

Abstract
Recent advances in microfluidics have enabled the molecular-level study of polymer dynamics using single DNA chains. Single polymer studies based on fluorescence microscopy allow for the direct observation of non-equilibrium polymer conformations and dynamical phenomena such as diffusion, relaxation, and molecular stretching pathways in flow. Microfluidic devices have enabled the precise control of model flow fields to study the non-equilibrium dynamics of soft materials, with device geometries including curved channels, cross-slots, and microfabricated obstacles and structures. This review explores recent microfluidic systems that have advanced the study of single polymer dynamics, while identifying new directions in the field that will further elucidate the relationship between polymer microstructure and bulk rheological properties.
Introduction
Microfluidics has revolutionized the study of biological systems and soft materials. Recent advances in microfabrication have enabled the precise control of small-scale flows in microfluidic devices, which provides a powerful platform to study the dynamics of soft materials at the molecular level. In particular, single polymer dynamics provides a direct view of polymer solution microstructure, including non-equilibrium polymer chain dynamics, polymer-solvent interactions, and macromolecular transport phenomena. In this way, single polymer dynamics holds strong potential to tremendously benefit the field of molecular rheology by providing a direct link between polymer microstructure, molecular-based constitutive models, and bulk rheological properties of polymeric materials. A comprehensive understanding of single polymer dynamics is crucial for applications including materials processing, non-Newtonian rheology, electrophoresis, DNA manipulation for genomics, biosensing, and lab-on-chip devices.
Single polymer studies have primarily focused on semi-flexible polymers using fluorescently-labelled DNA, but recent interest has turned toward flexible polymer chains. For the last two decades, double stranded deoxyribonucleic acid (DNA) has served as a model system for single molecule polymer dynamics.1, 2 Monodisperse samples of double stranded DNA can be readily synthesized via molecular cloning or polymerase chain reactions, and DNA molecules can be fluorescently labelled for direct observation with videomicroscopy.3 Early single polymer studies of DNA relied on optical tweezers and bead-tethered DNA to study chain relaxation,4 entanglement,5 and the stretching dynamics of single DNA molecules in uniform flow.6 Other techniques including magnetic tweezers and atomic force microscopy have been used to study the mechanical properties of DNA.7 More recently, single polymer studies of DNA have involved surface tethering, flow channels, and microfabricated structures and devices.
Although DNA has been extensively used as a model polymer, double stranded DNA is a semi-flexible polymer chain with distinct molecular properties compared to flexible chains, including a significantly larger persistence length (~65 nm for fluorescently-labelled DNA) relative to truly flexible polymers (~0.6 nm for many synthetic polymers).8 Recently, our group developed a new system that has enabled single molecule studies of flexible polymer chains based on fluorescently-labelled single stranded DNA, which has a bare persistence length of ~0.65 nm.9
For nearly all single polymer studies, microfabricated flow geometries are coupled with fluorescence microscopy, which allows for direct observation of non-equilibrium polymer microstructure. The main purpose of this review is to explore the use of recent microfluidic platforms in the field of single polymer dynamics. Microfluidic systems have also played a key role in measuring bulk-level rheological properties10, 11 and biosensing single molecules,12, 13 both of which have each been extensively reviewed elsewhere.
Using recent approaches in soft lithography, microfluidic devices can be fabricated inexpensively and reliably with tailored geometries to achieve well-defined microscale flows.14 Microdevices commonly rely on pressure-driven or electrokinetically-driven flows to stretch polymer chains far from equilibrium. Fundamental studies of single chain dynamics typically rely on simple device configurations to generate model flows, such as uniform, shear, or extensional flow, whereas increasingly complex flow networks can be achieved through interconnected channels, valves, and reservoirs. In microfluidic channels, the physics of viscous-dominated flows is characterized by principal dimensionless numbers.15 Dimensionless groups relevant to polymeric studies include Reynolds number , where ρ is the fluid density, U is the average velocity, L is a characteristic length scale, and η(γ̇) is the solution viscosity and Weissenberg number Wi = γ̇τ, where γ̇ is the shear rate and τ is the longest polymer relaxation time.
This review is organized to explore the study of single polymer dynamics using distinct microfluidic geometries, including channel-based flows, cross-slots, arrays, and nanofluidic confinement (Fig. 1). Each platform results from a different set of microfabrication methods, naturally lending to the study of polymer dynamics in tailored microscale flow fields. Prominent methodologies and unique architectures are highlighted for each system.
Fig. 1.

Schematics of several microfluidic platforms highlighted in this review. For each schematic, the direction of fluid flow is indicated by the arrows. (a) Straight channel; (b) planar micro-contraction; (c) planar 90° bend; (d) channel-based micro-curvilinear flow device; (e) hyperbolic contraction; (f) linear converging channels; (g) cross-slot geometry; (h) single obstacle; (i) ordered array of obstacles; and (j) slit-like confinement.
1. Channels
A single, straight channel is the fundamental element of a microfluidic device. Channels form the basis for more complex microfluidic designs, such as converging channels, micro-contractions, cross-slots, and post arrays. Microscale flows differ from macroscopic fluid flows because high surface-to-volume ratios enhance polymer-wall interactions, and small length scales result in laminar flow conditions within microchannels. In some cases, the dimensions of macromolecules can approach the dimensions of microchannels. For example, single λ-DNA molecules (contour length L = 16.3 μm and radius of gyration Rg = ~0.7 μm) may be confined in channels with dimensions ranging from ~100 μm to sub-micron scales.16, 17 Increased interactions between polymers and device surfaces can result in non-Newtonian flow phenomena, even at dilute polymer concentrations (Section 4).
Moving boundaries, pressure gradients, or electric field gradients are commonly used to drive fluid flows in microchannels. Channel-based studies generally aim to measure polymer conformations in flow, polymer effects on fluid flow, and single polymer transport mechanisms.
1.1. Moving boundaries
Surfaces can be used generate well-defined fluid flows in microfluidic channels. For example, simple shear flow can be generated near the vicinity of a moving boundary in a channel. In some cases, surfaces can play additional roles in polymer chain dynamics, e.g. adsorbing polymer chains or screening intramolecular hydrodynamic and/or excluded volume interactions. Under strong confinement, surface effects play a key role in polymer chain dynamics (Section 4).
Parallel plates
Shear-induced migration and the non-equilibrium tumbling dynamics of polymer chains in shear flow have been studied using single polymer dynamics. In quiescent conditions, polymer molecules adopt random coil configurations, but polymers are known to deform into highly stretched conformations in steady shear flow. Initial studies of single DNA molecules in shear flow revealed a rich spectrum of molecular conformations (Fig. 2).18, 19 In this work, shear flow was generated in the gap between two translating parallel glass plates. Polymers in dilute and semi-dilute solutions were observed to stretch during the start-up of shear flow, and flows above a critical Wi resulted in an overshoot in fractional extension, coupled with an overshoot in shear viscosity.20 Interestingly, the overshoot in 'bulk' shear viscosity was directly related to molecular stretching conformations in shear flow.21
Fig. 2.

Conformations of single DNA molecules in steady shear flow at Wi = 19. Time between images is (A) 6 seconds, (B) 0.84 seconds, or (C) 6 seconds. Scale bar: 5 μm. From D. E. Smith, H. P. Babcock and S Chu, Science, 1999, 283, 1724-1727. Reprinted with permission from AAAS.
DNA stretching dynamics were also studied in the flow-gradient plane of shear flow, where polymers were imaged in a thin gap between a pair of parallel quartz slides mounted upright with respect to the microscope objective.22 Using this approach, the tumbling dynamics of single DNA molecules in shear flow was directly observed for the first time.22, 23 In shear flow, it was observed that polymers undergo periodic end-over-end tumbling events such that the frequency of characteristic periodic motion scales sublinearly with flow rate.24
The dynamics of semi-dilute and concentrated solutions were also characterized in the flow-gradient plane of shear flow using single molecule studies. Entangled DNA solutions were studied at concentrations up to 35 c*, where c* is the overlap concentration, which revealed two distinct relaxation time scales following shear deformation. Teixeira et al. also observed molecular individualism, wherein molecules exhibited vastly different conformations despite exposure to identical flow histories.25
Torsional flow cell
Larson and co-workers developed a torsional flow cell to generate shear flow between a rotating circular glass plate and a fixed coverslip. In the vicinity of the fixed boundary (distances < ~0.3 L from the surface), DNA stretching was reduced from that observed in bulk shear flow, especially near adsorbing walls.26 Dilute and concentrated DNA solutions in torsional shear flow also exhibited shear-migration behaviour, wherein flow-stretched molecules drifted toward the centreline of the channel.27, 28 Shear migration resulted in depleted DNA concentrations in the near-surface region compared to bulk concentrations. Moreover, compact molecular extensions and weakened shear-induced migration were also observed in the near-surface region of flow in a microfluidic channel, and these phenomena were attributed to an upward drift velocity from surface-induced reduction of hydrodynamic interactions within the polymer chain.29 Importantly, shear migration and shear-induced extension appear to play significant roles on the flow profiles and molecular configurations of polymers in microfluidic platforms.
Translating tubes
Chu and co-workers constructed a novel microdevice that creates well-defined mixed flows with dominant elongational or rotational flow character. In this device, teflon tubes and rods are translated in opposing directions to explore the coil-stretch transition of DNA molecules for mixed flows in the neighbourhood of simple shear flow. As extension-dominated mixed flows approached simple shear flow, the transition between coiled and extended states partially softened, such that extended polymers were occasionally "kicked" out of alignment by Brownian fluctuations, resulting in a collapse to a coiled state. In rotation-dominated mixed flows, polymer conformations exhibited periodic deformations.30
1.2. Pressure-driven flow
Straight channels
Pressure gradients in channels generate Poiseuille (parabolic) flow, such that shear flow develops near channel walls. Microfluidic pressure-driven oscillatory flow, in which fluid flow alternates directions at a given frequency, primarily generates a shear flow profile.31 Oscillating flows allow for prolonged observation of DNA molecules, which directly enabled additional dynamical studies. The oscillatory flow resulted in shear-migration-induced focusing, in which molecules migrated toward the centreline and formed depletion layers at channel walls. Jo et al. observed that high Weissenberg number flows enhanced stretching and focusing, but larger oscillation frequencies reduced both of these effects.31
Converging/diverging flows
Microfluidic devices frequently contain converging and/or diverging features. A decrease in the cross-sectional area of a channel accumulates fluid strain and stretches incompressible fluid elements to conserve mass (volume), thereby resulting in an extensional flow. Several groups have studied the response of soft materials in the context of converging or diverging channels, which are typically fabricated in silicon16, 32 or quartz33 devices for structural precision.
Shrewsbury et al. characterized the effects of converging flow on DNA conformations in dilute (0.1% c*)16 and semi-dilute (10% c*)32 solutions. DNA molecules were observed to stretch in extensional flow generated in a contraction flow. Conversely, partially stretched molecules relaxed while moving through the channel and into diverging flow upon exiting the channel (Fig. 3). Near the device floor and ceiling, DNA molecules were observed in partially stretched conformations, resulting from near-surface effects of shearing flow. In dilute solution, molecules near walls had longer median lengths at the channel entrance than at the exit, suggestive of chain scission under high shear flow.16 Higher concentrations suppressed both DNA molecular stretching and median length variations between the channel entrance and exit.32 Overall, these results suggest that entanglements dissipate stress and prevent chain scission. Clearly, solution concentrations play a key role in DNA transport and should be considered when designing microfluidic devices.
Fig. 3.

DNA conformation along the channel centreline as a function of axial position in the device. The representative images indicate the dramatic stretching of the molecule due to the elongational flow at the channel entrance, and the recovery of its equilibrium conformation as it travels through the channel and into the downstream reservoir. From Ref. 16, with kind permission from Springer Science + Business Media.
DNA stretching was also studied using pressure-driven flow in continuously converging channels.33 Researchers at U.S. Genomics33 studied DNA dynamics in channels with funnel geometries with specific dimensions to tailor strain rate profiles. DNA molecules were pre-stretched in shear flow prior to transport through the funnels in order to minimize the effects of molecular individualism on chain stretching dynamics. High and low strain funnels were observed to fully extend DNA molecules, but a funnel with increasing strain rate yielded the most probable extended conformation, which is important for applications of DNA stretching such as genomic mapping. Kim et al. found similar results through numerical simulation of the same geometries, even though transport was modelled using an electric field instead of a pressure gradient.34
Curved streamlines and elastic secondary flows
Viscoelastic solutions significantly influence flow fields in a wide array of channel geometries, particularly planar micro-contractions and 90° corners (Fig. 1b, c). Curved geometries can be fabricated from polydimethylsiloxane (PDMS) or poly(methyl methacrylate) (PMMA), which are inexpensive and allow for reliable replication of micron-scale features using soft lithography techniques. The abrupt onset of fluid flow in a micro-contraction geometry results in elastic secondary flows, producing vortices or other instabilities depending on the contraction type (planar, square, or axisymmetric), contraction ratio, and fluid rheological properties.35
Rodd et al.36, 37 used polyethylene oxide solutions to study the effect of elasticity on vortex formation and growth in a PDMS micro-fabricated planar 16:1:16 contraction-expansion device. This microdevice permitted the study of flow regimes within a parameter space spanning several orders of magnitude of the elasticity number ( ), which is a function of fluid properties, flow rates, and characteristic length scales.
Flow through curved geometries is relevant to understand polymer transport in microfluidic lab-on-chip devices. DNA solutions were used to observe viscoelastic flows in abrupt planar 2:138 and 4:139 micro-contractions (silicon and PMMA devices, respectively). Hemminger et al.39 used fluorescently-labelled tracer DNA molecules in a 1% DNA solution to visualize vortex formation, transitional behaviour, and "jerky" shear banding due to slipping of DNA entanglements. Analogous to micro-contractions, flow of polymeric solutions around corners and bends can generate elastic secondary flows. Gulati et al.40 observed polystyrene tracer particles in shear-thinning DNA and polyethylene oxide solutions to visualize vortex development at the inner upstream corner of a 90° micro-bend.
Cheng et al.41 developed a channel-based PDMS device to manipulate DNA molecules with a micro-curvilinear flow (Fig. 1d). This work demonstrated the ability to induce radial acceleration in a circular side chamber and to observe simultaneous elongation and curvature of a single DNA molecule tethered to the chamber entrance.
1.3. Electrokinetic flow
Channel-based electrokinetic studies of single polymer molecules focus on migration mechanisms and molecular conformations, both of which are central to understanding and studying dynamics of single molecules in confinement, arrays of obstacles, and electrophoretic separation. Negatively charged phosphate groups along the backbone of DNA molecules allow for uniform macromolecular transport in the presence of an external electric field. Electric fields can be applied across various media, ranging from buffered solutions to linear polymer gels, which can be incorporated into microfabricated devices in order to investigate the response of single polymers to an electric field. In microchannels, electro-osmosis results in uniform plug-flow velocity profiles, such that shear flow is localized to a thin layer at the channel walls in the vicinity of the Debye layer. Electrophoretic flows have been extensively reviewed elsewhere.42
Straight channels
Chiesl et al.43, 44 used channel-like PDMS flow cells to study modes of electrophoretic DNA migration in capillaries containing various polymer solutions. Single molecule videomicroscopy revealed three different modes of migration for DNA in these solutions: stationary entanglement coupling, transient entanglement coupling (TEC), and reptation (Fig. 4), each of which affects the performance of electrophoretic separation.45 Stationary entanglement coupling was observed in a co-polymer network, such that the mechanism resembles a collision between a DNA molecule and a single post (Section 3).43 Transient entanglement coupling occurs in dilute polymer solutions, wherein the mechanism consists of entanglement of a DNA molecule with a nearby polymer, thereby resulting in stretching of the DNA molecule around the polymer, followed by disentanglement and contraction into a globular conformation. The migration behaviour of DNA transitions to reptation for polymer concentrations approaching the polymer entanglement concentration (c*). In this regime, DNA molecules move through an entangled polymer matrix by motion parallel to the backbone conformation.44
Fig. 4.

Single molecule images show distinct modes of DNA migration through polymer solutions in the presence of electric fields. White dots are superimposed as reference points for entanglement coupling mechanisms. Adapted with permission from T. N. Chiesl, K. W. Putz, M. Babu, P. Mathias, K. A. Shaikh, E. D. Goluch, C. Liu and A. E. Barron, Analytical Chemistry, 2006, 78, 4409-4415. Copyright 2006 American Chemical Society.
Straight channels allow for the study of DNA conformation in free solution in the presence of electric fields. Tang et al. studied DNA conformations in straight channels using a uniform field.46 Upon exposure to electric fields, DNA conformation was found to depend on field strength and ionic strength of the solution, rather than DNA location in the channel. Interestingly, uniform direct current (DC) electric fields were observed to compress DNA molecules, with coils appearing increasingly isotropic and more compact upon increasing the field strength. Alternating current (AC) at a moderate frequency (up to 100 Hz) was observed to lessen the degree of compressive behaviour. Following DNA molecule compression, this study also observed the expansion process for single DNA molecules, postulating disentanglement from knotted conformations upon chain extension from the compact state.
Converging channels
Randall et al. observed the transport of single DNA molecules in the presence of a uniform electric field in PDMS devices containing a region of cross-linked porous gel and/or a hyperbolic channel (Fig. 1e), seeking a method to uniformly stretch molecules.47 The hyperbolic contraction device imposed only moderate strain on molecules, thereby resulting in a broad probability distribution of chain extension due to molecular individualism. The cross-linked porous gel was observed to pre-condition an ensemble of DNA molecules, which migrated through the gel by reptation and further stretched upon exiting the gel region. DNA molecules exiting the gel exhibited a broad extension probability distribution, but the addition of a hyperbolic channel downstream a cross-linked gel resulted in uniform stretching.
DNA extension was also studied by imposing electric fields over converging channels of various geometries, analogous to pressure-driven funnels (Section 1.2). Liao et al. investigated the effect of DNA conformation on electrophoretic mobility in a PMMA hyperbolic channel, where it was found that electric field gradients produced by the converging channel locally stretched and increased electrophoretic mobility of molecules.48 Lee and co-workers fabricated a linear converging channel (Fig. 1f), which yielded similar results, resulting in rapid chain relaxation and compression in an expansion zone following the channel outlet.49 Geometrical variations indicated that shorter channels and channels with smaller contraction outlets enhance stretching. Lee and co-workers also investigated transport mechanisms of polymers and hard spheres through converging and diverging nozzles, finding distinct migration behaviours for the two materials.50 Interestingly, in converging nozzles, nanoparticles were observed to associate and block the nozzle outlet, while single DNA molecules migrated through the nozzle outlet; in diverging nozzles, opposite behaviour occurred.
2. Cross-slot devices
Microfluidic devices with cross-slot geometries have been instrumental in studying single polymer dynamics in extensional flow. Cross-slot devices have been extensively used to generate planar extensional flows, and other applications have included flow-focusing, filament thinning, and microfluidic adaptations of the four-roll mill. Pressure gradients or electric field gradients are often used to drive flow through cross-slot geometries.
2.1. Pressure-driven flow
Stagnation point flow
Cross-slot geometries can be used to generate planar extensional flows by directing two opposing inlet streams to converge near the centre of the cross-slot, with two opposing outlet streams emerging in orthogonal directions to the inlet streams (Fig. 1g). The velocity profile (νx, νy,) in planar extensional flow is given by Equations (1) and (2), where ε̇ is the strain rate of the fluid:
| (1) |
| (2) |
Near the centre of the cross-slot, the flow exhibits a stagnation point or point of zero velocity (Fig. 5, inset).
Fig. 5.

DNA stretching in a planar extensional flow uncovered the effects of initial polymer conformation on the transient extension of single DNA molecules, thereby revealing "molecular individualism." Top to bottom: dumbbell, kinked, half-dumbbell, and folded conformations (sketches of molecular configurations are included on the left). Time between images is 0.13 seconds. Inset: planar extensional flow. From T. T. Perkins, D. E. Smith and S. Chu, Science, 1997, 276, 2016-2021. Reprinted with permission from AAAS.
Chu and co-workers performed the earliest studies of single polymer dynamics in microfluidic devices using a stagnation point flow.51, 52 Single polymer studies of fluorescently-labelled λ-DNA molecules showed that transient and steady molecular extension were strong functions of the flow strength, characterized by the Deborah number (De = ε′τ), molecular residence time, and the initial conformation of individual molecules. Upon analysing the subset molecules that had reached a steady state extension for a given flow strength, the authors provided clear experimental evidence of a sharp coil-stretch transition near De = 0.5. These results underscored the importance of "molecular individualism" (Fig. 5) and highlighted challenges associated with interpreting bulk or ensemble-averaged measurements in complex fluids.
Single polymer experiments and Brownian dynamics simulations were further used to study the effects of intramolecular hydrodynamic interactions (HI) on polymer chain dynamics in extensional flow. For DNA molecules with large contour lengths (L = 1.3 mm), Schroeder et al. observed hysteresis in the coil-stretch transition for polymers in extensional flow.53, 54 More recently, we developed a new system to study single polymer dynamics using fluorescently-labelled ssDNA, which are truly flexible polymer chains with dominant HI and excluded volume interactions.9
In planar extensional flow, the stagnation point is a semi-stable point for centre-of-mass confinement in the flow field. Active control over the stagnation point position allows for single polymers to be confined in the extensional flow field for extremely long residence times. Manual control over the stagnation point was used to observe polymer hysteresis,53 to stretch molecules for DNA sequence detection,55 and to characterize salt-induced compaction of DNA from a stretched state.56 For DNA sequence detection, a fluorescently-labelled restriction endonuclease was bound to one of five target sites along λ-DNA, and the positions of the target binding sites were determined to within 1.5 kilobases.55 Salt-induced compaction was studied by directing two different solutions with variable salt concentrations in the opposing inlet streams. Stretched DNA molecules exposed to high salt concentrations in the presence of polyethylene glycol resulted in molecular compaction into a coiled state on a time scale much shorter than the longest relaxation time of the molecules.
Hydrodynamic trap
Recently, our group developed an automated feedback control system to trap single particles, single cells, or single polymer molecules for extended periods of time.57-59 The automated hydrodynamic trap will enable long time measurements of single polymer molecules over a wide range of flow rates, including large fluid strain rates that are not accessible by manual trapping.
Microfluidic four-roll mill
Aside from purely extensional flow, linear mixed flows with varying amounts of extensional and rotational components are useful for studying polymer dynamics. In order to generate linear mixed flows ranging in character from purely rotational to purely extensional to simple shear flow (50% rotation/50% extension), several groups have designed and fabricated microfluidic analogues to the four-roll mill using modified cross-slot configurations. Hudson et al. developed the first microfluidic four-roll mill using six inlet/outlet channels arranged in a cross configuration.60 This device was capable of producing a range of flow types, including purely extensional and simple shear flows. Muller and co-workers further developed a four-roll mill analogue with a cross-slot geometry capable of producing extensional-dominated flows as well as purely rotational flow.61 DNA tumbling dynamics in shear and rotational flows was studied using this device.62
Flow focusing
In addition to planar extensional flow, cross-slot microfluidic devices can also generate uniaxial extensional flow via flow focusing, whereby two opposing inlet streams impinge on a third, middle inlet stream. Flow focusing was used to study the deformation and relaxation of individual T2 DNA molecules, demonstrating that focusing flow streams relax the no-slip condition present in other extensional flow fields and provide a more homogeneous extensional profile.63
Filament thinning
Arratia and co-workers used microfluidic devices to simultaneously measure bulk flow properties and single molecule stretching in DNA suspensions.64 Dilute and semi-dilute DNA solutions with varying molecular weights were compared, and it was observed that larger polymer molecular weights and semi-dilute DNA solutions resulted in non-Newtonian extensional strain rate thinning extensional viscosities. Bulk flow measurements were compared to the behaviour of individual molecules inside the fluid filament using fluorescence microscopy, where it was found that the extensional flow strength inside the thinning filaments was sufficient to extend the molecules far from their equilibrium coiled state, while maintaining a broad distribution of molecular extensions.64
2.2. Electrokinetic flow
Stagnation point flow
Dynamic measurements of "free-solution" polymer stretching are typically made near the centreline of microfluidic devices, in order to minimize wall effects. In an alternative approach, the Lee group used electrokinetic-driven flows in a cross-slot device. In this work, microdevices exhibited plug-like flow profiles due to minimal pressure drops and were relatively simple to fabricate. Experiments with λ-DNA molecules resulted in similar behaviour in extensional flow in comparison to previous hydrodynamic measurements, demonstrating the viability of electrokinetic devices.65 An improved device that employed a five cross design with twelve independent electrodes was capable of generating a wide variety of flow types, including extensional, mixed linear flows (near shear), and rotational flows.66
Doyle and co-workers used electric fields to generate an extensional flow in microscale T-junctions67 and in nanoslit cross-slot channels.68 The work resulting from nanoslit confinement showed evidence of an extension-dependent longest relaxation time (Section 4). More recently, the effects of strong electric fields on DNA molecules revealed that molecules will form self-entanglements under some conditions.46 The presence of self-entanglements was verified by stretching compressed DNA molecules in a cross-slot channel, where compressed molecules required much larger forces to extend, and bright spots indicating knot formation persisted along stretched molecules.
ABEL trap
Moerner and co-workers developed a method for trapping individual particles using feedback-controlled electric fields in a quadrupole configuration. Using the Anti-Brownian ELectrophoretic (ABEL) Trap, particle position was determined via image processing, and a feedback voltage was applied to a quadrupole electrode setup such that electrophoretic drift in solution cancelled a particle's Brownian motion.69 An extensive review of the trap and applications can be found elsewhere.70 Using the ABEL trap, Cohen et al. studied the equilibrium motion and fluctuations of single λ-DNA molecules for extended periods of time without perturbing internal dynamics, observing evidence of internal hydrodynamic interactions that were not observed in previous measurements of freely diffusing DNA molecules.71, 72
3. Obstacles
Gel electrophoresis is commonly used for DNA separation and analysis. Initial observations of individual DNA molecules undergoing gel electrophoresis revealed a hairpin hooking and unhooking mechanism as molecules travel through the polymer network.73, 74 The transient hooking/unhooking mechanism sparked interest in fabricating devices that are capable of controlling the mobility of DNA fragments of variable length, thereby enabling separation of DNA based on molecular weight. DNA separation devices are designed to contain arrays of posts, such that DNA molecules collide with microfabricated obstacles, which increases the residence time of DNA in the arrays.75, 76
A fundamental understanding of single polymer dynamics and interactions with obstacles allows for devices to be engineered for electrophoresis and other applications. A recent review by Dorfman provides a comprehensive overview of microfabricated devices for electrophoresis, including theory, fabrication, simulations, and experiments.77 Therefore, in this discussion, we simply highlight influential work on dynamics and interactions of DNA molecules with single obstacles and arrays of obstacles.
3.1. Single obstacles
Upon collision with a single obstacle, DNA molecules may roll off the obstacle or engage in a hooking event.78 For hooking events, a molecule extends and slides around the obstacle until one end unhooks, and the entire molecule subsequently relaxes to a coiled state. For collisions with a single post, the probability (Phook), time (tH), and molecular configuration throughout the hooking event have been systematically studied.
Doyle and co-workers extensively studied the collision of DNA molecules with a single post in a large body of experimental work.78-80 Three main effects were observed for impact dynamics (Fig. 6): obstacle size Robs, offset between the DNA molecule and obstacle's centres of masses b, and electric field strength. The parameters were non-dimensionalized as a ratio of length scales , an "impact parameter" , and Deborah number , based on the strain rate for a given electrophoretic mobility μ and electric field strength E.
Fig. 6.
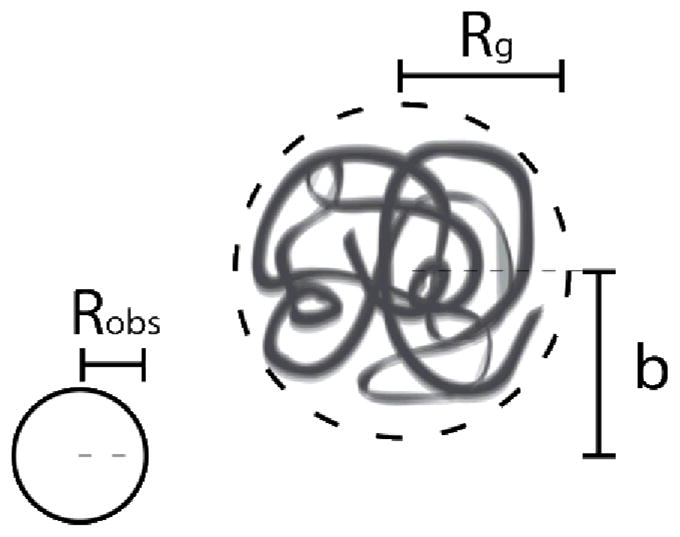
Schematic of a single DNA-post collision, with parameters including the polymer radius of gyration Rg, obstacle size Robs, and offset between centres of masses b.
For centreline collisions (b = 0) in the context of a large insulating post (fabricated with PDMS, ), pre-impact configuration of DNA significantly affected extension upon contacting the obstacle.78, 79 Furthermore, DNA molecules compressed affinely in the region near the rear of the obstacle.79 Insulated posts were observed to induce electric field gradients that extended DNA prior to and following the collision event.
For obstacles with dimensions on the same order as the coil size ( ), the probability of hooking Phook was observed to increase upon increasing the electric field strength and decreasing the impact parameter.78 Araki et al. observed the same dependence for Phook on De and for zinc oxide nanowires for features approaching the size limit of a point obstacle ( ). Moreover, these authors developed scaling arguments for the time of hooking around a thin post, defining tH as the time elapsed between the leading edge of DNA molecule reaching the post and the trailing end unhooking from the post, such that .81
Randall and Doyle further classified hooking collisions with small obstacles into four types (Fig. 7): U, J, W, and X (for "extending").80 In U and J collisions, DNA molecules follow rope-on-a-pulley behaviour, maintaining constant extension while unhooking. U and J collisions are distinguished by symmetric (U?shaped) or asymmetric (J?shaped) "arms" around the post. W collisions result from two ends of a molecule on the same side of the obstacle after impact. X collisions are similar to J collisions, except that the long arm continues extending while the short arm relaxes and retracts. Single molecule experiments were used to develop detailed models of the unhooking dynamics for each collision type.80
Fig. 7.

Classifications for hooking collisions of DNA molecules with a single post: (a) U symmetric hooks, (b) J asymmetric hooks, (c) W entangled hooks, and (d) X continuously extending hooks. Time between images is 1.33 seconds. Adapted with permission from G. C. Randall and P. S. Doyle, Macromolecules, 2006, 39, 7734-7745. Copyright 2006 American Chemical Society.
3.2. Array of obstacles
Early studies of DNA separation in microfabricated devices focused on post arrays75, 76 rather than single posts. Obstacle size, obstacle shape, and electric field strength were found to affect the dynamics of collisions with single posts. Moreover, obstacle spacing (including frequency, orientation, and regularity) introduces another set of parameters that significantly affect the dynamics of polymer motion through an obstacle array.
A systematic experimental evaluation of the full parameter space of post arrays has not yet been carried out.77 Challenges in fabrication methods have driven recent work focused on fabrication of nanoscale devices for biomolecule separation.82 The ability to easily tune the aforementioned parameters may enable a complete systematic study of dynamics. Due to fabrication challenges and a myriad of mechanisms for DNA motion through obstacle arrays, recent work has focused on the use of post arrays for DNA separations.77
Several groups studied the dynamics of single DNA molecules in post arrays using experiments and simulations. Doyle et al.83 and Minc et al.84, 85 varied the density of disordered post arrays with columns of self-assembled magnetic beads. Using electrophoretic flow, they achieved rapid separation of large fragment DNA84 and measured DNA velocity and dispersivity of the same order of magnitude as lattice Monte Carlo simulations.85 Ou et al. highlighted the importance of accounting for electric field gradients when considering DNA motion through a sparse ordered post array.86 Teclemariam et al. compared experiments and simulations of DNA transport in a post array with pressure-driven flow. Interestingly, this study demonstrated the ability to control DNA conformation and guide the location of hooking events by designing appropriate array geometries.87
4. Confinement
Nanoscale devices are often used to study single polymer molecules under confinement. Fundamental investigations of confined polymer behaviour are motivated by practical applications including polymer separation in porous media, nanofluidic sensors, and DNA sequencing. In devices with nanoscale dimensions, entropic forces significantly affect molecule conformation and dynamics. Polymers are "confined" when placed in geometries with at least one dimension d between the persistence length p and equilibrium coil size Rg of the polymer (e.g., p < d <Rg).
The degree of confinement influences the dynamical behaviour of polymers and is classified by the scale of d relative to Rg and p.88-91 The regimes of confinement include weak (bulk), moderate (de Gennes), and strong (Odijk) confinement. In weak confinement, the confining dimension is larger than the equilibrium coil size (d > Rg). In the de Gennes regime, the confining dimension is larger than the persistence length, but smaller than the radius of gyration (p ≪ d <Rg) . Finally, in the Odijk regime, the confining dimension is smaller than the persistence length (d < p).
Features with the required dimensions for confinement can be incorporated into nanofluidic devices. Fabrication generally involves lithography or etching on silicon or glass-like materials in order to maintain precise control over the structure.92 Lee and co-workers have also developed a patterning technique that involves DNA combing and imprinting, which holds promise for lowering the cost of controlled, nanoscopic fabrication.93
The fundamental behaviour of confined polymers has been reviewed elsewhere. 94, 92 Here, we focus on two types of confinement with particular attention to microfluidic platforms (Fig. 8): slit-like (one confining dimension) and tube-like (two confining dimensions). Experimental challenges limited early studies of single polymers in confinement to computational and theoretical methods,95 but recent advances in nanofabrication96, 97 and single molecule microscopy have allowed for direct visualization of confinement phenomena.
Fig. 8.
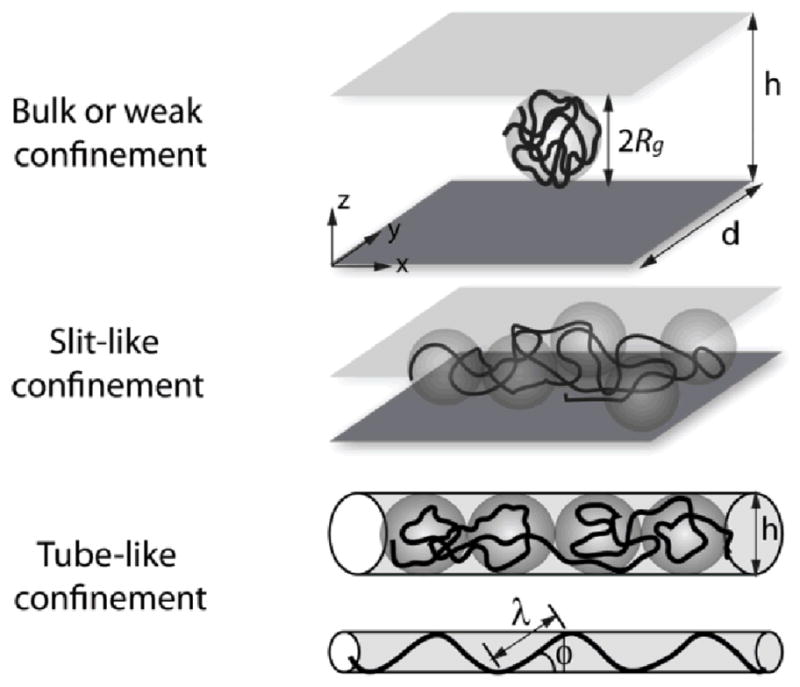
Schematics comparing polymer conformations in bulk solution, slit-like confinement, and tube-like confinement given polymer radius of gyration Rg and confinement dimensions h and d. Adapted from Ref. 94, with kind permission from Springer Science + Business Media and the Korean Society of Rheology.
4.1. Slit-like confinement
Microfluidic devices imposing slit-like confinement on molecules generally involve a microchannel with one sub-micrometre dimension, typically channel height h. As described, the strength of the confining region on a polymer chain depends on the polymer equilibrium radius of gyration and persistence length.
Early single molecule experiments revealed dramatic effects of confinement on polymer behaviour using arrays of posts in nanofabricated slits of varying heights.98 Bakajin et al. used electrophoretic forces to drive DNA molecules through an array and observed longer relaxation times of extended DNA molecules in shallower slits, suggesting an increase in hydrodynamic drag under confinement.98 Doyle and co-workers also studied DNA dynamics in slit-like confinement, including scaling of mobility, diffusion, and relaxation, and qualitative observations such as molecular conformation. Quantitative analyses of slit-like confinement by several groups have led to inconsistencies between numerical values for scaling exponents and transitional behaviour between regimes of confinement, which clearly motivated future studies in the field.92, 94, 99
Scaling in regimes of confinement
In the de Gennes regime (p ≪ h <Rg), a confined polymer can be described as a chain of self-avoiding blobs with diameter h.88, 89 Balducci et al.100 and Lin et al.101 measured an inverse scaling relation between DNA diffusivity and molecular weight in this regime, suggesting that the surfaces of the slit screened intramolecular hydrodynamic interactions. Balducci et al. also observed two relaxation regimes in moderate confinement,102 where the faster relaxation regime corresponds to conformational relaxation98 and the slower relaxation regime agrees with rotational relaxation times. 101,103
Additional work has focused on polymer behaviour in the transition between de Gennes (p ≪ h < Rg) and Odijk p ≈ h ≪Rg) confinement. In this regime, diffusivity D was observed to scale with height such that D ≈ h0.5, with no observable sharp transition between confinement regimes, even for h ≫ p. 100, 103, 104 In contrast to gradual scaling observed for diffusivity, Bonthuis et al. observed sharp transitions in relaxation time τ and radius of gyration between the de Gennes and Odijk regimes, such that Rg was independent of h and τ decreased with h in the Odijk regime.105
Two recent studies revisited this transition, focusing on conformation, diffusion, and relaxation in slit-like devices.99, 106 Tang et al. observed a broad, gradual transition of radius of gyration, diffusivity, and relaxation time,99 in contrast to the findings of Bonthuis et al.105 but in agreement with most other results.10, 103, 104 The gradual transition was further supported by Strychalski et al.106, 107 using a novel device consisting of a nanofluidic staircase108 that entropically drives transport of a single DNA molecule from strong to moderate confinement. The staircase varied stepwise in height to allow for observation of a single molecule under slit-like confinement at varying heights while maintaining all other conditions.106 This work is also one of few studies to explore the dynamics of nonlinear polymers using circular DNA.106, 107
The effect of confinement on DNA mobility was characterized in slits with varying height.17, 109 Stein et al. observed two distinct regimes of transport in pressure-driven flow, such that mobility increased with molecular weight in weak confinement (h > 3Rg), and mobility had no dependence on molecular weight under moderate confinement (h < Rg).17 Interestingly, electrophoretic mobility showed the opposite effect, decreasing with increasing molecular weight in strong confinement but showing no substantial difference in moderate confinement.109
A nanofluidic cross-slot device was also used to characterize relaxation times68 and the coil-stretch transition110 of electrophoretically-stretched DNA molecules in weak confinement. Similarly to studies of the de Gennes regime, two relaxation regimes were observed.68 Furthermore, the onset of stretching began earlier, and the coil-stretch transition occurred gradually with two distinct critical strain rates.110 Confinement enhanced stretching, trapping, and imaging focus of the molecules in comparison to other cross-slot devices (Section 2).
DNA conformation in nanofabricated structures
A remarkable feature of polymer behaviour under confinement is shape anisotropy,101, 105-107 which is manifested by spontaneous chain extension.111 In free solution, polymer molecules generally form isotropic coils. In confinement, conformational dynamics are attributed to the balance between entropic compression by the slit and electrostatic repulsion within the charged DNA backbone. Electrostatic repulsion serves as an opposing force on DNA conformation in confinement, and this phenomenon has been investigated in both slit- and channel-like confinement by observing conformation in solutions of varying ionic strengths. In slit-like confinement, lower ionic strengths resulted in increased extension of both linear and circular DNA molecules, which was attributed to fewer ions available to screen electrostatic repulsions within DNA.112 Interestingly, scaling exponents for radius of gyration and relaxation time with varying ionic strength and slit height were the same for confined linear and circular molecules.113
Studies of entropic effects in slit-like confinement have involved a wide array of artificial topographies. Nanopillar array devices generally consist of an entropic interface between regions of slit-like confinement with and without nanopillars. DNA molecules can be driven across the interface using an electric field. Upon removal of the field, molecules stretched across the interface undergo entropic recoil into the region without nanopillars, whereas molecules fully incorporated into the array would remain stationary.114 Such devices have also been used to separate DNA molecules of different lengths115 (Section 3).
Entropic interfaces have also been studied using arrays of nanopits. A proof-of-principle experiment by Reisner et al. demonstrated the ability to influence position and conformation of DNA molecules, which spontaneously filled entropically favourable nanopits.116 This system has been further extended to tune dynamic properties of single DNA molecules such as diffusion.117 Stein et al. also utilized pressure-driven flow over nanopits, wherein DNA molecules exhibited single- or multiple-pit occupancy behaviour based on dimensions, and DNA were transported through the device by "hopping" between pits.118 Similar behaviour was observed using fabricated channels in which one or both lateral walls took the shape of a periodic waveform.119, 120 A uniform electric field induced DNA migration, such that molecules stretched over protruding wave structures before contracting into globular conformation between protrusions.119
Finally, an entropic trap has been developed by introducing an close-fitting oil droplet into a microfluidic device (Fig. 9).121 The trap allowed for observation of extension and self-assembly of DNA molecules in slit-like confinement without fabrication of a nanoscale device.
Fig. 9.
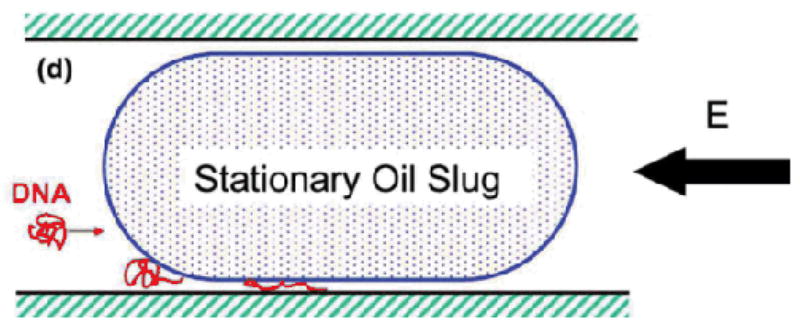
Deformation of a DNA molecule that is electrokinetically driven into confinement between a stationary oil "slug" and microchannel wall. Reprinted figure with permission from S.-F. Hsieh and H.-H. Wei, Physical Review E, 79, 021901, 2009. Copyright (2009) by The American Physical Society.
4.2. Tube-like confinement
Experimental studies of tube-like confinement of single polymers require fabrication of devices with two sub-micrometre dimensions, typically the height and width of rectangular channels. Guo et al. pioneered this fabrication process using nanoimprint lithography,122 and entropic forces in the resulting devices with tube-like confinement further constrain polymer molecules than in slit-like confinement. The increased entropic effects ultimately influence the natural extension of DNA driven through nanotubes by capillary force.122
Several groups characterized the extension of DNA molecules in tube-like confinement, measuring linear scaling between end-to-end distance and contour length123 and power law scaling with channel size.124, 125 The power law scaling of linear molecules deviated from predictions of blob theory; however, the scaling of circular DNA molecules appears to agree with blob theory,124, 125 which clearly motivates additional computational studies and/or experimental work.92, 94 Reisner et al. also observed a qualitative shift in extension for channels with smaller dimensions, possibly corresponding to a transition from the de Gennes to Odijk confinement regimes.124 Fluctuations of internal DNA segments were also probed in the transitional regime, revealing folded, blob-like structures separated by relatively straight "Odijk-like" segments with a mean average extension of 10 μm.126 Locally compact, non-stretched regions of DNA could significantly impact the resolution of genomic mapping applications that rely on DNA confinement.
Craighead and co-workers conducted pioneering work on tube-like confinement, reporting the first measurements of relaxation from a compressed state, rather than an extended state127 and observing conformational effects on electrokinetic transport through nanochannels.128 The group also studied entropic forces acting on polymers in tube-like confinement by stretching DNA across an interface between slit-like confinement and nanochannels,129 in similar fashion to the entropic studies of slit-like confinement using nanopillars.114, 115 The nanochannel study simplified the optical analysis in comparison to the nanopillar study and revealed several types of DNA motion in tube-like confinement, including relaxation, unfolding, and ultimately entropic recoil (Fig. 10).129 Further investigation of folded DNA molecules in tube-like confinement indicate increased extension of folded portions and entropically-induced unfolding, likely due to excluded volume interactions.130
Fig. 10.

Schematics of polymer behaviour in tube-like confinement: (A) a relaxed polymer molecule is recoils by entropic forces across interface at nanochannel entrance; (B) a folded molecule unfolds and recoils simultaneously; (C) a molecule driven electrophoretically into the nanochannel relaxes to an equilibrium extension length; (D) a molecule driven partially into the nanochannel relaxes and recoils simultaneously. Reprinted from Biophysical Journal, 90 (12), J. T. Mannion, C. H. Reccius, J. D. Cross and H. G. Craighead, Conformational Analysis of Single DNA Molecules Undergoing Entropically Induced Motion in Nanochannels, 4538-4545, Copyright (2006), with permission from Elsevier.
Studies of electrostatic repulsion on DNA molecules in tube-like confinement have reported an increase in DNA extension with reduced ionic concentrations.131-133 Optimized experimental conditions allowed for stretching a DNA molecule to nearly ninety per cent of its contour length.133 Reisner et al. determined that the increase in persistence length was too drastic to be explained by only the change in salt concentration, suggesting an increase in self-avoidant chain behaviour due to reduced screening of electrostatic interactions within the charged DNA backbone.131 Interestingly, the introduction of a neutral crowding agent, such as dextran nanoparticles, can lead to both extension and compaction of DNA molecules in confinement at low salt concentrations.134, 135 Prior to compaction, the crowding agent promoted extension by reducing the effective channel diameter. Upon reaching a threshold volume of dextran, the DNA molecules collapsed into a globular state.134 While this behaviour paralleled DNA response to dextran in nanoslit confinement, it appears to deviate from the behaviour in bulk solution.135
Perspectives
Recent advances in microfabrication and novel microfluidic geometries and have directly facilitated the study of polymer dynamics at the molecular level. Microfluidic platforms have enabled forays into challenging areas of polymer physics, providing new perspectives in single polymer dynamics and opening the door to further explorations. As discussed in this review, double stranded DNA has served as the "model macromolecule" for single polymer studies in non-equilibrium fluid flows. In the near future, the horizons of this field will be expanded to allow for the study of a wide variety soft materials at the molecular level, including flexible polymers, branched polymers, and copolymers. Furthermore, the study of entangled solutions and/or complex flows will allow for improved connections between polymer microstructure, macroscopic flow properties, and processing of industrially relevant materials.
Previous attempts at single molecule studies of fluorescently-labelled synthetic organic polymers have been limited.136 However, there is a strong need to study new materials at the molecular level, thereby broadening the range of material properties for single molecule studies. Recently, our group synthesized and observed long molecules of single stranded DNA,9 which is a promising platform for flexible polymers. In addition, a few studies have explored architectural effects using star-branched DNA137, 138 or DNA nanostructures,139 though these investigations often focus on the synthesis and biological applications of these materials.
A second key need in the field is in-depth studies of the dynamics of entangled polymer solutions in flow. Smith and co-workers have extensively investigated the diffusion of entangled linear and circular DNA molecules.140-142 These findings clearly motivate additional studies of extensional and mixed flows, which hold the potential to enable development of new molecular-based models and constitutive relations. A recent and interesting example of single polymer studies in entangled conditions involved uniaxial extension of a film, which was imaged using scanning near-field optical microscopy.143 Advanced imaging techniques such as high-resolution and/or super-resolution fluorescence imaging methods have also begun to appear in single polymer studies,144 potentially leading to exciting discoveries by imaging at the nanoscale (20 nm), far below the diffraction limit for optical microscopy (~250 nm).
Finally, we envision the convergence of single polymer studies with "bulk" rheometry in the context of microfluidic devices, as recently demonstrated by coupling confocal microscopy with a torsional flow cell rheometer.145 Such work can be further aided by automating microfluidic devices, which would further enable the study of soft materials in well-defined ways.57-59 Microfluidic trapping and lab-on-chip devices demonstrate the ability to exert precise control over nanoparticle or polymer dynamics, all of which will be useful in making the connection between microstructure and bulk material properties.
Acknowledgments
This work was supported by a Packard Fellowship from the David and Lucille Packard Foundation and an NIH Pathway to Independence Award (4R00HG004183-03).
References
- 1.Pecora R. Science. 1991;251:893–898. doi: 10.1126/science.2000490. [DOI] [PubMed] [Google Scholar]
- 2.Shaqfeh ESG. Journal of Non-Newtonian Fluid Mechanics. 2005;130:1–28. [Google Scholar]
- 3.Ladoux B, Quivy J-P, Doyle PS, Almouzni G, Viovy J-L. Science Progress. 2001;84:267–290. doi: 10.3184/003685001783238961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Perkins TT, Quake SR, Smith DE, Chu S. Science. 1994;264:822–826. doi: 10.1126/science.8171336. [DOI] [PubMed] [Google Scholar]
- 5.Perkins TT, Smith DE, Chu S. Science. 1994;264:819–822. doi: 10.1126/science.8171335. [DOI] [PubMed] [Google Scholar]
- 6.Perkins TT, Smith DE, Larson RG, Chu S. Science. 1995;268:83–87. doi: 10.1126/science.7701345. [DOI] [PubMed] [Google Scholar]
- 7.Bustamante C, Smith SB, Liphardt J, Smith D. Current Opinion in Structural Biology. 2000;10:279–285. doi: 10.1016/s0959-440x(00)00085-3. [DOI] [PubMed] [Google Scholar]
- 8.Latinwo F, Schroeder CM. Soft Matter. 2011;7:7907–7913. doi: 10.1039/c1sm05298e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Brockman C, Kim SJ, Schroeder CM. Soft Matter. 2011;7:8005–8012. doi: 10.1039/C1SM05297G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hu X, Boukany PE, Hemminger OL, Lee LJ. Macromolecular Materials and Engineering. 2011;296:308–320. [Google Scholar]
- 11.Pipe CJ, McKinley GH. Mechanics Research Communications. 2009;36:110–120. [Google Scholar]
- 12.Lin XQ, Li GX. Prog Chem. 2011;23:800–809. [Google Scholar]
- 13.Howorka S, Siwy Z. Chem Soc Rev. 2009;38:2360–2384. doi: 10.1039/b813796j. [DOI] [PubMed] [Google Scholar]
- 14.Whitesides GM. Nature. 2006;442:368–373. doi: 10.1038/nature05058. [DOI] [PubMed] [Google Scholar]
- 15.Squires TM, Quake SR. Reviews of Modern Physics. 2005;77:977–1026. [Google Scholar]
- 16.Shrewsbury PJ, Muller SJ, Liepmann D. Biomedical Microdevices. 2001;3:225–238. [Google Scholar]
- 17.Stein D, van der Heyden FHJ, Koopmans WJA, Dekker C. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:15853–15858. doi: 10.1073/pnas.0605900103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Smith DE, Babcock HP, Chu S. Science. 1999;283:1724–1727. doi: 10.1126/science.283.5408.1724. [DOI] [PubMed] [Google Scholar]
- 19.LeDuc P, Haber C, Bao G, Wirtz D. Nature. 1999;399:564–566. doi: 10.1038/21148. [DOI] [PubMed] [Google Scholar]
- 20.Babcock HP, Smith DE, Hur JS, Shaqfeh ESG, Chu S. Physical Review Letters. 2000;85:2018–2021. doi: 10.1103/PhysRevLett.85.2018. [DOI] [PubMed] [Google Scholar]
- 21.Hur JS, Shaqfeh ESG, Babcock HP, Smith DE, Chu S. Journal of Rheology. 2001;45:421–450. [Google Scholar]
- 22.Teixeira RE, Babcock HP, Shaqfeh ESG, Chu S. Macromolecules. 2005;38:581–592. [Google Scholar]
- 23.Schroeder CM, Teixeira RE, Shaqfeh ESG, Chu S. Macromolecules. 2005;38:1967–1978. [Google Scholar]
- 24.Schroeder CM, Teixeira RE, Shaqfeh ESG, Chu S. Physical Review Letters. 2005:95. doi: 10.1103/PhysRevLett.95.018301. [DOI] [PubMed] [Google Scholar]
- 25.Teixeira RE, Dambal AK, Richter DH, Shaqfeh ESG, Chu S. Macromolecules. 2007;40:2461–2476. [Google Scholar]
- 26.Li L, Hu H, Larson RG. Rheologica Acta. 2004;44:38–46. [Google Scholar]
- 27.Fang L, Larson RG. Macromolecules. 2007;40:8784–8787. [Google Scholar]
- 28.Fang L, Hsieh C-C, Larson RG. Macromolecules. 2007;40:8490–8499. [Google Scholar]
- 29.Fang L, Hu H, Larson RG. Journal of Rheology. 2005;49:127–138. [Google Scholar]
- 30.Babcock HP, Teixeira RE, Hur JS, Shaqfeh ESG, Chu S. Macromolecules. 2003;36:4544–4548. [Google Scholar]
- 31.Jo K, Chen Y-L, de Pablo JJ, Schwartz DC. Lab on a Chip. 2009;9:2348–2355. doi: 10.1039/b902292a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shrewsbury PJ, Liepmann D, Muller SJ. Biomedical Microdevices. 2002;4:17–26. [Google Scholar]
- 33.Larson JW, Yantz GR, Zhong Q, Charnas R, D'Antoni CM, Gallo MV, Gillis KA, Neely LA, Phillips KM, Wong GG, Gullans SR, Gilmanshin R. Lab on a Chip. 2006;6:1187–1199. doi: 10.1039/b602845d. [DOI] [PubMed] [Google Scholar]
- 34.Kim JM, Doyle PS. Lab on a Chip. 2007;7:213–225. doi: 10.1039/b612021k. [DOI] [PubMed] [Google Scholar]
- 35.Shaqfeh ESG. Annu Rev Fluid Mech. 1996;28:129–185. [Google Scholar]
- 36.Rodd LE, Scott TP, Boger DV, Cooper-White JJ, McKinley GH. Journal of Non-Newtonian Fluid Mechanics. 2005;129:1–22. [Google Scholar]
- 37.Rodd LE, Cooper-White JJ, Boger DV, McKinley GH. Journal of Non-Newtonian Fluid Mechanics. 2007;143:170–191. [Google Scholar]
- 38.Gulati S, Muller SJ, Liepmann D. Journal of Non-Newtonian Fluid Mechanics. 2008;155:51–66. [Google Scholar]
- 39.Hemminger OL, Boukany PE, Wang S-Q, Lee LJ. Journal of Non-Newtonian Fluid Mechanics. 2010;165:1613–1624. [Google Scholar]
- 40.Gulati S, Dutcher CS, Liepmann D, Muller SJ. Journal of Rheology. 2010;54:375–392. [Google Scholar]
- 41.Cheng C-M, Kim Y, Yang J-M, Leuba SH, LeDuc PR. Lab on a Chip. 2009;9:2339–2347. doi: 10.1039/b907860f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Viovy J-L. Reviews of Modern Physics. 2000;72:813–872. [Google Scholar]
- 43.Chiesl TN, Putz KW, Babu M, Mathias P, Shaikh KA, Goluch ED, Liu C, Barron AE. Analytical Chemistry. 2006;78:4409–4415. doi: 10.1021/ac060193u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chiesl TN, Forster RE, Root BE, Larkin M, Barron AE. Analytical Chemistry. 2007;79:7740–7747. doi: 10.1021/ac071160x. [DOI] [PubMed] [Google Scholar]
- 45.Forster RE, Hert DG, Chiesl TN, Fredlake CP, Barron AE. Electrophoresis. 2009;30:2014–2024. doi: 10.1002/elps.200900264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tang J, Du N, Doyle PS. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:16153–16158. doi: 10.1073/pnas.1105547108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Randall GC, Schultz KM, Doyle PS. Lab on a Chip. 2006;6:516–525. doi: 10.1039/b515326c. [DOI] [PubMed] [Google Scholar]
- 48.Liao W-C, Watari N, Wang S, Hu X, Larson RG, Lee LJ. Electrophoresis. 2010;31:2813–2821. doi: 10.1002/elps.201000081. [DOI] [PubMed] [Google Scholar]
- 49.Hu X, Wang S, Lee LJ. Physical Review E. 2009;79:041911. doi: 10.1103/PhysRevE.79.041911. [DOI] [PubMed] [Google Scholar]
- 50.Wang S, Hu X, Lee LJ. Lab on a Chip. 2008;8:573–581. doi: 10.1039/b719410b. [DOI] [PubMed] [Google Scholar]
- 51.Perkins TT, Smith DE, Chu S. Science. 1997;276:2016–2021. doi: 10.1126/science.276.5321.2016. [DOI] [PubMed] [Google Scholar]
- 52.Smith DE, Chu S. Science. 1998;281:1335–1340. doi: 10.1126/science.281.5381.1335. [DOI] [PubMed] [Google Scholar]
- 53.Schroeder CM, Babcock HP, Shaqfeh ESG, Chu S. Science. 2003;301:1515–1519. doi: 10.1126/science.1086070. [DOI] [PubMed] [Google Scholar]
- 54.Schroeder CM, Shaqfeh ESG, Chu S. Macromolecules. 2004;37:9242–9256. [Google Scholar]
- 55.Dylla-Spears R, Townsend JE, Jen-Jacobson L, Sohn LL, Muller SJ. Lab on a Chip. 2010;10:1543–1549. doi: 10.1039/b926847b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Xu W, Muller SJ. Lab on a Chip. 2012;12:647–651. doi: 10.1039/c2lc20880f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tanyeri M, Johnson-Chavarria EM, Schroeder CM. Applied Physics Letters. 2010:96. doi: 10.1063/1.3431664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Johnson-Chavarria EM, Tanyeri M, Schroeder CM. J Vis Exp. 2011:e2517. doi: 10.3791/2517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Tanyeri M, Ranka M, Sittipolkul N, Schroeder CM. Lab on a Chip. 2011:11. doi: 10.1039/c1lc20604d. [DOI] [PubMed] [Google Scholar]
- 60.Hudson SD, Phelan FR, Handler MD, Cabral JT, Migler KB, Amis EJ. Applied Physics Letters. 2004;85:335–337. [Google Scholar]
- 61.Lee JS, Dylla-Spears R, Teclemariam NP, Muller SJ. Applied Physics Letters. 2007:90. [Google Scholar]
- 62.Lee JS, Shaqfeh ESG, Muller SJ. Physical Review E. 2007:75. doi: 10.1103/PhysRevE.75.040802. [DOI] [PubMed] [Google Scholar]
- 63.Wong PK, Lee Y-K, Ho C-M. Journal of Fluid Mechanics. 2003;497:55–65. [Google Scholar]
- 64.Juarez G, Arratia PE. Soft Matter. 2011;7:9444–9452. [Google Scholar]
- 65.Juang YJ, Wang S, Hu X, Lee LJ. Physical Review Letters. 2004;93:268105. doi: 10.1103/PhysRevLett.93.268105. [DOI] [PubMed] [Google Scholar]
- 66.Hu X, Wang SN, Juang YJ, Lee LJ. Applied Physics Letters. 2006:89. [Google Scholar]
- 67.Tang J, Doyle PS. Applied Physics Letters. 2007:90. [Google Scholar]
- 68.Balducci AG, Tang J, Doyle PS. Macromolecules. 2008;41:9914–9918. [Google Scholar]
- 69.Cohen AE, Moerner WE. Applied Physics Letters. 2005:86. [Google Scholar]
- 70.Fields AP, Cohen AE. Methods in enzymology. 2010;475:149–174. doi: 10.1016/S0076-6879(10)75007-2. [DOI] [PubMed] [Google Scholar]
- 71.Cohen AE, Moerner WE. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:12622–12627. doi: 10.1073/pnas.0610396104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Cohen AE, Moerner WE. Physical Review Letters. 2007:98. doi: 10.1103/PhysRevLett.98.116001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Smith SB, Aldridge PK, Callis JB. Science. 1989;243:203–206. doi: 10.1126/science.2911733. [DOI] [PubMed] [Google Scholar]
- 74.Schwartz DC, Koval M. Nature. 1989;338:520–522. doi: 10.1038/338520a0. [DOI] [PubMed] [Google Scholar]
- 75.Volkmuth WD, Austin RH. Nature. 1992;358:600–602. doi: 10.1038/358600a0. [DOI] [PubMed] [Google Scholar]
- 76.Volkmuth WD, Duke T, Wu MC, Austin RH, Szabo A. Physical Review Letters. 1994;72:2117–2120. doi: 10.1103/PhysRevLett.72.2117. [DOI] [PubMed] [Google Scholar]
- 77.Dorfman KD. Reviews of Modern Physics. 2010;82:2903–2947. [Google Scholar]
- 78.Randall GC, Doyle PS. Physical Review Letters. 2004:93. doi: 10.1103/PhysRevLett.93.058102. [DOI] [PubMed] [Google Scholar]
- 79.Randall GC, Doyle PS. Macromolecules. 2005;38:2410–2418. [Google Scholar]
- 80.Randall GC, Doyle PS. Macromolecules. 2006;39:7734–7745. [Google Scholar]
- 81.Araki N, Aydil ES, Dorfman KD. Electrophoresis. 2010;31:3675–3680. doi: 10.1002/elps.201000331. [DOI] [PubMed] [Google Scholar]
- 82.Kaji N, Okamoto Y, Tokeshi M, Baba Y. Chem Soc Rev. 2010;39:948–956. doi: 10.1039/b900410f. [DOI] [PubMed] [Google Scholar]
- 83.Doyle PS, Bibette J, Bancaud A, Viovy JL. Science. 2002;295:2237–2237. doi: 10.1126/science.1068420. [DOI] [PubMed] [Google Scholar]
- 84.Minc N, Futterer C, Dorfman KD, Bancaud A, Gosse C, Goubault C, Viovy JL. Analytical Chemistry. 2004;76:3770–3776. doi: 10.1021/ac035246b. [DOI] [PubMed] [Google Scholar]
- 85.Minc N, Bokov P, Zeldovich KB, Futterer C, Viovy JL, Dorfman KD. Electrophoresis. 2005;26:362–375. doi: 10.1002/elps.200410115. [DOI] [PubMed] [Google Scholar]
- 86.Ou J, Cho J, Olson DW, Dorfman KD. Physical Review E. 2009:79. doi: 10.1103/PhysRevE.79.061904. [DOI] [PubMed] [Google Scholar]
- 87.Teclemariam NP, Beck VA, Shaqfeh ESG, Muller SJ. Macromolecules. 2007;40:3848–3859. [Google Scholar]
- 88.Brochard F, Degennes PG. Journal of Chemical Physics. 1977;67:52–56. [Google Scholar]
- 89.Brochard F. Journal De Physique. 1977;38:1285–1291. [Google Scholar]
- 90.Odijk T. Macromolecules. 1983;16:1340–1344. [Google Scholar]
- 91.Odijk T. Physical Review E. 2008:77. doi: 10.1103/PhysRevE.77.060901. [DOI] [PubMed] [Google Scholar]
- 92.Levy SL, Craighead HG. Chem Soc Rev. 2010;39:1133–1152. doi: 10.1039/b820266b. [DOI] [PubMed] [Google Scholar]
- 93.Guan J, Boukany PE, Hemminger O, Chiou N-R, Zha W, Cavanaugh M, Lee LJ. Advanced Materials. 2010;22:3997–4001. doi: 10.1002/adma.201000136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Hsieh C-C, Doyle PS. Korea-Australia Rheology Journal. 2008;20:127–142. [Google Scholar]
- 95.Jendrejack RM, Schwartz DC, Graham MD, de Pablo JJ. Journal of Chemical Physics. 2003;119:1165–1173. doi: 10.1063/1.1637331. [DOI] [PubMed] [Google Scholar]
- 96.Chantiwas R, Park S, Soper SA, Kim BC, Takayama S, Sunkara V, Hwang H, Cho Y-K. Chem Soc Rev. 2011;40:3677–3702. doi: 10.1039/c0cs00138d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Abgrall P, Nguyen NT. Analytical Chemistry. 2008;80:2326–2341. doi: 10.1021/ac702296u. [DOI] [PubMed] [Google Scholar]
- 98.Bakajin OB, Duke TAJ, Chou CF, Chan SS, Austin RH, Cox EC. Physical Review Letters. 1998;80:2737–2740. [Google Scholar]
- 99.Tang J, Levy SL, Trahan DW, Jones JJ, Craighead HG, Doyle PS. Macromolecules. 2010;43:7368–7377. [Google Scholar]
- 100.Balducci A, Mao P, Han J, Doyle PS. Macromolecules. 2006;39:6273–6281. [Google Scholar]
- 101.Lin P-K, Fu C-C, Chen YL, Chen Y-R, Wei P-K, Kuan CH, Fann WS. Physical Review E. 2007;76:011806. doi: 10.1103/PhysRevE.76.011806. [DOI] [PubMed] [Google Scholar]
- 102.Balducci A, Hsieh CC, Doyle PS. Physical Review Letters. 2007:99. doi: 10.1103/PhysRevLett.99.238102. [DOI] [PubMed] [Google Scholar]
- 103.Hsieh C-C, Balducci A, Doyle PS. Macromolecules. 2007;40:5196–5205. [Google Scholar]
- 104.Strychalski EA, Levy SL, Craighead HG. Macromolecules. 2008;41:7716–7721. [Google Scholar]
- 105.Bonthuis DJ, Meyer C, Stein D, Dekker C. Physical Review Letters. 2008:101. doi: 10.1103/PhysRevLett.101.108303. [DOI] [PubMed] [Google Scholar]
- 106.Strychalski EA, Geist J, Gaitan M, Locascio LE, Stavis SM. Macromolecules. 2012;45:1602–1611. [Google Scholar]
- 107.Stavis SM, Geist J, Gaitan M, Locascio LE, Strychalski EA. Lab on a Chip. 2012;12:1174–1182. doi: 10.1039/c2lc21152a. [DOI] [PubMed] [Google Scholar]
- 108.Stavis SM, Strychalski EA, Gaitan M. Nanotechnology. 2009;20:165302. doi: 10.1088/0957-4484/20/16/165302. [DOI] [PubMed] [Google Scholar]
- 109.Cross JD, Strychalski EA, Craighead HG. J Appl Phys. 2007:102. [Google Scholar]
- 110.Tang J, Trahan DW, Doyle PS. Macromolecules. 2010;43:3081–3089. doi: 10.1021/ma902689c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Krishnan M, Monch I, Schwille P. Nano Letters. 2007;7:1270–1275. doi: 10.1021/nl0701861. [DOI] [PubMed] [Google Scholar]
- 112.Hsieh C-C, Balducci A, Doyle PS. Nano Letters. 2008;8:1683–1688. doi: 10.1021/nl080605+. [DOI] [PubMed] [Google Scholar]
- 113.Lin P-k, Hsieh C-C, Chen Y-L, Chou C-F. Macromolecules. 2012 [Google Scholar]
- 114.Turner SWP, Cabodi M, Craighead HG. Physical Review Letters. 2002:88. [Google Scholar]
- 115.Cabodi M, Turner SWP, Craighead HG. Analytical Chemistry. 2002;74:5169–5174. doi: 10.1021/ac025879a. [DOI] [PubMed] [Google Scholar]
- 116.Reisner W, Larsen NB, Flyvbjerg H, Tegenfeldt JO, Kristensen A. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:79–84. doi: 10.1073/pnas.0811468106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Klotz AR, Brandao HB, Reisner WW. Macromolecules. 2012;45:2122–2127. [Google Scholar]
- 118.Del Bonis-O'Donnell JT, Reisner W, Stein D. New Journal of Physics. 2009:11. [Google Scholar]
- 119.Duong TT, Kim G, Ros R, Streek M, Schmid F, Brugger J, Anselmetti D, Ros A. Microelectronic Engineering. 2003;67-8:905–912. [Google Scholar]
- 120.Streek M, Schmid F, Duong TT, Anselmetti D, Ros A. Physical Review E. 2005;71:011905. doi: 10.1103/PhysRevE.71.011905. [DOI] [PubMed] [Google Scholar]
- 121.Hsieh S-F, Wei H-H. Physical Review E. 2009:79. [Google Scholar]
- 122.Guo LJ, Cheng X, Chou C-F. Nano Letters. 2003;4:69–73. [Google Scholar]
- 123.Tegenfeldt JO, Prinz C, Cao H, Chou S, Reisner WW, Riehn R, Wang YM, Cox EC, Sturm JC, Silberzan P, Austin RH. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:10979–10983. doi: 10.1073/pnas.0403849101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Reisner W, Morton KJ, Riehn R, Wang YM, Yu Z, Rosen M, Sturm JC, Chou SY, Frey E, Austin RH. Physical Review Letters. 2005;94:196101. doi: 10.1103/PhysRevLett.94.196101. [DOI] [PubMed] [Google Scholar]
- 125.Persson F, Utko P, Reisner W, Larsen NB, Kristensen A. Nano Letters. 2009;9:1382–1385. doi: 10.1021/nl803030e. [DOI] [PubMed] [Google Scholar]
- 126.Su T, Das SK, Xiao M, Purohit PK. PLoS ONE. 2011;6:e16890. doi: 10.1371/journal.pone.0016890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Reccius CH, Mannion JT, Cross JD, Craighead HG. Physical Review Letters. 2005:95. doi: 10.1103/PhysRevLett.95.268101. [DOI] [PubMed] [Google Scholar]
- 128.Reccius CH, Stavis SM, Mannion JT, Walker LP, Craighead HG. Biophysical Journal. 2008;95:273–286. doi: 10.1529/biophysj.107.121020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Mannion JT, Reccius CH, Cross JD, Craighead HG. Biophysical Journal. 2006;90:4538–4545. doi: 10.1529/biophysj.105.074732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Levy SL, Mannion JT, Cheng J, Reccius CH, Craighead HG. Nano Letters. 2008;8:3839–3844. doi: 10.1021/nl802256s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Reisner W, Beech JP, Larsen NB, Flyvbjerg H, Kristensen A, Tegenfeldt JO. Physical Review Letters. 2007;99:058302. doi: 10.1103/PhysRevLett.99.058302. [DOI] [PubMed] [Google Scholar]
- 132.Zhang C, Zhang F, van Kan JA, van der Maarel JRC. Journal of Chemical Physics. 2008:128. doi: 10.1063/1.2937441. [DOI] [PubMed] [Google Scholar]
- 133.Kim Y, Kim KS, Kounovsky KL, Chang R, Jung GY, dePablo JJ, Jo K, Schwartz DC. Lab on a Chip. 2011;11:1721–1729. doi: 10.1039/c0lc00680g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Zhang C, Shao PG, van Kan JA, van der Maarel JRC. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:16651–16656. doi: 10.1073/pnas.0904741106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Jones JJ, van der Maarel JRC, Doyle PS. Nano Letters. 2011;11:5047–5053. doi: 10.1021/nl203114f. [DOI] [PubMed] [Google Scholar]
- 136.Wang Y, Warshawsky A, Wang CY, Kahana N, Chevallard C, Steinberg V. Macromol Chem Phys. 2002;203:1833–1843. [Google Scholar]
- 137.Heuer DM, Yuan CL, Saha S, Archer LA. Electrophoresis. 2005;26:64–70. doi: 10.1002/elps.200406128. [DOI] [PubMed] [Google Scholar]
- 138.Freedman KO, Lee J, Li YG, Luo D, Skobeleva VB, Ke PC. J Phys Chem B. 2005;109:9839–9842. doi: 10.1021/jp0444924. [DOI] [PubMed] [Google Scholar]
- 139.Seeman NC. Biochemistry. 2003;42:7259–7269. doi: 10.1021/bi030079v. [DOI] [PubMed] [Google Scholar]
- 140.Smith DE, Perkins TT, Chu S. Physical Review Letters. 1995;75:4146–4149. doi: 10.1103/PhysRevLett.75.4146. [DOI] [PubMed] [Google Scholar]
- 141.Robertson RM, Smith DE. Proceedings of the National Academy of Sciences. 2007;104:4824–4827. doi: 10.1073/pnas.0700137104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Robertson RM, Smith DE. Macromolecules. 2007;40:3373–3377. [Google Scholar]
- 143.Ube T, Aoki H, Ito S, Horinaka J-i, Takigawa T, Masuda T. Macromolecules. 2011;44:4445–4451. [Google Scholar]
- 144.Aoki H, Mori K, Ito S. Soft Matter. 2012;8:4390–4395. [Google Scholar]
- 145.Boukany PE, Hemminger O, Wang SQ, Lee LJ. Physical Review Letters. 2010;105:027802. doi: 10.1103/PhysRevLett.105.027802. [DOI] [PubMed] [Google Scholar]