

Abstract
Calcium is an extraordinarily versatile signaling ion, encoding cellular responses to a wide variety of external stimuli. In neurons, mitochondria can accumulate enormous amounts of calcium, with the consequence that mitochondrial calcium uptake, sequestration and release play pivotal roles in orchestrating calcium-dependent responses as diverse as gene transcription and cell death. In this review, we consider the basic chemistry of calcium as a ‘sticky’ cation, which leads to extremely high bound/free ratios, and discuss areas of current interest or controversy. Topics addressed include methodologies for measuring local intracellular calcium, mitochondrial calcium buffering and loading capacity, mitochondrially directed spatial calcium gradients, and the role of calcium overload-dependent mitochondrial dysfunction in glutamate-evoked excitotoxic injury and neurodegeneration. Finally, we consider the relationship between delayed calcium de-regulation, the mitochondrial permeability transition and the generation of reactive oxygen species, and propose a unified view of the ‘source specificity’ and ‘calcium overload’ models of N-methyl-D-aspartate (NMDA) receptor-dependent excitotoxicity. Non-NMDA receptor mechanisms of excitotoxicity are discussed briefly.
Keywords: calcium buffering, calcium deregulation, cell death, electron microscopy, electron probe microanalysis, energy filtering electron microscopy, excitotoxicity, hippocampal neurons, mitochondria, permeability transition
Introduction
Neurons respond to activating stimuli by initiating calcium (Ca2+) entry through plasma membrane channels, but the consequent increase in free cytosolic Ca2+ is strongly modulated by the activity of intracellular calcium stores [1]. In particular, Ca2+ uptake, sequestration and release by the endoplasmic reticulum and mitochondria – the two major Ca2+-regulating organelles – play essential roles in modulating and interpreting Ca2+ signals [2,3]. Of special interest in this context is a renewed focus on mitochondrial calcium handling [4], and the role that this plays in bioenergetics, organelle communication, organelle dynamics and trafficking, cell death signaling, and other equally important aspects of cell signaling. As in other cell types, mitochondria play a pivotal role in neuronal Ca2+ signaling [5]. In addition, mitochondrial calcium overload and subsequent dysfunction are thought to be critically important for triggering the cell death that follows ischemic and traumatic brain injury [6–8], as well as in several neurodegenerative disorders including Alzheimer’s, Parkinson’s, Huntington’s and amyotrophic lateral sclerosis (ALS) [9,10]. An excellent review volume addressing several general aspects of mitochondrial function and calcium signaling has recently been published [11, and the other 13 reviews in that issue]. Here we consider selected topics that are timely, mutually complementary and sometimes controversial, focusing particularly on the influence that the large pool of bound calcium has on mitochondrial function and dysfunction in neurons.
Physiological calcium-regulated mitochondrial function
Distribution of intracellular calcium: free versus total calcium
Under physiological conditions, intracellular Ca2+ is tightly regulated, not only in the cytosol but also within organelles, by distinct collections of Ca2+ transporters, i.e. channels, pumps and exchangers. The resting total cellular calcium concentration in neurons is typically about 1 mM, but the vast majority of intracellular calcium, > 99.9%, is bound to cytosolic proteins or sequestered in the endoplasmic reticulum. Consequently, baseline free cytosolic Ca2+ is usually maintained at approximately 100 nM, with stimulation causing global increases to approximately 1 μM; local increases may be substantially higher [12,13]. Because the cytosolic and intra-organelle pools of bound calcium are so large, they have a profound influence on the magnitude, shape and time course of evoked Ca2+ transients. Although cytosolic Ca2+ elevations are the direct effectors of function, the bound calcium pool nevertheless comes into play, for example by siphoning large proportions of Ca2+ currents away from the signaling pool or by sequestering significant amounts of calcium within non-cytosolic compartments, in which different but characteristic free/bound equilibria affect intraluminal Ca2+ signaling. The magnitude and importance of such effects vary widely, depending on the concentrations, distributions, affinities and kinetics of Ca2+-binding proteins, pumps and leaks.
There are large concentration gradients across both plasma membranes and organelle membranes, both at rest and after Ca2+ entry and elevation. In resting neurons, both the total and free mitochondrial calcium levels are quite low, estimated as approximately 0.1 mM and 100 nM, respectively [14,15]. Thus, in contrast to the endoplasmic reticulum, mitochondria do not generally serve as a calcium ‘store’. However, after stimulation, mitochondria are capable of accumulating enormous amounts of calcium [16,17]. The accumulation and sequestration of calcium within mitochondria is thought to be profoundly important for processes ranging from synaptic transmission to ischemic brain injury.
Quantitative analysis of intracellular and intra-organelle calcium levels
Given the functional importance of free intracellular Ca2+ ions, and the sheer size of the bound calcium pool, it is clearly useful to know how concentrations of total calcium (which are essentially equivalent to bound calcium concentrations) in specific subcellular locations change in parallel with free Ca2+. We therefore briefly review the various methods for quantitative measurement of cellular calcium, noting the strengths and weaknesses of each method. Measurements of free cytosolic Ca2+ are now routinely and almost universally obtained by means of Ca2+-specific fluorescent probes [18]. A variety of ratiometric probes suitable for quantitative assays are commercially available. However, there are a few caveats that should be mentioned. First, the probe molecules themselves act as Ca2+ buffers, and as such perturb the underlying equilibria between Ca2+ and endogenous buffers. Second, it is important to choose probes with a Kd appropriate for the expected concentration range, and this can be problematic in cases where changes in Ca2+ concentration covers two or more orders of magnitude. Third, the specificity of a given probe must be kept in mind. This type of problem is exemplified by the evidence for crosstalk between Ca2+ and Zn2+ when using fura-2 [19] or calcium green-1 [20]. Options for quantitative intra-organelle Ca2+ measurements are much more limited, and the spatial resolution of most Ca2+ probes precludes single-organelle analysis [21]. However, recent advances in genetically encoded, quantitative calcium sensors, such as ratiometric pericam or fluorescence resonance energy transfer (FRET)-based cameleons that can be targeted to specific organelles or subcellular locations, have opened up new lines of investigation [22].
Compared to the advanced state of methodologies for free Ca2+ analysis, the determination of total calcium concentrations is more problematic. The classical 45Ca2+ isotope uptake method is still sometimes used, but this is decidedly inconvenient and suffers from the fact that only population-averaged data are obtained. This approach is therefore not applicable when, as is often the case, single-cell or even single-organelle data are required. An alternative, well-established quantitative method is electron probe microanalysis (EPMA), a technique that uses the electron microscope as a spectrometer by parking a stationary, focused, nanometer-sized electron beam on a subcellular region of interest and using an energy-dispersive X-ray detector to collect the emitted, calcium-specific X-rays (see [23,24] for technical reviews). EPMA provides resolution down to the single-organelle level, i.e. is not limited to ensemble averages. It also provides sub-millimolar sensitivity. However, EPMA also has significant limitations. Specimen preparation requires extensive and elaborate cryo-techniques, the instrumentation is specialized and expensive, and spectrum collection is time-consuming.
Energy-filtered transmission electron microscopy (EFTEM), a technique that provides high-resolution, quantitative element-specific images, may in the near future ameliorate several of the problems that plague EPMA. EFTEM is now widely used in materials science [25], but so far has rarely been used in biology. For calcium detection, the main problem was extracting the weak calcium L2,3 signal from the high mass thickness-dependent background. However, a recent report [26] has described a novel approach to this problem, requiring only four images, each containing over one million pixels. This represents a major improvement in throughput and collection efficiency compared to EPMA. At present, the achievable resolution and sensitivity are adequate for measurements of high physiological and pathophysiological calcium concentrations at the single-organelle level. Perhaps most promisingly, the entire method can be implemented using off-the-shelf microscope packages from several manufacturers, although cryospecimen preparation is still required. Application of this EFTEM technique to mapping calcium in individual mitochondria of frog sympathetic neurons is described below.
Mitochondrial calcium uptake and buffering
Neuronal mitochondria take up Ca2+ through the so-called uniporter [27,28], a channel that is itself Ca2+-sensitive, and which, when opened by elevated cytosolic Ca2+, allows Ca2+ to flow into the matrix down the mitochondrion’s steep electrochemical gradient. Mitochondrial Ca2+ release in neurons is regulated primarily by a Na+/Ca2+ exchanger [29]. The maximal rate of release is much lower than the maximal rate of uptake, which is why continuous mitochondrial calcium accumulation is observed when the cytosolic Ca2+ is high. The net effect of the mitochondrial Ca2+ transport pathways is that this organelle contains little calcium in resting cells, but abruptly begins to accumulate large amounts of calcium during stimulated Ca2+ entry, and to release this calcium load during recovery [30]. (More information on mitochondrial Ca2+ transport is provided in the reviews by Starkov [31] and by Chinopoulos and Adam-Vizi [32]). The physiological effects of elevated intramitochondrial calcium are numerous and significant, and include adjusting aerobic ATP production, modulating the effects of elevated cytosolic Ca2+ on transmitter release, synaptic transmission and excitability, regulating organelle dynamics and trafficking, mediating signaling to the nucleus, regulating the generation of reactive oxygen species (ROS), and activating the release of death signals [30–34]. Nonetheless, the mechanisms by which mitochondrial calcium accumulation influences global and local Ca2+ signals remain incompletely understood, and this continues to be a field of active investigation.
As noted, mitochondria have an enormous capacity to accumulate and store calcium, most of it in a bound form; estimates of the bound/free ratio following stimulation are as high as 4000 [30,35]. What accounts for the large proportion of bound calcium? It has been known for decades that intramitochondrial calcium-and phosphorus-rich deposits of uncertain stoichiometry form under conditions that produce large mitochondrial calcium loads [36,37]. Although historically this situation has been viewed as lethal, it is now clear that localized, nanometer-sized inclusions that are chemically similar to the well-known larger deposits are observed almost from the outset of physiological calcium accumulation [16,37–40]. Certain key aspects of these precipitates, including their heterogeneous distribution both within and among mitochondria, are illustrated by the ‘calcium map’ in Fig. 1, which was generated using the new EFTEM approach described above. Precipitate formation is thought to represent an important, high-capacity calcium storage mechanism that blunts cytosolic Ca2+ elevations during Ca2+ entry while providing a reserve pool of Ca2+ that extends post-stimulus recovery [16,37]. It also clamps an upper limit on free mitochondrial Ca2+ [37,39]. This mechanism has implications not only for normal responses to stimulated Ca2+ entry, but also for conditions such as ischemic or traumatic brain injury. One possibility is that the high capacity of the mitochondrial Ca/P buffering system represents a major line of defense against excitotoxic calcium overload [41,42]. Evidence supporting this idea is discussed below.
Fig. 1.
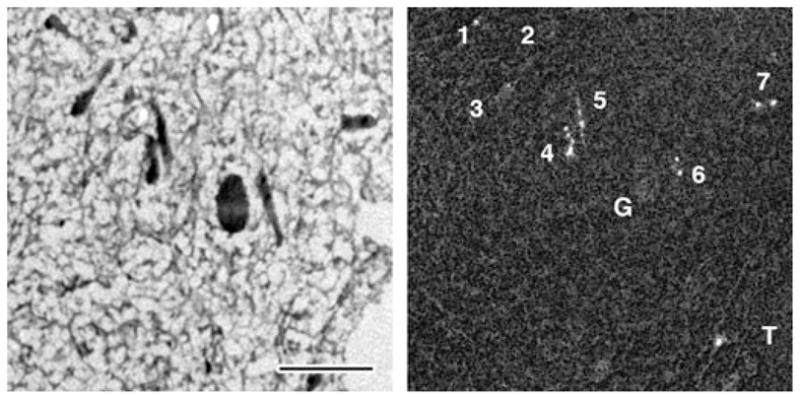
Energy-filtered TEM (EFTEM) map of mitochondrial calcium distribution in a depolarization activated frog sympathetic neuron. Left panel: zero-loss structural EFTEM image of an ultra-thin cryosection prepared from a rapidly frozen frog sympathetic neuron 5 min after termination of a 2 min depolarization with 50 mM K+ [16]. Right panel: quantitative EFTEM map of the mitochondrial calcium distribution, recorded as described previously [26]. The field shown contains seven mitochondria, illustrating the general heterogeneity of mitochondrial calcium accumulation, i.e. some mitochondria (1, 2 and 3) have taken up little if any calcium, while others (4, 5, 6 and 7) have accumulated much more. The resolution of the map is high enough to reveal the punctate nature of mitochondrial calcium sequestration e.g. in mitochondria 6 and 7. Note that neither the dense pigment granule (G) nor the tear in the section (T) generate mass thickness artifacts in the calcium map. Note also the large field of view. Scale bar = 1 μm.
Spatial dependence of mitochondrial calcium uptake
Broad variability between individual mitochondria with regard to calcium uptake and sequestration in individual cells is a general characteristic of in situ mitochondrial populations [16]. The basis for this variability is unknown, but probable factors include the following: bioenergetic status, substrate availability, the activity of regulatory proteins such as Bcl-2, and spatial proximity to a Ca2+ source. The last factor has been elegantly and extensively documented in non-neuronal cells, for which mitochondrial/endoplasmic reticulum communication via local Ca2+ transients has been firmly established [43,44]. Analogous mechanisms undoubtedly also operate in neurons, but the situation is more complicated because Ca2+ elevations in neurons are typically initiated by Ca2+ entry through plasma membrane channels, which adds a geometric component to the controlling factors.
The importance of location is well illustrated in the isolated frog sympathetic neuron, a cell type that serves as an excellent biophysical model on account of its large size (approximately 40 μm diameter), spherical geometry, and the reliable response of plasma membrane voltage-gated Ca2+ channels to depolarization [45]. In these cells, brief depolarization induces rapidly dissipating cytosolic Ca2+ gradients, which begin just under the plasma membrane and diffuse inwardly, decaying in <1 s [46]. Depolarization-induced mitochondrial Ca2+ uptake reflects the radial dependence of these cytosolic gradients, i.e. calcium accumulation is overwhelmingly restricted to those peripheral mitochondria exposed to the earliest and highest cytosolic Ca2+ transients [47]. Mitochondria near the cell center accumulate virtually no calcium. The net effect is that the peripheral mitochondria act as ‘firewalls’ [48], damping or eliminating Ca2+ signaling to centrally located organelles.
As this mechanism appears to be a general feature of Ca2+ signaling [49], it may well have broad implications for neuronal function. For example, it seems likely that appropriately positioned mitochondria may serve to discriminate surface-localized signaling events, such as L-channel signaling to the nucleus [50], from global signaling. Given the potential significance of location-dependent Ca2+ signaling, additional direct data demonstrating this type of mitochondrial regulation in a more relevant experimental context, for example dendritic shafts, would be welcome.
Mitochondrial calcium in neuronal injury and degeneration
NMDAR-mediated excitotoxicity
Glutamate is the major excitatory neurotransmitter in the brain, but one can have too much of a good thing – exposure of central neurons to excessive glutamate leads to excitotoxic death [51,52]. Necrotic or apoptotic-like excitotoxic death is implicated in the pathophysiology of several neurological disorders, including various dementias, stroke, central nervous system trauma, Parkinsons’s, Huntington’s, and other neurodegenerative diseases. The N-methyl-D-aspartate (NMDA) subtype of the glutamate receptor (NMDAR) plays a central role in excitotoxic injury. Physiological activation of these receptors permits the flow of cations, primarily Na+ and Ca2+, through their ion channel in a process that is essential for normal synaptic transmission as well as for a variety of Ca2+-dependent signaling pathways. However, massively elevated levels of glutamate, such as occur in the ischemic core after a stroke, trigger overwhelming NMDAR stimulation, leading to loss of ion homeostasis, cell swelling and necrotic death [53]. In contrast, moderate NMDAR hyperactivity, such as that occurring in the ischemic penumbra of a stroke and in many neurodegenerative diseases, results in somewhat less excessive Ca2+ influx, which can initiate apoptotic-like damage [9,54].
The ability of mitochondria to accumulate enormous amounts of calcium in situ plays an important role in excitotoxic injury. There is compelling evidence that excessive Ca2+ influx through NMDARs targets mitochondria, leading to mitochondrial calcium overload that in turn triggers mitochondrial dysfunction and activation of death signals [7,33,55]. However, the precise cellular response to mitochondrial injury is variable, often unclear and controversial. Current models of excitotoxicity implicate one or more of the following mitochondria-related events: uncoupling of oxidative phosphorylation [56,57], activation of the mitochondrial permeability transition [58,59], release of pro-apoptotic proteins [60,61], activation of poly(ADP-ribose) polymerase-1 [62] and proteases such as calpain [63,64], increased production of ROS [65,66] and delayed Ca2+ de-regulation [67], ultimately resulting in necrotic or apoptotic-like cell death [54]. Although the contribution of each of these processes to the activation of death pathways is firmly established, the sequence of events, as well as the significance of various mechanisms, is less clear. Several of these issues are discussed below.
The mitochondrial permeability transition in excitotoxic cell death
The mitochondrial permeability transition (MPT) pore is a voltage- and Ca2+-dependent high-conductance channel breaching the inner mitochondrial membrane [59,68]. Although the structure and molecular composition of the MPT pore remain elusive, activation of this complex is recognized as a major cause of ischemia/reperfusion injury in the heart, where it is a prime target for cardioprotection [69,70]. (The molecular nature of the MPT complex is discussed elsewhere in this review series [31]). However, the possible involvement of MPT in excitotoxic neurodegeneration has been a matter of debate for over a decade. A significant body of evidence, both clinical and experimental, supports a role for MPT in ischemic injury [6,8,71,72], as well as in neurodegenerative diseases, such as Huntington’s, Parkinson’s and Alzheimer’s [9]. Nonetheless, uncertainty remains, at least partly due to the inconsistent effects of cyclosporin A, the gold standard of MPT inhibitors, on neuronal mitochondria [55,67,73]. Apparently, cyclosporin A is not reliably diagnostic of MPT in neuronal mitochondria, possibly because its protective effect can be overcome by high calcium loads [59,74].
In isolated mitochondria, MPT activation is characterized by loss of the mitochondrial membrane potential, swelling of the mitochondrial matrix, and outer membrane rupture, followed by release of internalized Ca2+ and apoptogenic proteins [59]. However, it has proven difficult to establish that MPT occurs in in situ mitochondria during neurodegeneration of intact neurons. We reported that cultured hippocampal neurons display several hallmarks of MPT after episodes of excitotoxicity-induced mitochondrial Ca2+ loading [38] that were quantitatively comparable to those that induce MPT in isolated brain mitochondria [37]. However, retention of high levels of calcium in the form of calcium- and phosphate-rich precipitates (discussed above and also below) in swollen mitochondria after presumptive pore opening (Fig. 2, upper panel) seemed to be inconsistent with the expectation that mitochondrial Ca2+ should equilibrate with cytosolic Ca2+ after MPT. This paradox was resolved by experiments using isolated brain and liver mitochondria to demonstrate that neither MPT induction nor additional mitochondrial depolarization (with protonophore) led to complete release of accumulated calcium [75] (Fig. 2, lower panel). Indeed, significant amounts of accumulated calcium were retained for many minutes after MPT activation.
Fig. 2.
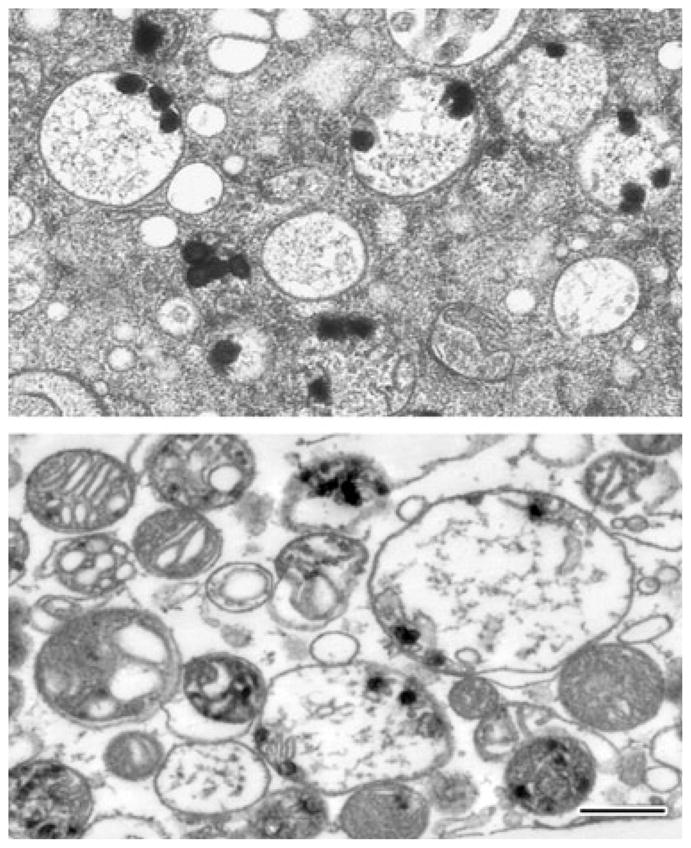
Mitochondrial calcium loads persist long after activation of the mitochondrial permeability transition. Electron micrographs of typical damaged in situ mitochondria of NMDA-treated cultured hippocampal neurons (upper panel) and calcium-loaded isolated rat brain mitochondria (lower panel). Isolated mitochondria were calcium-loaded in the presence of ATP using a continuous infusion protocol. After the abrupt onset of MPT, mitochondria were further exposed to the protonophore carbonyl cyanide 4-(trifluoromethoxy)-phenylhydrazone (FCCP) and frozen 200 s later (see [75] for experimental details). Both preparations were high pressure-frozen and freeze-substituted in order to preserve the calcium-rich precipitates, whose continued presence demonstrates the longevity of mitochondrial calcium loads. The lower panel also illustrates the variable response of individual mitochondria to calcium challenge. Scale bar = 500 nm (both panels).
Several recent studies offer further evidence for the involvement of MPT in excitotoxic cell death. For example, it has been demonstrated that glutamate exposure leads to transient matrix alkalinization, followed by acidification; this effect is absent in neurons that are deficient in cyclophilin D, a putative modulatory pore component [76]. Similarly, a deficiency in the ADP/ATP translocator (which is not essential for MPT but does regulate pore opening [77]) has been reported to be neuroprotective against glutamate excitotoxicity [78]. Interestingly, MPT activation may underlie increased excitotoxic vulnerability attributable to dysfunction of the mitochondrial Na+/Ca2+ exchanger, for example as seems to occur in cells carrying a mutation in the Parkinson’s-associated mitochondrial gene PINK-1 [79]. To summarize, despite remaining controversies, the weight of current evidence appears to favor an important role for MPT in excitotoxic injury [55,80].
Delayed calcium de-regulation
Delayed Ca2+ de-regulation (DCD) generally refers to the second phase of a typical biphasic cytosolic Ca2+ response to glutamate exposure. DCD was first described over 10 years ago in hippocampal [81], spinal [82] and cerebellar granule neurons [83], but the exact cause and significance of DCD is still unknown and is actively debated. During chronic glutamate exposure, DCD is postulated to indicate the point of no return, i.e. it is the first indication of cell demise when Ca2+ regulation is irreversibly impaired. On the other hand, certain large pyramidal neurons, e.g. cortical and hippocampal neurons, exhibit fast, DCD-like but reversible secondary Ca2+ elevations [56,76,84,85] that become irreversible during longer glutamate or NMDA exposures [56]. For these cell types, primary DCD by itself is apparently not sufficient to indicate impending cell death.
The relationship between DCD and the activation of MPT is also unclear. Mitochondrial calcium overload-induced MPT is implicated in both reversible and irreversible DCD [76,86]. Correlation between the onset of depolarization of the mitochondrial membrane potential and DCD [56], as well as blocking DCD with cyclosporin A [57], indicate to a role for MPT, especially at later stages. However, in neurons from cyclophilin D knockout mice, mitochondrial membrane potential depolarization and matrix acidification – both presumptive indicators of MPT – were only delayed relative to DCD, but not abolished, when stimulated with low concentrations of glutamate [76]. Another recent knockout study linked MPT to DCD only after prolonged glutamate exposure [87].
These knockout studies illustrate well the variable relationship between cytosolic Ca2+ overload, mitochondrial calcium loading and MPT, and, if considered from the viewpoint of the highly variable responses of individual mitochondria within the same cell, suggest an explanation for one of the most puzzling but clinically relevant features of excitotoxicity, namely that cell death often does not occur until 24–48 h after exposure to a toxic insult. For example, in cultured hippocampal neurons, toxic over-stimulation of NMDARs for periods of up to 30 min induces apparent DCD together with massive mitochondrial calcium accumulation and swelling, the latter presumably indicating MPT [38]. However, because of the functional variability between mitochondria [88], only a fraction of the mitochondria were irreversibly damaged, becoming dysfunctional and releasing cytochrome c (Fig. 3); the remainder rapidly accumulated calcium and became swollen and depolarized, but these steps were all reversible, such that the majority retained intermembrane proteins and recovered normal function after stimulus removal. If the permanently damaged fraction exceeded approximately 35%, the cell ultimately died [38]. This heterogeneity, in which only a sub-population of mitochondria releases death signals, while others respire and produce ATP normally, provides an attractive explanation for the delayed nature of excitotoxic death.
Fig. 3.

Heterogeneous calcium accumulation within and among individual mitochondria. Electron micrograph of a high pressure-frozen, freeze-substituted, cultured hippocampal neuron demonstrating that neighboring mitochondria respond differently to NMDA exposure (100 μM for 30 min). Although all mitochondria took up significant amounts of calcium, as indicated by the electron-dense calcium- and phosphorus-rich precipitates, some (arrowheads) have presumably undergone MPT, becoming swollen and releasing matrix material and apoptogenic proteins, while others (arrows) have not. Scale bar = 500 nm. Modified from [38]. The diagram below illustrates schematically how this heterogeneity can account for delayed cell death. Specifically, damaged mitochondria early on release factors necessary to activate downstream death signaling, but undamaged mitochondria that are not dysfunctional maintain energy production and other essential mitochondrial functions for the time period between injury and death.
There is much current interest in the role that ROS play in cell physiology. Much of the interest derives from the dual nature of ROS signaling. There is little doubt that various ROS are important modulators of redox-sensitive cell signaling pathways [89,90], but they also contribute to oxidative stress during and following excitotoxic injury. Mitochondria are a major source of ROS, particularly the superoxide radical anion, which is generated as a result of ‘leakage’ from the respiratory chain [91] and perhaps by other mechanisms [92]. Consequently, mitochondrial ROS production is up-regulated in parallel with ATP generation when mitochondria take up calcium. However, whether mitochondrial sources of ROS are a significant component of excitotoxic death mechanisms is controversial [73,93]. It has been argued that, at least for cerebellar granule neurons, glutamate-evoked mitochondrial calcium uptake per se does not increase ROS production, elevation of which is only observed after DCD [94,95]. Recent studies concerning the role of oxidative stress in ischemic and/or reperfusion injury have clarified the time course of the various sources and mechanisms of ROS generation [65,96]. The emerging view is that mitochondrial sources are significant only in terms of providing a burst of ROS shortly after insults such as hypoxia. This phase terminates upon mitochondrial depolarization. Later phases of ROS production are mediated by xanthine oxidase and NADPH oxidase. The latter may be especially important as its activity is ramped up during reperfusion.
A role for calcium-rich mitochondrial precipitates
One feature of stimulated NMDA-treated neurons is a dramatic increase in the number and size of the electron-dense, calcium-rich precipitates within the mitochondrial matrix, even in unswollen (undamaged) mitochondria. It has been known for decades that mitochondria are prone to form such precipitates in response to strong calcium loading [36], and that the chemistry of these precipitates is quite complex, especially with regard to the counter-ions, e.g. various phosphates, adenine nucleotides, phosphocitrate, and other candidates [32,97]. For many years, the significance of this process has remained unclear, but recent studies [37–40] have provided new insight into the physical basis and functional consequences of this phenomenon.
In the context of excitotoxicity, the ability of mitochondria to tolerate huge amounts of calcium –precisely because it can be immobilized as a precipitate – allows the mitochondria to function as a protective ‘firewall’, accumulating and sequestering larger calcium loads before overload triggers mitochondrial dysfunction. However, the amount of calcium that mitochondria can tolerate, i.e. the calcium loading capacity, essentially defines a mitochondrion’s injury resistance, is variable and depends, at a minimum, on the activity history and energetic state of the cell, as well as the conditions of calcium loading. Why is this? Almost 50 years ago, it was found that ADP or ATP increase the stability of mitochondrial precipitates [98]. More recent work has shown that, for both isolated and in situ brain mitochondria, changing the conditions of precipitate formation affects their composition and stability. Thus, the amount of precipitate formed and the calcium loading capacity are both greatly increased in the presence of endogenous MPT inhibitors such as ADP and ATP [75], while the mitochondrial calcium loading capacity is much larger in hippocampal neurons that have been ‘pre-conditioned’ to tolerate stronger glutamate challenges than in naïve neurons (Fig. 4) [84]. Interestingly, in both cases the high Ca/P ratio of the precipitates, implying a different and more insoluble chemical form, correlates with increased mitochondrial calcium sequestration capacity. It may be inferred from these observations that formation of relatively stable precipitates retards MPT, thereby improving cell survival.
Fig. 4.
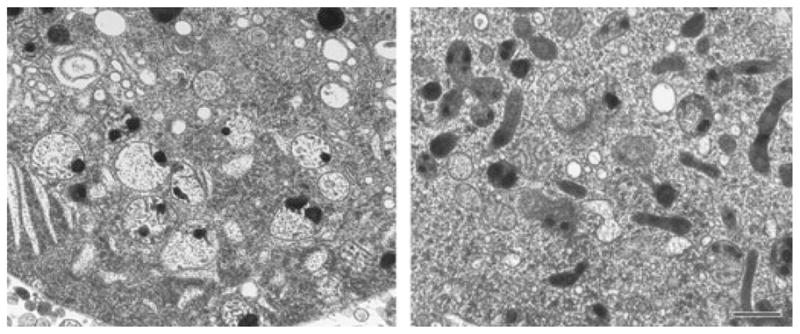
The variability of mitochondrial calcium loading capacity. Electron micrographs of high pressure-frozen, freeze-substituted, hippocampal neurons from naïve (left panel) and NMDA-tolerant (‘pre-conditioned’ [84]) cultures (right panel) after exposure to 100 μM NMDA. There are few swollen, damaged mitochondria in the tolerant cell, even though the calcium load is large and comparable to that in naive cells. This damage resistance reflects a general, experimentally induced increase in the calcium loading capacity. Scale bar = 500 nm (both panels).
The physiological impact of precipitate solubility manifests itself in another way. Mobilization of precipitated calcium and its release, even from damaged mitochondria, is a slow process [75]. Given that mitochondrial Ca2+ release significantly modifies the timing and spatial characteristics of cytosolic Ca2+ elevations, it seems likely that prolonged release of precipitated calcium will influence Ca2+ signaling during post-stimulus recovery periods, i.e. mitochondria may well utilize the precipitates to retain a historical record of previous activity.
Reconciling the ‘calcium load’ versus ‘route specificity’ hypotheses
There is unequivocal evidence that excitotoxicity is mediated by elevation of cytosolic Ca2+, and that the level of acute death correlates with the absolute amount of calcium taken up [99–101]. This information forms the basis of the ‘calcium load’ hypothesis. Other experiments – for example, studies showing that neurotoxicity triggered by Ca2+ influx through NMDARs exceeds that triggered by other routes of Ca2+ entry [102] – indicated that toxicity depends on the activation of specific NMDAR-dependent routes of Ca2+ entry. Such observations gave rise to the ‘source specificity’ or ‘route specificity’ hypothesis, i.e. the idea that lethality depends more on how or where calcium enters the cell rather than on how much enters [102–104].
Over the subsequent 20 years, much additional evidence has emerged in support of both concepts. For example, concerning route specificity, strong activation of only extra-synaptic NMDARs in hippocampal neurons has been linked to mitochondrial dysfunction and excitotoxic death, while synaptic stimulation was found to promote survival [105]. This information suggest that NMDAR location plays an important role in specifying the induction of death versus survival pathways. It has also been suggested that subunit composition, specifically the presence of the NR2B subunit, determines excitotoxicity [104,106], but this seems to be a developmental phenomenon rather than a manifestation of route specificity [107–110]. An additional aspect of route specificity is illustrated by the accumulating evidence that disrupting the post-synaptic density signaling complex in cortical neurons is modestly neuroprotective against NMDAR-mediated excitotoxicity [103,111,112]. This observation was recently explained by showing that there exists a synaptically located injury pathway that is coupled to the post-synaptic density through Ca2+-activated, neuronal nitric oxide synthetase (nNOS)-dependent p38 mitogen-activated protein kinase (MAPK) signaling [113].
Until recently, it has been difficult to relate excitotoxicity mechanisms that depend on generalized calcium overload and mitochondrial dysfunction to route-specific mechanisms. In particular, the situation was complicated by apparent discrepancies regarding the size of calcium loads mediated by various routes of entry [19,101–103,105,109,114]. Clarity has come from the realization that there are multiple pathways for glutamate-stimulated Ca2+ entry, but toxic stimuli are nonetheless uniformly characterized by an obligatory link to massive mitochondrial calcium loading, mainly, but not necessarily only, through NMDARs [109]. These features are illustrated by the data in Fig. 5, which compares calcium loading via NMDARs and voltage-gated Ca2+ channels (unpublished observations). Application of fluorescent probes with appropriately low affinities revealed that NMDARs mediate much higher cytosolic Ca2+ elevations [19,101,114], while direct EMPA measurements in hippocampal neurons showed that this translates into much higher and injurious mitochondrial calcium loading (unpublished observations). Nonetheless, Ca2+ entry through voltage-gated Ca2+ channels can still contribute to excitotoxicity by synergistically adding to the overall calcium load, especially in cells in which voltage-gated Ca2+ channel expression is fully developed [109]. All things considered, it appears that calcium loading and route specificity can be reconciled by invoking the unifying principle that a given route is toxic precisely because it mediates larger calcium loads.
Fig. 5.

Ca2+ entry and cell death are much higher after NMDAR activation than after depolarization-evoked VGCC activation. The traces show the dose–response of cytosolic Ca2+ in cultured hippocampal neurons to increasing concentrations of NMDA (μM, as indicated) in comparison with the strong depolarization-induced Ca2+ entry via VGCCs (90 mM K+ plus 1 μM Bay K 8644 (a calcium channel activator) in 10 mM Ca2+ saline). The relative death rates at 24 h are given in parentheses. Free Ca2+ was measured using the low-affinity ratiometric probe fura-4FF. Near-maximal VGCC activation and NMDA at the lowest dose used (10 μM) elicit similarly small Ca2+ elevations and minimal cell death.
However, important questions remain. For example, do mitochondria establish privileged access to Ca2+ entering through (presumably extra-synaptic) NMDARs, as proposed some time ago [115] and consistent with current concepts of functional Ca2+ signaling microdomains [4,116]? Given that synaptic and extra-synaptic receptors may have differential exposure to glutamate under different injury scenarios, how much do NMDAR-mediated synaptic pathways contribute to cell death in various clinical settings? Do Ca2+-dependent synaptic death pathways involve mitochondrial dysfunction? The last question is of some interest in considering how to reconcile the dual toxic and pro-survival activities of synaptic NMDARs. Soriano et al. [113] suggest that NO-linked synaptic NMDAR signaling to p38 MAPK is tolerated during normal synaptic activity, but harmful when induced by chronic over-stimulation. This idea offers an opportunity to test the calcium load hypothesis in a well-defined route-specific context by asking whether the strength and outcome of synaptic stimulation are reflected in differential calcium loading and concomitant mitochondrial dysfunction.
Other mechanisms of injurious calcium entry
As Ca2+ entry through NMDARs is the major trigger for excitotoxic injury, it is disappointing that anti-excitotoxic therapies targeted to glutamate receptors have so far failed to protect against ischemic injuries [117,118]. This has led to suggestions that NMDA receptor-independent pathways of Ca2+ loading might be involved in ischemic death. Currently attractive candidate pathways include Ca2+-permeable α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptors (AMPARs) [119], acid-sensing channels [120] and transient receptor potential channels [121]. With regard to hippocampal injury, there is much current interest in contributions from AMPARs. The strong neuroprotective effect of AMPAR antagonists on pyramidal neurons in the CA1 region of the hippocampus days after global ischemia suggests that selective injury might be mediated by Ca2+ entry via Ca2+-permeable AMPARs [122]. Presumably, the responsible AMPARs are those in which RNA editing of GluR2 subunits has not occurred, or those lacking this subunit altogether [123–125]. In either case, the result is increased Ca2+ permeability of receptors that are normally Ca2+-impermeable [119,126]. With regard to potential injury mechanisms, there are some parallels between NMDARs and calcium-permeable AMPARs. For example, both respond to excessive stimulation by mediating calcium loading and consequent ROS production [101]. On the other hand, and in contrast to NMDARs, Ca-permeable AMPARs are not blocked by Mg2+ and are highly permeable to another endogenous cation, Zn2+, which can be highly toxic [126].
Finally, it has been reported that mitochondrial dysfunction-dependent kainate excitotoxicity in cerebellar neurons is mediated by Na+ elevation [127]. Elevated Na+ was also proposed to be responsible for metabolic compromise and DCD during extended exposures to low concentrations of NMDA [128]. In addition to mitochondrial calcium loading and oxidative stress during glutamate exposure, ATP insufficiency due to reduced spare respiratory capacity has been suggested to play a primary role in excitotoxic neuronal death [129]. We anticipate that future studies will elucidate the relative contributions and interdependence of these potentially important mechanisms, especially with regard to clarifying their relationships to NMDAR-driven excitotoxicity.
Concluding remarks
The ability of in situ mitochondria to accumulate calcium plays an important role in health and disease. Tightly regulated and finely tuned changes in intracellular Ca2+ concentrations regulate a host of essential neuronal functions. On the other hand, disruption of Ca2+ homeostasis is a primary mechanism of injury and disease. Neurodegeneration is currently one of the most intensely investigated areas in the neurosciences, and it has become increasingly clear that Ca2+ de-regulation, leading to pathologically elevated cytosolic Ca2+, underlies numerous degenerative conditions. Mitochondrial calcium overload and subsequent dysfunction are arguably the most important injury processes triggered by excessively high cytosolic Ca2+. However, while the central role of mitochondrial dysfunction in degenerative disease is clear, many of the underlying mechanisms are not. Thus, an important goal of future research will be to obtain a better understanding of how mitochondria come to be at risk in the first place, and how to reduce this risk.
Acknowledgments
The authors are indebted to the staff of the National Institute of Neurological Disorders and Stroke Electron Microscopy facility (Director Dr Jung-Hwa Tao-Cheng) for excellent technical assistance. This research was supported by the Intramural Research Program of the National Institute of Neurological Disorders and Stroke, National Institutes of Health.
Abbreviations
- AIF
apoptosis-inducing factor
- AMPAR
α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor
- cyt c
cytochrome c
- DCD
delayed calcium de-regulation
- EFTEM
energy-filtered transmission electron microscopy
- EPMA
electron probe microanalysis
- Pi
inorganic phosphate
- MPT
mitochondrial permeability transition
- NMDAR
N-methyl-D-aspartate receptor
- ROS
reactive oxygen species
- [Ca]m
total intramitochondrial calcium
- VGCC
voltage-gated Ca2+ channel
Footnotes
The notation ‘Ca2+’ is used conventionally throughout to indicate the free ion in solution; ‘calcium’ is spelled out when referring to bound or total calcium, or when the physical state is unspecified.
References
- 1.Clapham DE. Calcium signaling. Cell. 2007;131:1047–1058. doi: 10.1016/j.cell.2007.11.028. [DOI] [PubMed] [Google Scholar]
- 2.Friel DD. Mitochondria as regulators of stimulus-evoked calcium signals in neurons. Cell Calcium. 2000;28:307–316. doi: 10.1054/ceca.2000.0172. [DOI] [PubMed] [Google Scholar]
- 3.Verkhratsky A. Physiology and pathophysiology of the calcium store in the endoplasmic reticulum of neurons. Physiol Rev. 2005;85:201–279. doi: 10.1152/physrev.00004.2004. [DOI] [PubMed] [Google Scholar]
- 4.Rizzuto R, Pozzan T. Microdomains of intracellular Ca2+: molecular determinants and functional consequences. Physiol Rev. 2006;86:369–408. doi: 10.1152/physrev.00004.2005. [DOI] [PubMed] [Google Scholar]
- 5.Berridge MJ. Neuronal calcium signaling. Neuron. 1998;21:13–26. doi: 10.1016/s0896-6273(00)80510-3. [DOI] [PubMed] [Google Scholar]
- 6.Friberg H, Wieloch T. Mitochondrial permeability transition in acute neurodegeneration. Biochimie. 2002;84:241–250. doi: 10.1016/s0300-9084(02)01381-0. [DOI] [PubMed] [Google Scholar]
- 7.Starkov AA, Chinopoulos C, Fiskum G. Mitochondrial calcium and oxidative stress as mediators of ischemic brain injury. Cell Calcium. 2004;36:257–264. doi: 10.1016/j.ceca.2004.02.012. [DOI] [PubMed] [Google Scholar]
- 8.Norenberg MD, Rao KV. The mitochondrial permeability transition in neurologic disease. Neurochem Int. 2007;50:983–997. doi: 10.1016/j.neuint.2007.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bezprozvanny I. Calcium signaling and neurodegenerative diseases. Trends Mol Med. 2009;15:89–100. doi: 10.1016/j.molmed.2009.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gibson GE, Starkov A, Blass JP, Ratan RR, Beal MF. Cause and consequence: mitochondrial dysfunction initiates and propagates neuronal dysfunction, neuronal death and behavioral abnormalities in age-associated neurodegenerative diseases. Biochim Biophys Acta. 2010;1802:122–134. doi: 10.1016/j.bbadis.2009.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Halestrap AP. Mitochondrial calcium in health and disease. Biochim Biophys Acta. 2009;1787:1289–1290. doi: 10.1016/j.bbabio.2009.07.011. [DOI] [PubMed] [Google Scholar]
- 12.Meldolesi J, Pozzan T. The endoplasmic reticulum Ca2+ store: a view from the lumen. Trends Biochem Sci. 1998;23:10–14. doi: 10.1016/s0968-0004(97)01143-2. [DOI] [PubMed] [Google Scholar]
- 13.Pozzan T, Rizzuto R. High tide of calcium in mitochondria. Nat Cell Biol. 2000;2:E25–E27. doi: 10.1038/35000095. [DOI] [PubMed] [Google Scholar]
- 14.Pozzo-Miller LD, Pivovarova NB, Leapman RD, Buchanan RA, Reese TS, Andrews SB. Activity-dependent calcium sequestration in dendrites of hippocampal neurons in brain slices. J Neurosci. 1997;17:8729–8738. doi: 10.1523/JNEUROSCI.17-22-08729.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Babcock DF, Hille B. Mitochondrial oversight of cellular Ca2+ signaling. Curr Opin Neurobiol. 1998;8:398–404. doi: 10.1016/s0959-4388(98)80067-6. [DOI] [PubMed] [Google Scholar]
- 16.Pivovarova NB, Hongpaisan J, Andrews SB, Friel DD. Depolarization-induced mitochondrial Ca accumulation in sympathetic neurons: spatial and temporal characteristics. J Neurosci. 1999;19:6372–6384. doi: 10.1523/JNEUROSCI.19-15-06372.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Montero M, Alonso MT, Carnicero E, Cuchillo-Ibanez I, Albillos A, Garcia AG, Garcia-Sancho J, Alvarez J. Chromaffin-cell stimulation triggers fast millimolar mitochondrial Ca2+ transients that modulate secretion. Nat Cell Biol. 2000;2:57–61. doi: 10.1038/35000001. [DOI] [PubMed] [Google Scholar]
- 18.Rudolf R, Mongillo M, Rizzuto R, Pozzan T. Looking forward to seeing calcium. Nat Rev Mol Cell Biol. 2003;4:579–586. doi: 10.1038/nrm1153. [DOI] [PubMed] [Google Scholar]
- 19.Cheng C, Reynolds IJ. Calcium-sensitive fluorescent dyes can report increases in intracellular free zinc concentration in cultured forebrain neurons. J Neurochem. 1998;71:2401–2410. doi: 10.1046/j.1471-4159.1998.71062401.x. [DOI] [PubMed] [Google Scholar]
- 20.Stork CJ, Li YV. Intracellular zinc elevation measured with a ‘calcium-specific’ indicator during ischemia and reperfusion in rat hippocampus: a question on calcium overload. J Neurosci. 2006;26:10430–10437. doi: 10.1523/JNEUROSCI.1588-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pozzan T, Rudolf R. Measurements of mitochondrial calcium in vivo. Biochim Biophys Acta. 2009;1787:1317–1323. doi: 10.1016/j.bbabio.2008.11.012. [DOI] [PubMed] [Google Scholar]
- 22.McCombs JE, Palmer AE. Measuring calcium dynamics in living cells with genetically encodable calcium indicators. Methods. 2008;46:152–159. doi: 10.1016/j.ymeth.2008.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Roomans GM, Von Euler A. X-ray microanalysis in cell biology and cell pathology. Cell Biol Int. 1996;20:103–109. doi: 10.1006/cbir.1996.0014. [DOI] [PubMed] [Google Scholar]
- 24.Fernandez-Segura E, Warley A. Electron probe X-ray microanalysis for the study of cell physiology. Methods Cell Biol. 2008;88:19–43. doi: 10.1016/S0091-679X(08)00402-0. [DOI] [PubMed] [Google Scholar]
- 25.Egerton RF. New techniques in electron energy-loss spectroscopy and energy-filtered imaging. Micron. 2003;34:127–139. doi: 10.1016/s0968-4328(03)00023-4. [DOI] [PubMed] [Google Scholar]
- 26.Aronova MA, Kim YC, Pivovarova NB, Andrews SB, Leapman RD. Quantitative EFTEM mapping of near physiological calcium concentrations in biological specimens. Ultramicroscopy. 2009;109:201–212. doi: 10.1016/j.ultramic.2008.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nicholls DG, Budd SL. Mitochondria and neuronal survival. Physiol Rev. 2000;80:315–360. doi: 10.1152/physrev.2000.80.1.315. [DOI] [PubMed] [Google Scholar]
- 28.Gunter TE, Sheu SS. Characteristics and possible functions of mitochondrial Ca2+ transport mechanisms. Biochim Biophys Acta. 2009;1787:1291–1308. doi: 10.1016/j.bbabio.2008.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Crompton M, Moser R, Ludi H, Carafoli E. The interrelations between the transport of sodium and calcium in mitochondria of various mammalian tissues. Eur J Biochem. 1978;82:25–31. doi: 10.1111/j.1432-1033.1978.tb11993.x. [DOI] [PubMed] [Google Scholar]
- 30.Nicholls DG. Mitochondria and calcium signaling. Cell Calcium. 2005;38:311–317. doi: 10.1016/j.ceca.2005.06.011. [DOI] [PubMed] [Google Scholar]
- 31.Starkov AA. The molecular identity of the mitochondrial Ca2+ sequestration system. FEBS J. 2010;277:3652–3663. doi: 10.1111/j.1742-4658.2010.07756.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chinopoulos C, Adam-Vizi V. Mitochondrial Ca2+ sequestration and precipitation revisited. FEBS J. 2010;277:3637–3651. doi: 10.1111/j.1742-4658.2010.07755.x. [DOI] [PubMed] [Google Scholar]
- 33.Duchen MR. Mitochondria in health and disease: perspectives on a new mitochondrial biology. Mol Aspects Med. 2004;25:365–451. doi: 10.1016/j.mam.2004.03.001. [DOI] [PubMed] [Google Scholar]
- 34.Mattson MP, Gleichmann M, Cheng A. Mitochondria in neuroplasticity and neurological disorders. Neuron. 2008;60:748–766. doi: 10.1016/j.neuron.2008.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Horikawa Y, Goel A, Somlyo AP, Somlyo AV. Mitochondrial calcium in relaxed and tetanized myocardium. Biophys J. 1998;74:1579–1590. doi: 10.1016/S0006-3495(98)77869-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Carafoli E. Historical review: mitochondria and calcium: ups and downs of an unusual relationship. Trends Biochem Sci. 2003;28:175–181. doi: 10.1016/S0968-0004(03)00053-7. [DOI] [PubMed] [Google Scholar]
- 37.Chalmers S, Nicholls DG. The relationship between free and total calcium concentrations in the matrix of liver and brain mitochondria. J Biol Chem. 2003;278:19062–19070. doi: 10.1074/jbc.M212661200. [DOI] [PubMed] [Google Scholar]
- 38.Pivovarova NB, Nguyen HV, Winters CA, Brantner CA, Smith CL, Andrews SB. Excitotoxic calcium overload in a subpopulation of mitochondria triggers delayed death in hippocampal neurons. J Neurosci. 2004;24:5611–5622. doi: 10.1523/JNEUROSCI.0531-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.David G, Talbot J, Barrett EF. Quantitative estimate of mitochondrial [Ca2+] in stimulated motor nerve terminals. Cell Calcium. 2003;33:197–206. doi: 10.1016/s0143-4160(02)00229-4. [DOI] [PubMed] [Google Scholar]
- 40.Panov AV, Andreeva L, Greenamyre JT. Quantitative evaluation of the effects of mitochondrial permeability transition pore modifiers on accumulation of calcium phosphate: comparison of rat liver and brain mitochondria. Arch Biochem Biophys. 2004;424:44–52. doi: 10.1016/j.abb.2004.01.013. [DOI] [PubMed] [Google Scholar]
- 41.Nicholls DG. A role for the mitochondrion in the protection of cells against calcium overload? Prog Brain Res. 1985;63:97–106. doi: 10.1016/S0079-6123(08)61978-0. [DOI] [PubMed] [Google Scholar]
- 42.Dubinsky JM, Brustovetsky N, LaFrance R. Protective roles of CNS mitochondria. J Bioenerg Biomembr. 2004;36:299–302. doi: 10.1023/B:JOBB.0000041757.68148.3c. [DOI] [PubMed] [Google Scholar]
- 43.Rizzuto R, Marchi S, Bonora M, Aguiari P, Bononi A, De Stefani D, Giorgi C, Leo S, Rimessi A, Siviero R, et al. Ca2+ transfer from the ER to mitochondria: when, how and why. Biochim Biophys Acta. 2009;1787:1342–1351. doi: 10.1016/j.bbabio.2009.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Csordas G, Hajnoczky G. SR/ERmitochondrial local communication: calcium and ROS. Biochim Biophys Acta. 2009;1787:1352–1362. doi: 10.1016/j.bbabio.2009.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Friel DD, Tsien RW. An FCCP-sensitive Ca2+ store in bullfrog sympathetic neurons and its participation in stimulus-evoked changes in [Ca2+]i. J Neurosci. 1994;14:4007–4024. doi: 10.1523/JNEUROSCI.14-07-04007.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hernandez-Cruz A, Sala F, Adams PR. Subcellular calcium transients visualized by confocal microscopy in a voltage-clamped vertebrate neuron. Science. 1990;247:858–862. doi: 10.1126/science.2154851. [DOI] [PubMed] [Google Scholar]
- 47.Hongpaisan J, Pivovarova NB, Colegrove SL, Leapman RD, Friel DD, Andrews SB. Multiple modes of calcium-induced calcium release in sympathetic neurons II: a [Ca2+]i- and location-dependent transition from endoplasmic reticulum Ca accumulation to net Ca release. J Gen Physiol. 2001;118:101–112. doi: 10.1085/jgp.118.1.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Barrow SL, Sherwood MW, Dolman NJ, Gerasimenko OV, Voronina SG, Tepikin AV. Movement of calcium signals and calcium-binding proteins: firewalls, traps and tunnels. Biochem Soc Trans. 2006;34:381–384. doi: 10.1042/BST0340381. [DOI] [PubMed] [Google Scholar]
- 49.Collins TJ, Lipp P, Berridge MJ, Bootman MD. Mitochondrial Ca2+ uptake depends on the spatial and temporal profile of cytosolic Ca2+ signals. J Biol Chem. 2001;276:26411–26420. doi: 10.1074/jbc.M101101200. [DOI] [PubMed] [Google Scholar]
- 50.Deisseroth K, Mermelstein PG, Xia H, Tsien RW. Signaling from synapse to nucleus: the logic behind the mechanisms. Curr Opin Neurobiol. 2003;13:354–365. doi: 10.1016/s0959-4388(03)00076-x. [DOI] [PubMed] [Google Scholar]
- 51.Olney JW, Sharpe LG. Brain lesions in an infant rhesus monkey treated with monosodium glutamate. Science. 1969;166:386–388. doi: 10.1126/science.166.3903.386. [DOI] [PubMed] [Google Scholar]
- 52.Choi DW. Excitotoxic cell death. J Neurobiol. 1992;23:1261–1276. doi: 10.1002/neu.480230915. [DOI] [PubMed] [Google Scholar]
- 53.Choi DW. Ionic dependence of glutamate neurotoxicity. J Neurosci. 1987;7:369–379. doi: 10.1523/JNEUROSCI.07-02-00369.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Orrenius S, Zhivotovsky B, Nicotera P. Regulation of cell death: the calcium–apoptosis link. Nat Rev Mol Cell Biol. 2003;4:552–565. doi: 10.1038/nrm1150. [DOI] [PubMed] [Google Scholar]
- 55.Nicholls DG. Mitochondrial calcium function and dysfunction in the central nervous system. Biochim Biophys Acta. 2009;1787:1416–1424. doi: 10.1016/j.bbabio.2009.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Schinder AF, Olson EC, Spitzer NC, Montal M. Mitochondrial dysfunction is a primary event in glutamate neurotoxicity. J Neurosci. 1996;16:6125–6133. doi: 10.1523/JNEUROSCI.16-19-06125.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Vergun O, Keelan J, Khodorov BI, Duchen MR. Glutamate-induced mitochondrial depolarisation and perturbation of calcium homeostasis in cultured rat hippocampal neurones. J Physiol. 1999;519:451–466. doi: 10.1111/j.1469-7793.1999.0451m.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Brustovetsky N, Brustovetsky T, Jemmerson R, Dubinsky JM. Calcium-induced cytochrome c release from CNS mitochondria is associated with the permeability transition and rupture of the outer membrane. J Neurochem. 2002;80:207–218. doi: 10.1046/j.0022-3042.2001.00671.x. [DOI] [PubMed] [Google Scholar]
- 59.Bernardi P, Krauskopf A, Basso E, Petronilli V, Blachly-Dyson E, Di Lisa F, Forte MA. The mitochondrial permeability transition from in vitro artifact to disease target. FEBS J. 2006;273:2077–2099. doi: 10.1111/j.1742-4658.2006.05213.x. [DOI] [PubMed] [Google Scholar]
- 60.Budd SL, Tenneti L, Lishnak T, Lipton SA. Mitochondrial and extramitochondrial apoptotic signaling pathways in cerebrocortical neurons. Proc Natl Acad Sci USA. 2000;97:6161–6166. doi: 10.1073/pnas.100121097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Jemmerson R, Dubinsky JM, Brustovetsky N. Cytochrome c release from CNS mitochondria and potential for clinical intervention in apoptosis-mediated CNS diseases. Antioxid Redox Signal. 2005;7:1158–1172. doi: 10.1089/ars.2005.7.1158. [DOI] [PubMed] [Google Scholar]
- 62.Andrabi SA, Dawson TM, Dawson VL. Mitochondrial and nuclear cross talk in cell death: parthanatos. Ann NY Acad Sci. 2008;1147:233–241. doi: 10.1196/annals.1427.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Bano D, Nicotera P. Ca2+ signals and neuronal death in brain ischemia. Stroke. 2007;38:674–676. doi: 10.1161/01.STR.0000256294.46009.29. [DOI] [PubMed] [Google Scholar]
- 64.Bevers MB, Neumar RW. Mechanistic role of calpains in postischemic neurodegeneration. J Cereb Blood Flow Metab. 2008;28:655–673. doi: 10.1038/sj.jcbfm.9600595. [DOI] [PubMed] [Google Scholar]
- 65.Abramov AY, Scorziello A, Duchen MR. Three distinct mechanisms generate oxygen free radicals in neurons and contribute to cell death during anoxia and reoxygenation. J Neurosci. 2007;27:1129–1138. doi: 10.1523/JNEUROSCI.4468-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Forder JP, Tymianski M. Postsynaptic mechanisms of excitotoxicity: involvement of postsynaptic density proteins, radicals, and oxidant molecules. Neuroscience. 2009;158:293–300. doi: 10.1016/j.neuroscience.2008.10.021. [DOI] [PubMed] [Google Scholar]
- 67.Nicholls DG. Mitochondrial dysfunction and glutamate excitotoxicity studied in primary neuronal cultures. Curr Mol Med. 2004;4:149–177. doi: 10.2174/1566524043479239. [DOI] [PubMed] [Google Scholar]
- 68.Halestrap AP. What is the mitochondrial permeability transition pore? J Mol Cell Cardiol. 2009;46:821–831. doi: 10.1016/j.yjmcc.2009.02.021. [DOI] [PubMed] [Google Scholar]
- 69.Di Lisa F, Canton M, Menabo R, Kaludercic N, Bernardi P. Mitochondria and cardioprotection. Heart Fail Rev. 2007;12:249–260. doi: 10.1007/s10741-007-9028-z. [DOI] [PubMed] [Google Scholar]
- 70.Halestrap AP, Pasdois P. The role of the mitochondrial permeability transition pore in heart disease. Biochim Biophys Acta. 2009;1787:1402–1415. doi: 10.1016/j.bbabio.2008.12.017. [DOI] [PubMed] [Google Scholar]
- 71.Baines CP, Kaiser RA, Purcell NH, Blair NS, Osinska H, Hambleton MA, Brunskill EW, Sayen MR, Gottlieb RA, Dorn GW, et al. Loss of cyclophilin D reveals a critical role for mitochondrial permeability transition in cell death. Nature. 2005;434:658–662. doi: 10.1038/nature03434. [DOI] [PubMed] [Google Scholar]
- 72.Schinzel AC, Takeuchi O, Huang Z, Fisher JK, Zhou Z, Rubens J, Hetz C, Danial NN, Moskowitz MA, Korsmeyer SJ. Cyclophilin D is a component of mitochondrial permeability transition and mediates neuronal cell death after focal cerebral ischemia. Proc Natl Acad Sci USA. 2005;102:12005–12010. doi: 10.1073/pnas.0505294102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Chinopoulos C, Adam-Vizi V. Calcium, mitochondria and oxidative stress in neuronal pathology. Novel aspects of an enduring theme. FEBS J. 2006;273:433–450. doi: 10.1111/j.1742-4658.2005.05103.x. [DOI] [PubMed] [Google Scholar]
- 74.Brustovetsky N, Dubinsky JM. Limitations of cyclosporin A inhibition of the permeability transition in CNS mitochondria. J Neurosci. 2000;20:8229–8237. doi: 10.1523/JNEUROSCI.20-22-08229.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kristian T, Pivovarova NB, Fiskum G, Andrews SB. Calcium-induced precipitate formation in brain mitochondria: composition, calcium capacity, and retention. J Neurochem. 2007;102:1346–1356. doi: 10.1111/j.1471-4159.2007.04626.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Li V, Brustovetsky T, Brustovetsky N. Role of cyclophilin D-dependent mitochondrial permeability transition in glutamate-induced calcium deregulation and excitotoxic neuronal death. Exp Neurol. 2009;218:171–182. doi: 10.1016/j.expneurol.2009.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kokoszka JE, Waymire KG, Levy SE, Sligh JE, Cai J, Jones DP, MacGregor GR, Wallace DC. The ADP/ATP translocator is not essential for the mitochondrial permeability transition pore. Nature. 2004;427:461–465. doi: 10.1038/nature02229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Lee J, Schriner SE, Wallace DC. Adenine nucleotide translocator 1 deficiency increases resistance of mouse brain and neurons to excitotoxic insults. Biochim Biophys Acta. 2009;1787:364–370. doi: 10.1016/j.bbabio.2009.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Gandhi S, Wood-Kaczmar A, Yao Z, Plun-Favreau H, Deas E, Klupsch K, Downward J, Latchman DS, Tabrizi SJ, Wood NW, et al. PINK1-associated Parkinson’s disease is caused by neuronal vulnerability to calcium-induced cell death. Mol Cell. 2009;33:627–638. doi: 10.1016/j.molcel.2009.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Duchen MR, Verkhratsky A, Muallem S. Mitochondria and calcium in health and disease. Cell Calcium. 2008;44:1–5. doi: 10.1016/j.ceca.2008.02.001. [DOI] [PubMed] [Google Scholar]
- 81.Randall RD, Thayer SA. Glutamate-induced calcium transient triggers delayed calcium overload and neurotoxicity in rat hippocampal neurons. J Neurosci. 1992;12:1882–1895. doi: 10.1523/JNEUROSCI.12-05-01882.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Tymianski M, Charlton MP, Carlen PL, Tator CH. Secondary Ca2+ overload indicates early neuronal injury which precedes staining with viability indicators. Brain Res. 1993;607:319–323. doi: 10.1016/0006-8993(93)91523-u. [DOI] [PubMed] [Google Scholar]
- 83.Budd SL, Nicholls DG. Mitochondria, calcium regulation, and acute glutamate excitotoxicity in cultured cerebellar granule cells. J Neurochem. 1996;67:2282–2291. doi: 10.1046/j.1471-4159.1996.67062282.x. [DOI] [PubMed] [Google Scholar]
- 84.Pivovarova NB, Stanika RI, Watts CA, Brantner CA, Smith CL, Andrews SB. Reduced calcium-dependent mitochondrial damage underlies the reduced vulnerability of excitotoxicity-tolerant hippocampal neurons. J Neurochem. 2008;104:1686–1699. doi: 10.1111/j.1471-4159.2007.05080.x. [DOI] [PubMed] [Google Scholar]
- 85.Gerencser AA, Mark KA, Hubbard AE, Divakaruni AS, Mehrabian Z, Nicholls DG, Polster BM. Real-time visualization of cytoplasmic calpain activation and calcium deregulation in acute glutamate excitotoxicity. J Neurochem. 2009;110:990–1004. doi: 10.1111/j.1471-4159.2009.06194.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Shalbuyeva N, Brustovetsky T, Bolshakov A, Brustovetsky N. Calcium-dependent spontaneously reversible remodeling of brain mitochondria. J Biol Chem. 2006;281:37547–37558. doi: 10.1074/jbc.M607263200. [DOI] [PubMed] [Google Scholar]
- 87.Abramov AY, Duchen MR. Mechanisms underlying the loss of mitochondrial membrane potential in glutamate excitotoxicity. Biochim Biophys Acta. 2008;1777:953–964. doi: 10.1016/j.bbabio.2008.04.017. [DOI] [PubMed] [Google Scholar]
- 88.Collins TJ, Berridge MJ, Lipp P, Bootman MD. Mitochondria are morphologically and functionally heterogeneous within cells. EMBO J. 2002;21:1616–1627. doi: 10.1093/emboj/21.7.1616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Droge W. Free radicals in the physiological control of cell function. Physiol Rev. 2002;82:47–95. doi: 10.1152/physrev.00018.2001. [DOI] [PubMed] [Google Scholar]
- 90.Hongpaisan J, Winters CA, Andrews SB. Strong calcium entry activates mitochondrial superoxide generation, upregulating kinase signaling in hippocampal neurons. J Neurosci. 2004;24:10878–10887. doi: 10.1523/JNEUROSCI.3278-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Lambert AJ, Brand MD. Reactive oxygen species production by mitochondria. Methods Mol Biol. 2009;554:165–181. doi: 10.1007/978-1-59745-521-3_11. [DOI] [PubMed] [Google Scholar]
- 92.Starkov AA, Fiskum G, Chinopoulos C, Lorenzo BJ, Browne SE, Patel MS, Beal MF. Mitochondrial α-ketoglutarate dehydrogenase complex generates reactive oxygen species. J Neurosci. 2004;24:7779–7788. doi: 10.1523/JNEUROSCI.1899-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Feissner RF, Skalska J, Gaum WE, Sheu SS. Crosstalk signaling between mitochondrial Ca2+ and ROS. Front Biosci. 2009;14:1197–1218. doi: 10.2741/3303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Vesce S, Kirk L, Nicholls DG. Relationships between superoxide levels and delayed calcium deregulation in cultured cerebellar granule cells exposed continuously to glutamate. J Neurochem. 2004;90:683–693. doi: 10.1111/j.1471-4159.2004.02516.x. [DOI] [PubMed] [Google Scholar]
- 95.Johnson-Cadwell LI, Jekabsons MB, Wang A, Polster BM, Nicholls DG. ‘Mild uncoupling’ does not decrease mitochondrial superoxide levels in cultured cerebellar granule neurons but decreases spare respiratory capacity and increases toxicity to glutamate and oxidative stress. J Neurochem. 2007;101:1619–1631. doi: 10.1111/j.1471-4159.2007.04516.x. [DOI] [PubMed] [Google Scholar]
- 96.Brennan AM, Suh SW, Won SJ, Narasimhan P, Kauppinen TM, Lee H, Edling Y, Chan PH, Swanson RA. NADPH oxidase is the primary source of superoxide induced by NMDA receptor activation. Nat Neurosci. 2009;12:857–863. doi: 10.1038/nn.2334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Lehninger AL, Reynafarje B, Vercesi A, Tew WP. Transport and accumulation of calcium in mitochondria. Ann NY Acad Sci. 1978;307:160–176. doi: 10.1111/j.1749-6632.1978.tb41941.x. [DOI] [PubMed] [Google Scholar]
- 98.Carafoli E, Rossi CS, Lehninger AL. Uptake of adenine nucleotides by respiring mitochondria during active accumulation of Ca++ and phosphate. J Biol Chem. 1965;240:2254–2261. [PubMed] [Google Scholar]
- 99.Hartley DM, Kurth MC, Bjerkness L, Weiss JH, Choi DW. Glutamate receptor-induced 45Ca2+ accumulation in cortical cell culture correlates with subsequent neuronal degeneration. J Neurosci. 1993;13:1993–2000. doi: 10.1523/JNEUROSCI.13-05-01993.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Eimerl S, Schramm M. The quantity of calcium that appears to induce neuronal death. J Neurochem. 1994;62:1223–1226. doi: 10.1046/j.1471-4159.1994.62031223.x. [DOI] [PubMed] [Google Scholar]
- 101.Carriedo SG, Yin HZ, Sensi SL, Weiss JH. Rapid Ca2+ entry through Ca2+-permeable AMPA/kainate channels triggers marked intracellular Ca2+ rises and consequent oxygen radical production. J Neurosci. 1998;18:7727–7738. doi: 10.1523/JNEUROSCI.18-19-07727.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Tymianski M, Charlton MP, Carlen PL, Tator CH. Source specificity of early calcium neurotoxicity in cultured embryonic spinal neurons. J Neurosci. 1993;13:2085–2104. doi: 10.1523/JNEUROSCI.13-05-02085.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Sattler R, Xiong Z, Lu WY, Hafner M, MacDonald JF, Tymianski M. Specific coupling of NMDA receptor activation to nitric oxide neurotoxicity by PSD-95 protein. Science. 1999;284:1845–1848. doi: 10.1126/science.284.5421.1845. [DOI] [PubMed] [Google Scholar]
- 104.Liu Y, Wong TP, Aarts M, Rooyakkers A, Liu L, Lai TW, Wu DC, Lu J, Tymianski M, Craig AM, et al. NMDA receptor subunits have differential roles in mediating excitotoxic neuronal death both in vitro and in vivo. J Neurosci. 2007;27:2846–2857. doi: 10.1523/JNEUROSCI.0116-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Hardingham GE, Fukunaga Y, Bading H. Extrasynaptic NMDARs oppose synaptic NMDARs by triggering CREB shut-off and cell death pathways. Nat Neurosci. 2002;5:405–414. doi: 10.1038/nn835. [DOI] [PubMed] [Google Scholar]
- 106.Zhou M, Baudry M. Developmental changes in NMDA neurotoxicity reflect developmental changes in subunit composition of NMDA receptors. J Neurosci. 2006;26:2956–2963. doi: 10.1523/JNEUROSCI.4299-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Sinor JD, Du S, Venneti S, Blitzblau RC, Leszkiewicz DN, Rosenberg PA, Aizenman E. NMDA and glutamate evoke excitotoxicity at distinct cellular locations in rat cortical neurons in vitro. J Neurosci. 2000;20:8831–8837. doi: 10.1523/JNEUROSCI.20-23-08831.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.von Engelhardt J, Coserea I, Pawlak V, Fuchs EC, Kohr G, Seeburg PH, Monyer H. Excitotoxicity in vitro by NR2A- and NR2B-containing NMDA receptors. Neuropharmacology. 2007;53:10–17. doi: 10.1016/j.neuropharm.2007.04.015. [DOI] [PubMed] [Google Scholar]
- 109.Stanika RI, Pivovarova NB, Brantner CA, Watts CA, Winters CA, Andrews SB. Coupling diverse routes of calcium entry to mitochondrial dysfunction and glutamate excitotoxicity. Proc Natl Acad Sci USA. 2009;106:9854–9859. doi: 10.1073/pnas.0903546106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Martel MA, Wyllie DJ, Hardingham GE. In developing hippocampal neurons, NR2B-containing N-methyl-D-aspartate receptors (NMDARs) can mediate signaling to neuronal survival and synaptic potentiation, as well as neuronal death. Neuroscience. 2009;158:334–343. doi: 10.1016/j.neuroscience.2008.01.080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Aarts M, Liu Y, Liu L, Besshoh S, Arundine M, Gurd JW, Wang YT, Salter MW, Tymianski M. Treatment of ischemic brain damage by perturbing NMDA receptor–PSD-95 protein interactions. Science. 2002;298:846–850. doi: 10.1126/science.1072873. [DOI] [PubMed] [Google Scholar]
- 112.Cui H, Hayashi A, Sun HS, Belmares MP, Cobey C, Phan T, Schweizer J, Salter MW, Wang YT, Tasker RA, et al. PDZ protein interactions underlying NMDA receptor-mediated excitotoxicity and neuroprotection by PSD-95 inhibitors. J Neurosci. 2007;27:9901–9915. doi: 10.1523/JNEUROSCI.1464-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Soriano FX, Martel MA, Papadia S, Vaslin A, Baxter P, Rickman C, Forder J, Tymianski M, Duncan R, Aarts M, et al. Specific targeting of pro-death NMDA receptor signals with differing reliance on the NR2B PDZ ligand. J Neurosci. 2008;28:10696–10710. doi: 10.1523/JNEUROSCI.1207-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Hyrc KL, Bownik JM, Goldberg MP. Ionic selectivity of low-affinity ratiometric calcium indicators: mag-Fura-2, Fura-2FF and BTC. Cell Calcium. 2000;27:75–86. doi: 10.1054/ceca.1999.0092. [DOI] [PubMed] [Google Scholar]
- 115.Peng TI, Greenamyre JT. Privileged access to mitochondria of calcium influx through N-methyl-D-aspartate receptors. Mol Pharmacol. 1998;53:974–980. [PubMed] [Google Scholar]
- 116.Wheeler DG, Barrett CF, Groth RD, Safa P, Tsien RW. Distinct Strategies for Excitation – Transcription Coupling Engaged by Specific Calcium Channel Types. 2009 http://www.abstractsonline.com/Plan/ViewAbstract.aspx?sKey=a32ddce5-f184-41ab-9770-275b8f07cddf&cKey=72954a75-042a-4673-a6bd-1cea09312d56&mKey={081F7976-E4CD-4F3D-A0AF-E8387992A658}
- 117.Ikonomidou C, Turski L. Why did NMDA receptor antagonists fail clinical trials for stroke and traumatic brain injury? Lancet Neurol. 2002;1:383–386. doi: 10.1016/s1474-4422(02)00164-3. [DOI] [PubMed] [Google Scholar]
- 118.Wahlgren NG, Ahmed N. Neuroprotection in cerebral ischaemia: facts and fancies – the need for new approaches. Cerebrovasc Dis. 2004;17(Suppl 1):153–166. doi: 10.1159/000074808. [DOI] [PubMed] [Google Scholar]
- 119.Liu SJ, Zukin RS. Ca2+-permeable AMPA receptors in synaptic plasticity and neuronal death. Trends Neurosci. 2007;30:126–134. doi: 10.1016/j.tins.2007.01.006. [DOI] [PubMed] [Google Scholar]
- 120.Xiong ZG, Chu XP, Simon RP. Acid sensing ion channels – novel therapeutic targets for ischemic brain injury. Front Biosci. 2007;12:1376–1386. doi: 10.2741/2154. [DOI] [PubMed] [Google Scholar]
- 121.Besancon E, Guo S, Lok J, Tymianski M, Lo EH. Beyond NMDA and AMPA glutamate receptors: emerging mechanisms for ionic imbalance and cell death in stroke. Trends Pharmacol Sci. 2008;29:268–275. doi: 10.1016/j.tips.2008.02.003. [DOI] [PubMed] [Google Scholar]
- 122.Noh KM, Yokota H, Mashiko T, Castillo PE, Zukin RS, Bennett MV. Blockade of calcium-permeable AMPA receptors protects hippocampal neurons against global ischemia-induced death. Proc Natl Acad Sci USA. 2005;102:12230–12235. doi: 10.1073/pnas.0505408102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Calderone A, Jover T, Noh KM, Tanaka H, Yokota H, Lin Y, Grooms SY, Regis R, Bennett MV, Zukin RS. Ischemic insults derepress the gene silencer REST in neurons destined to die. J Neurosci. 2003;23:2112–2121. doi: 10.1523/JNEUROSCI.23-06-02112.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Liu S, Lau L, Wei J, Zhu D, Zou S, Sun HS, Fu Y, Liu F, Lu Y. Expression of Ca2+-permeable AMPA receptor channels primes cell death in transient forebrain ischemia. Neuron. 2004;43:43–55. doi: 10.1016/j.neuron.2004.06.017. [DOI] [PubMed] [Google Scholar]
- 125.Peng PL, Zhong X, Tu W, Soundarapandian MM, Molner P, Zhu D, Lau L, Liu S, Liu F, Lu Y. ADAR2-dependent RNA editing of AMPA receptor subunit GluR2 determines vulnerability of neurons in forebrain ischemia. Neuron. 2006;49:719–733. doi: 10.1016/j.neuron.2006.01.025. [DOI] [PubMed] [Google Scholar]
- 126.Kwak S, Weiss JH. Calcium-permeable AMPA channels in neurodegenerative disease and ischemia. Curr Opin Neurobiol. 2006;16:281–287. doi: 10.1016/j.conb.2006.05.004. [DOI] [PubMed] [Google Scholar]
- 127.Rego AC, Ward MW, Nicholls DG. Mitochondria control AMPA/kainate receptor-induced cytoplasmic calcium deregulation in rat cerebellar granule cells. J Neurosci. 2001;21:1893–1901. doi: 10.1523/JNEUROSCI.21-06-01893.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Vander Jagt TA, Connor JA, Shuttleworth CW. Localized loss of Ca2+ homeostasis in neuronal dendrites is a downstream consequence of metabolic compromise during extended NMDA exposures. J Neurosci. 2008;28:5029–5039. doi: 10.1523/JNEUROSCI.5069-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Nicholls DG. Spare respiratory capacity, oxidative stress and excitotoxicity. Biochem Soc Trans. 2009;37:1385–1388. doi: 10.1042/BST0371385. [DOI] [PubMed] [Google Scholar]