks. Repeat injection of 50 μL two more times to get triplicates of each data point. Wash syringe after each injection as described in Note 19.
To acquire nitrite standards for the calibration curve, inject in triplicates of 100, 150, 200, 250, and 300 μL of 1 μM nitrite solution. In each case, take care to wait until the signal drops back to the baseline and then acquire 1–2 min of baseline. The calibration curve and its uses are described in Note 20.
Nitrite standards are usually measured before samples. If it later becomes apparent that nitrite levels of some measured samples are out of the range of injected standards (peak heights differ considerably), a second set of nitrite standards with appropriate amounts of nitrite should be injected after all samples have been measured. This set of injections is then used as the calibration curve.
If nitrite levels and other NOx combined levels (R-SNO + R-NNO + nitrosylHb (HbNO)) are expected to be comparable, pretreatment with acid sulfanilamide (AS, MW 172.21) and separate measurement of total NOx and nitrite-depleted sample is necessary – for AS treatment procedure and for general guidelines of data acquisition. We recommend use of AS treatment on the first trial for all samples; once the ratio of NOx/nitrite is determined to be reasonably low, neglecting the NOx contribution and skipping AS treatment is acceptable (see Note 9 and Fig. 4.2).
At a minimum, triplicates of all samples must be measured. Injection amounts may vary from 50 μL up to 300–500 μL, depending on expected nitrite content. All peaks must be clearly distinguished from baseline/noise variation. Multiple injections of 500 μL are not recommended, as this will quickly fill up the reaction vessel.
Each time an aliquot is injected into the reaction vessel; record the time of injection, what was injected and in what volume. If any dilutions or additional processing of the samples occurred, notes should be made to permit accurate tracking of all dilutions of the original sample.
3.4. Chemiluminescence – Data Processing
After all samples and nitrite standards are measured, nitrite levels in injected aliquots and original samples can be determined. We import the data into Origin software (OriginLab Corp. Northampton, MA, USA), but any other software capable of determining the area under peaks can be used Excel (Microsoft), Sigmaplot (Systat Software), Prizm (GraphPad Software) or similar.
Data acquired using CLD88Y are saved in an Excel file. This file does not contain any information about the origin of injection therefore separate records must be kept manually. Sievers NOA uses its proprietary Liquid software to acquire data into two separate files. “Filename.data” contains data and “filename.info” with time of mark and comments.
Both formats, Excel and “filename.data” are standard; most spreadsheet-based software will open them directly.
To process acquired data (Fig. 4.2).
Open data file in Origin (or other software).
Determine the area under the peaks using baseline markers placed at the peak take off and after the signal returns to baseline again.
If treatment with AS is used, correct the areas under the peaks for NOx contribution accordingly (see Note 9 and Fig. 4.2).
Using the calibration curve, determine the amount of nitrite in each aliquot injected into the reaction vessel, N (see Note 20).
Determine the total dilution factor of original sample in the aliquot, D. For example, if the sample was diluted in a 1:9 ratio (v/v, sample/buffer), D=1/10=0.1.
Calculate the fraction of the volume of original sample, VS that was present in the total volume of injected aliquot, VT.
Divide the amount of nitrite in aliquot N (determined in point 3) by the volume of the original sample: N/VS or N/(D/VT). This is the concentration of nitrite in the original sample.
Footnotes
The reaction of nitrite with heme-containing proteins represents the major route for nitrite destruction in any biological sample. This is especially important in blood, as hemoglobin is present in millimolar concentrations. The reaction of nitrite with ferrous hemoglobin, in its oxy- or deoxy- state, leads to oxidation of nitrite to nitrate and ferrous heme to ferric heme and, in the case of deoxyHb to a new reaction product, nitrosylHb (HbNO). Ferric heme in metHb does not react further with nitrite. Nitrite has a low affinity for ferric heme and the metHb–nitrite complex formation is accepted. However, this weakly bound complex is formed only in excess of nitrite and its formation will not affect results when chemiluminescence is used to measure nitrite levels.
| (reaction 1) |
| (reaction 2) |
| (reaction 3) |
| (reaction 4) |
“Stop” solution is a mixture of the following reagents: K3Fe(CN)6, NEM, NP-40, and water. K3Fe(CN)6 is the main “nitrite-preserving” ingredient. The product of its reaction with oxyHb and deoxyHb is metHb. NEM is a thiol reactive compound commonly used to modify cys-teine residues in proteins and peptides; here it is used to prevent additional R-SNO formation that would increase the signal detected by I3-based chemiluminescence. NP-40 is a nonionic detergent used to assure complete lysis of membranes and fast mixing of K3Fe(CN)6 and NEM with the entire contents of the cell. Triton X-100 can be used instead of NP-40.
Only water with low-nitrite content, such as Milli-Q treated water or molecular biology grade water, should be used for preparation of samples and solutions, as well as for final wash of syringes and all glassware.
Stop solution should be prepared daily by mixing 1.32 g K3Fe(CN)6 and 0.0665 g NEM in 4.5 mL water, with addition of 0.5 mL NP-40. The solution should be kept at 4°C and can be stored for a few days. However, for best results it is recommended to use fresh solution for sample preparation, whenever possible.
All necessary materials and stop solution should be prepared in advance and kept at hand – immediate access to a tabletop centrifuge cooled to 4°C and a vortex are strongly suggested if nitrite measurement in RBC’s are required. Once blood is collected, it is critical to work as fast as possible, as nitrite decays in the blood with half-life of ~11–13 min (6).
| (reaction 5) |
| (reaction 6) |
| (reaction 7) |
| (reaction 8) |
I3-based reducing solution is not selective for nitrite. NO will be released also from R-SNO, R-NNO, and Fe-NO functional groups. Nitrate will not be reduced in I3 solution. The “additional” or “false” nitrite signal can be accounted for by using nitrite-specific sample treatments with AS. Nitrite reacts with sulfanilamide in an acidic environment and irreversibly forms diazonium cation complex that is not reduced to NO in I3 solution (4, 6). It is also possible to separate and quantify contributions from R-SNO and Fe-NO using different targeted sample pre-treatments (1–4, 7).
Procedure to separate the nitrite-related chemiluminescence signal from total NOx signal using AS treatment:
- 5% solution of acidified sulfanilamide (5% wt/v in 1 M HCl or 290 mM solution of sulfanilamide) is prepared by dissolving 0.5 g of sulfanilamide in 10 mL of 1 M HCl.
- Divide the sample into two aliquots, A and B.
- Aliquot A is mixed well with AS solution in a ratio 1/9 (v/v, AS/sample) and incubated for 3 min to deplete free nitrite. NOx level is measured using the standard method.
- Water or buffer is added into aliquot B at the same ratio as above to account for dilution, and NOx levels are measured using the standard method.
- True nitrite signal is calculated by subtracting AS-treated sample A from non-treated sample B as shown in Fig. 4.2.
- At normal physiological conditions nitrite is present in blood in at least 10-fold excess over other NOx metabolites (excluding nitrate), therefore the AS treatment is necessary only if highly precise determination of nitrite concentration is required. We recommend first running a few pilot aliquots with and without AS treatment to estimate the contribution of other NOx metabolites and decide accordingly.
Best results are obtained with freshly prepared I3 solution used within 1 day and kept preferably in a dark bottle, as I-containing reagents are light sensitive. We prepared 90 mL of I3 solution by dissolving 1 g KI and 0.65 g I2 in 20 mL water with subsequent addition of 70 mL acetic acid. This amount is enough to refill the reaction vessel ~10 times.
Nitrite can be reduced to NO using a mixture of KI or NaI (sodium iodide, MW 149.89, light sensitive) and glacial acetic acid by the same mechanism as I3 solution. This original mixture was thought to be specific for nitrite. However, it has been shown that the I3− ion, which can also reduce R-SNO and R-NNO, forms in the reaction vessel over time and the real selectivity toward nitrite can be only achieved in freshly prepared KI/acetic acid solutions and is maintained only for the first few sample injections. These requirements make the use of KI/acetic acid solution impractical, and it is therefore currently not as widespread as the I3-based method (3).
- Reactants: KI, glacial acetic acid, H2O. NaI can be used instead of KI.
- Prepare 667.15 mM solution of KI in H2O. Mix this solution with glacial acetic acid in 1:9 proportions. This reaction mixture is dark yellow and is light sensitive. It can be kept in dark bottle at room temperature and used within 1 day. An easy way to prepare a sufficient supply for 1 day (50 mL) is to dissolve 553.7 mg of KI (or 500 mg of NaI) in 5 mL of H2O, then add 45 mL of acetic acid.
| (reaction 9) |
| (reaction 10) |
| (reaction 11) |
| (reaction 12) |
| (reaction 13) |
- Reactants: ascorbic acid, glacial acetic acid, and H2O.
- Prepare 500 mM ascorbic acid in water. Mix this solution with glacial acetic acid in a ratio 1:7 to prepare the reaction mixture.
- According to (5), completion of nitrite reduction depends on ascorbic acid concentration; and at least 50 mM ascorbic acid is recommended for complete reduction of plasma nitrite. However, we believe that few pilot experiments with different concentrations of ascorbic acid and nitrite standards in the expected nitrite concentration range are necessary prior to final sample measurements.
For deproteination, some authors use methanol in a ratio 1:1 (v/v) with multiple centrifugations at 750×g for 2 min each until the supernatant appears clear (5) or in a ratio of 1:2 (v/v, sample/methanol) with a single centrifugation at 21,000×g at 4°C for 15 min (8).
Deproteination step significantly dilutes the nitrite in samples, and it is not recommended when extremely low nitrite levels are expected. In such a case, we recommend the use of a twofold increase in antifoam and direct injection of samples containing protein. After each injection, frequent changes of the reaction mixture will be required.
If an error message “connection not found” is displayed and the Excel macro does not run, check the USB connector/replace cable and restart the computer. If the problem persists, the driver for the communication port needs to be reinstalled.
Biological samples containing large amounts of protein must be either completely deproteinated prior to injection into reaction vessel, or antifoaming agent needs to be added to the I3 mixture. Often samples are only partially deproteinated so there is still a need for antifoam. We use antifoam B emulsion with 50–100 μL of antifoam for 9 mL of reaction solution, added directly into reaction vessel (1). Others recommend antifoam 204 organic (3) or antifoam SE-15 (8). It is important to remember that antifoam itself may contain some nitrite impurities, so it is recommended to wait after antifoam addition until all nitrite is consumed and the baseline is stable again.
High baseline signals occur for several reasons: humidity –water droplets or dust in the gas line or filter, high NOx pollution in air, loose connections between reaction vessels and the instrument, cap of the reaction vessel not fully closed, or an old Teflon septum. From all listed, the most frequent reasons are loose connectors and an old Teflon septum.
Sievers NOA instrument set up:
All glassware is connected to Sievers NOA the same way as described for CLD 88Y in Section 3.3.1 and shown in Fig. 4.3. The acquired data are processed as described in Sections 3.3 and 3.4.
- Open O2 tank connected to the instrument. NOA derives O3 from the attached O2 tank and will give an error message if O2 supply is inadequate or missing.
- Choose option “analysis” from the main menu on the instrument panel by highlighting it and push “enter.” On the next screen choose “start” and press “enter.” The vacuum pump should start at this point. Wait for cooler of the photomultiplier to reach a temperature below −12°C and the vacuum in the reaction chamber to reach approximately 6 Torr. The baseline should stabilize itself below 50 mV – the absolute value depends on the starting state of the instrument and it takes between 30 min and several hours. The baseline has to be stable within 1–2 mV, the nominal value for the baseline is less important than its stability, as all measurements are relative to the baseline. However, the lower the initial baseline and the lower the cooler temperature will give more precision and more sensitivity.
- When temperature and pressure are stable, turn on the computer attached to the instrument and start Labview-based Liquid software. To achieve communication between the instrument and acquisition software, data output from the instrument has to be enabled as follows: press “enter” when in “data menu” – screen will read “output disabled” – press “enter” to change it to “enabled” and then press “clear” to return back to data screen. If preparation of a new sample is done while the instrument is running, data transfer should be disabled when a measurement is not taken – see point 7.b. below for a detailed explanation.
- Wait for a stable baseline and start injecting samples and nitrite standards into I3 reaction solution as described already for CLD 88Y. Always wait for a stable baseline after the peak. The Liquid software has an option to “mark” times of injection and it will export them into a separate file (filename.info) together with any written comments – this is a useful complement to the data file (filename.data).
- Once all samples and standards are measured, choose the “stop” option in the Liquid software menu – this will write data from a temporary file into a permanent file known as “filename.data” (see also point 7.b)
- To stop NOA analyzer, disconnect the He tank from the glass reaction vessel first, then press “clear” to get into main menu. Choose “analysis” option, then “stop” and confirm “stop” on the confirmation screen. Turn off supply of O2 from O2 tank.
-
NOA troubleshooting tips:
- Instrument should be always left in standby mode, unless physically moved to another location. After turning it off for a longer period of time (1–2 days), a few days of constant running are needed to stabilize the baseline again.
- It is possible to enable/disable the data output as many times as necessary during one session of Liquid software. The software will display an error message when communication is disabled, but it will continue running and writing into the same file after communication is enabled again. This feature is important because Liquid software runs only for a preset amount of time (maximum continuous run time is 1 h) and will discard all data unless “stop” button is chosen before the run time expires.
- If the “error” message in Liquid software persists after communication was enabled, check if the baud rate for the communication port, set for the instrument, and in the software match. To check baud rate on the instrument: from main menu, choose option “control,” and then “setup.” There are two options in this menu: “view” and “change.” Choosing “view” only shows the current setup, “change” will allow modifications. Choosing either of them will display the next menu with the option “configuration” and then “COM port.” This option displays the current baud rate and changes are made using “up” and “down” arrows keys. From the software side, baud rate is setup from the first pop-up window after the software starts.
We use Hamilton syringes (size 100–500 μL) with sharp needle tips to inject small aliquots of samples into I3 solution through the Teflon septum of the reaction vessel cap. For the best results use gastight syringes; however, good results are also obtained with ordinary Hamilton syringes that are new and still tight. It is necessary to thoroughly wash each syringe prior to use, as well as before and after sample injection. High-quality nitrite-free water, the same grade that is used to prepare all solutions and samples, is also used to wash syringes. We use two 50 mL Falcon centrifuge tubes filled with Milli-Q treated water to wash our syringes and control their cleanness as follows: use the first Falcon tube to wash the syringe by repeating a fill up/empty cycle at least 15–20 times. After cleaning, fill the syringe with 100 μL of water from the second tube and inject the water into I3 solution in the reaction vessel. If the running baseline of chemiluminescence signal changes, i.e., a peak appears, repeat the washing procedure until no peak or only a very small peak of residual nitrite is observed. In case of any residual peak, multiple water injections permit later estimation of nitrite contamination in the water. If the “water peak” persists, use a new bottle of water (verify that Millipore system measures conductivity of ~18 MΩcm, if not – change cartridges) and/or replace the Hamilton syringe. It is absolutely crucial for determination of precise nitrite levels to minimize nitrite contamination from external sources.
Determination of a calibration curve and its use:
- Measure the area under each peak for all nitrite standards.
- Plot these areas as a function of total amount of nitrite for each injection. Total amount of nitrite in 50, 100, 150, 200, 250, and 300 μL of 1 μM nitrite solution is 50, 100, 150, 200, 250, and 300 pM. Recalculate the amount of nitrite injected if different volumes were used.
- Calculate the slope, K, of the standard curve.
- To determine the total amount of nitrite in the sample, divide this area by the slope (total amount of nitrite in sample) = (area under peak)/K.
References
- 1.Pelletier MM, Kleinbongard P, Ring-wood L, Hito R, Hunter CJ, Schechter AN, et al. The measurement of blood and plasma nitrite by chemiluminescence: pitfalls and solutions. Free Radic Biol Med. 2006;41:541–548. doi: 10.1016/j.freeradbiomed.2006.05.001. [DOI] [PubMed] [Google Scholar]
- 2.Yang BK, Vivas EX, Reiter CD, Gladwin MT. Methodologies for the sensitive and specific measurement of S-nitrosothiols, iron-nitrosyls and Nitrite in biological samples. Free Radic Res. 2003;37:1–10. doi: 10.1080/1071576021000033112. [DOI] [PubMed] [Google Scholar]
- 3.Pinder AG, Rogers SC, Khalatbari A, Ingram TE, James PE. The measurement of nitric oxide and its metabolites in biological samples by ozone-based chemiluminescence. In: Hancock JT, editor. Methods in Molecular Biology, Redox-Mediated Signal Transduction. Vol. 476. Humana press; Totowa, NJ: 2008. pp. 11–28. [DOI] [PubMed] [Google Scholar]
- 4.Mac Arthur PH, Shiva S, Gladwin MT. Measurement of circulating nitrite and S-nitrosothiols by reductive chemiluminescence. J Chromatogr B. 2007;851:93–105. doi: 10.1016/j.jchromb.2006.12.012. [DOI] [PubMed] [Google Scholar]
- 5.Nagababu E, Rifkind JM. Measurement of plasma nitrite by chemiluminescence without interference of S-, N-nitroso and nitrated species. Free Radic Biol Med. 2007;42:1146–1154. doi: 10.1016/j.freeradbiomed.2006.12.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tsikas D. Analysis of nitrite and nitrate in biological fluids by assay based on Griess reaction: appraisal of the Griess reaction in the L-arginine/nitric oxide area of research. J Chromatogr B. 2007;851:51–70. doi: 10.1016/j.jchromb.2006.07.054. [DOI] [PubMed] [Google Scholar]
- 7.Bryan NS, Grisham MB. Methods to detect nitric oxide and its metabolites in biological samples. Free Radic Biol Med. 2007;43:645–657. doi: 10.1016/j.freeradbiomed.2007.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hendgen-Cotta U, Grau M, Rasaaf T, Gharinin P, Kelm M, Kleinbongard P. Reductive gas-phase chemiluminescence and flow injection analysis for measurement of nitric oxide pool in biological matrices. Method Enzymol. 2008;441:295–315. doi: 10.1016/S0076-6879(08)01216-0. [DOI] [PubMed] [Google Scholar]
- 9.Wang X, Bryan NS, MacArthur PH, Rodriguez J, Gladwin MT, Feel-isch M. Measurement of nitric oxide levels in the red cell. J Biol Chem. 2006;281:26994–27002. doi: 10.1074/jbc.M603953200. [DOI] [PubMed] [Google Scholar]
- 10.Dejam A, Kleinbongard P, Rasaaf T, Hamada S, Gharni P, Rodriguez J, Feel-isch M, Kelm M. Thiols enhance NO formation from nitrate photolysis. Free Radic Biol Med. 2003;35:1551–1559. doi: 10.1016/j.freeradbiomed.2003.09.009. [DOI] [PubMed] [Google Scholar]
- 11.Laver JR, Stevanin TM, Read RC. Chemiluminescence quantification of NO and its derivatives in liquid samples. Method Enzymol. 2008;436:113–127. doi: 10.1016/S0076-6879(08)36007-8. [DOI] [PubMed] [Google Scholar]
- 12.Rasaaf T, Feelisch M, Kelm M. Circulating NO pool: assessment of nitrite and nitroso species in blood and tissues. Free Radic Biol Med. 2004;36:413–422. doi: 10.1016/j.freeradbiomed.2003.11.011. [DOI] [PubMed] [Google Scholar]
- 13.Cornelius J, Tran T, Turner N, Piazza A, Mills L, Slack R, Hauser S, Alexander JS, Grisham M, Feelisch M, Rodriguez J. Isotope tracing enhancement of chemiluminescence assays for nitric oxide research. Biol Chem. 2009;390:181–189. doi: 10.1515/BC.2009.017. [DOI] [PubMed] [Google Scholar]