

Abstract
The glutamate-aspartate transporter GLAST is a radial glia marker that is highly expressed in GL261 stem-like cells (GSCs). To target GLAST, we treated glioma-bearing mice with three subcutaneous injections of four GLAST peptides emulsified with Montanide ISA-51 in association with granulocyte macrophage colony-stimulating factor (GM-CSF) injections. Vaccination with GLAST peptides significantly prolonged survival, effectively enhanced systemic T-cell and NK-cell responses and promoted robust antitumor cytotoxicity. GLAST expression significantly decreased in gliomas from immunized mice, as evaluated by histological analysis and real-time PCR (RT-PCR). Moreover, the immunization protocol led to the upregulation of interferonγ (IFNγ) and tumor necrosis factorα (TNFα) as well as to the downregulation of transforming growth factor (TGF) β1 and β2 in the tumor. Beyond these changes, gliomas from immunized mice exhibited an increased recruitment of NK cells and antigen-specific CD8+ T cells expressing the tumor homing molecule VLA-4, as well as a local chemotactic gradient featuring expression of CXCL10 (which may be responsible for the recruitment of CTLs), CCL3, CCL4 and CCL5 (which are involved in NK-cell migration), and NKG2D ligand on glioma cells. Importantly, although GLAST is expressed in the central nervous system, autoimmune reactions were not observed in immunized mice. Altogether, these results support the contention that GLAST may constitute a glioma antigen against which immune responses can be efficiently induced without major safety concerns.
Keywords: chemokines, GLAST, glioblastoma, immunotherapy, peptides, radial glia
Introduction
The current standard treatment for glioblastoma (GB) involves surgery, radiotherapy and chemotherapy with temozolomide (TMZ) and is associated with an overall survival (OS) of 14.6 mo.1,2 Phase I/II clinical trials of anti-GB immunotherapy have demonstrated the feasibility and the safety of this approach. Moreover, immune-mediated antitumor responses have been detected in these studies.3-10 Dendritic cells (DCs) loaded with multiple antigens (i.e., tumor cell lysates, tumor-eluted peptides or fusion products of DCs and GB cells) were used to reduce the risk of tumor escape due to antigen-loss variants. Interestingly, strategies focused on promoting antitumor immune responses against tumor-associated antigens in the central nervous system (CNS) have also been proposed.11,12 In addition, the identification of GB cell subpopulations displaying stem cell gene expression programs provided the foundation for immunological studies aimed at targeting these cells. Thus, murine and human GB cell populations contain a fraction of cells with stem cell-like features, and it has been proposed that only this population may be responsible for glioma recurrence. Several groups, including ours, found that GB populations enriched in glioma stem-like cells (GSCs) can give rise to gliomas that closely resemble the original tumor but that are rather different from the experimental gliomas generated by brain injections of serum-driven established cell lines.13,14 GSCs have been found to maintain the genetic alterations of their originating tumor and are tumorigenic in nude mice.15,16 We have found that GSCs can also be obtained from established cultures of murine gliomas, such as GL261 cells. DCs loaded with antigens from GL261 GSCs were significantly more effective than DCs loaded with antigens from serum-cultured GL261 cells in inducing immune rejection of highly malignant gliomas that were otherwise lethal in approximately one month. These findings provide a proof of principle that targeting cell populations enriched in GSCs may increase the efficacy of anti-glioma (and possibly anti-tumor) immunotherapy.17 Characterization of the gene expression profiles of these cells revealed five genes related to radial glia, a source of neural stem cells located in the subventricular zone (SVZ) of the adult brain, that were upregulated in GL261 GSCs.18 We focused our attention on the surface marker GLAST (astrocyte-specific glutamate-aspartate transporter), which plays an important role in glutamate uptake and the regulation of excitatory neurotransmission and prevents glutamate-mediated excitotoxicity in the CNS. GLAST is highly expressed in Bergmann glial cells in the cerebellum and less so in the forebrain and the spinal cord.19,20 The expression of GLAST on GB cells supports the idea that GLAST could be a good target for GB immunotherapy. In this study, we show that immunization with GLAST-derived peptides effectively promotes specific antitumor responses. Our data suggest that the absence of autoimmune reactions and toxicity in this experimental model is associated with a chemotactic gradient that facilitates the homing of immune cells to the tumor site.
Results
Vaccination with GLAST peptides results in long-term survival
To determine whether GLAST peptide-based immunotherapy is protective against GL261 gliomas, C57BL/6 syngeneic mice were vaccinated three times with GLAST-derived peptides beginning four days after intracranial injection of GL261 GSCs (Fig. 1A). Survival analysis demonstrated that GLAST peptides provide significant protection against GL261 GSCs. All control animals (glioma-bearing mice treated with vehicle only) died by day 25, whereas treatment with GLAST peptides emulsified with Montanide ISA-51 in combination with granulocyte macrophage colony-stimulating factor (GM-CSF) injections extended the survival of mice to 40% (p < 0.002 vs. control) (Fig. 1B). In a previous set of experiments, a group of mice received GLAST peptides with imiquimod (an agonist of toll-like receptor 7) as an adjuvant. This treatment only afforded partial protection against GL261 gliomas (24.10 ± 2.30 d for controls vs. 32.90 ± 9.60 d for immunized mice, mean ± SD; p = 0.02) or cytotoxicity (data not shown).

Figure 1. (A) Experimental schema of in vivo treatment. C57BL/6N mice were injected i.c. with GL261 GSCs. Immunized mice received three s.c. vaccinations on days 4, 11 and 18 with all peptides (15 μg/single peptide) emulsified in Montanide™ ISA 51 VG. A total of 3 μg of GM-CSF/mouse were administered during each treatment. Control mice were treated with vehicle only. On days 15 and 22, three mice/group were sacrificed for immune monitoring and histological analysis. (B) Kaplan-Meier survival analysis showed that immunized mice (n = 10) survived longer than control mice (vehicle, n = 10) (p < 0.002). (C) In vitro LDH cytotoxicity assay revealed that splenocytes from immunized mice recognize and lyse GL261 target cells but not NIH 3T3 cells. (D) Flow cytometry performed on splenocytes from immunized mice showed that the percentages of CD4+ and CD8+ T cells and NK cells were significantly increased when compared with controls (upper panels, CD4+ cells: 16.20 ± 0.60% vs. 8.20 ± 1.30%, p = 0.01; CD8+ cells: 13.50 ± 2.10% vs. 4.10 ± 0.07%, p = 0.02; lower panel, NKp46+ CD3- cells: 3.00 ± 0.30 vs. 0.90 ± 0.07, p = 0.009). Data were obtained from three different evaluations and are reported as the mean % ± SD of immunized vs. control mice. (E) RT-PCR analysis showed that the splenocytes of immunized mice express higher levels of IFNγ, TNFα and granzyme B. Infiltration of NK cells was investigated by analyzing the relative expression of the transcription factor E4BP4. ** p < 0.001; *** p < 0.0001.
Vaccination with GLAST peptides induces specific antitumor cytotoxicity
To determine whether GL261-specific effector cells were generated in response to peptide vaccination, pre-stimulated splenocytes were assayed for in vitro cytotoxicity 15 d after tumor implantation. The splenocytes were re-stimulated with peptides and tested 5 d later for cytotoxic activity against GL261 GSCs or NIH 3T3 cells as a negative control, using an LDH release assay. The splenocytes from mice treated with GLAST peptides displayed strong cytotoxic activity against GL261 GSCs when compared with those from control mice (Fig. 1C). The specificity of the cytotoxicity was confirmed by the total absence of reaction against NIH 3T3 cells.
To characterize the direct effects of peptide vaccination in association with GM-CSF treatment on T-cell function, spleens and cervical draining lymph nodes were harvested 15 d after tumor implantation, and multiple cell populations were quantified by flow cytometry. We observed significant increases in NK cells, CD8+ T cells and CD4+ T cells in the spleens and lymph nodes of immunized mice when compared with controls (Fig. 1D and S1). RT-PCR analysis of splenocytes revealed that GLAST peptide administration also enhanced the expression of interferon γ (IFNγ), tumor necrosis factor α (TNFα) granzyme B and the NK cell-specific transcription factor E4BP4 (Fig. 1E). These data support the specificity of the vaccine-induced effector response and the involvement of NK cells in the response to GLAST peptide immunization.
Immunization with peptides specifically targets GLAST-positive glioma cells and modulates the tumor microenvironment
Fifteen days after tumor implantation, GLAST expression was significantly decreased in gliomas from immunized mice. Histological analysis showed that GLAST expression disappeared during the treatment course (Fig. 2A). GLAST expression was 5.2 ± 2.1-fold lower in the tumors of immunized mice than in those of mice treated with vehicle only (Fig. 2B). We also observed a significant reduction in Ki67-positive cells (p < 0.0001) in immunized vs. control mice (Fig. 2C). Immunofluorescence analysis confirmed that GL261 gliomas from control mice strongly expressed GLAST and co-expressed nestin on day 22, whereas GLAST and nestin expression were not detected in gliomas from immunized mice (Fig. S2).

Figure 2. (A) Histological analysis showed strong and diffuse positivity for GLAST in gliomas from vehicle mice. GLAST expression was decreased in gliomas from immunized mice (magnification 2.5X and 20X). (B) RT-PCR confirmed the significant decrease in GLAST expression in gliomas from immunized mice when compared with control mice (n = 3/group). *** p < 0.0001. (C) Quantitative analysis of Ki67-positive cells summarized as the mean ± SD of the number of cells counted in 5 different 40X tumor sections. ** p < 0.001. (D) Relative expression of IFNγ, TNFα, granzyme B, the transcription factor E4BP4 and (E) TGBβ1 and TGFβ2 were investigated using RT-PCR on RNA from immunized and control mice. ** p < 0.001, *** p < 0.0001, **** p < 0.00001.
To characterize the effect of peptide vaccination on the tumor microenvironment, we investigated the expression of key molecules involved in immune activation (IFNγ and TNFα) or suppression (transforming growth factor, TGFβ1, and TGFβ2) by RT-PCR analysis of freshly harvested tumors from immunized and control mice (n = 3/group). IFNγ and TNFα expression levels were 9.60 ± 0.05- and 16.80 ± 0.03- fold higher (p < 0.0001), respectively, in immunized mice than in control mice. Moreover, the levels of granzyme B, the serine protease released by CTLs, and E4BP4 were 25.60 ± 0.12- and 15.00 ± 0.05-fold higher (p < 0.0001 and p < 0.01), respectively (Fig. 2D). In contrast, TGFβ1 and TGFβ2 expression levels were 3.4- and 2.6 ± 0.2-fold lower, respectively, in immunized mice than in control animals (p < 0.001) (Fig. 2E). Similar expression patterns were observed by RT-PCR analysis of paraffin-embedded gliomas (data not shown).
Peptide immunogenicity and efficacy are related to granuloma formation
To verify the immunogenicity of the GLAST-derived peptides, we first considered the appearance of potential local reactions during treatment. Only in immunized mice we observed the formation of a nodule at each injection site immediately after vaccination, enabling the slow, localized release of antigen. In immunized mice that displayed prolonged survival or specific immune reactions, we found four separate granulomas appearing as localized nodules with different sizes at the locations where each of the four peptides was administered.
Non-necrotizing granulomas showing a high proliferation index and lymphocyte accumulation associated with macrophages were found at the injection sites of peptide 1 (Fig. 3A). Granulomas that formed at the injection sites of peptides 2, 3 and 4 were characterized by the prevalence of macrophages, the total absence of proliferating cells, CD8+ and CD4+ T cells, and noticeable central necrosis possibly caused by the activity of Th1 cells (Fig. 3B). In particular, IFNγ is considered a major cause of necrosis in established granulomas as a consequence of exacerbated immune responses.21,22

Figure 3. (A) Granulomas derived from the injection site of peptide 1 appeared well developed and exhibited non-necrotic tissues. They were highly proliferative, as indicated by Ki67 staining and were infiltrated by CD4+, CD8+ and CD11b+ cells. (B) Granulomas from peptide 4 injection sites exhibited central necrosis, a total absence of Ki67-positive cells, CD8+ and CD4+ T lymphocytes and a massive CD11b+-cell infiltration. (C) Splenocytes derived from immunized mice proliferated significantly more than splenocytes derived from vehicle mice. This was particularly apparent in the presence of peptides 3 and 4. *** p < 0.0001, **** p < 0.00001.
To test the in vitro immunogenicity of the four peptides used for vaccinations, splenocytes from control and immunized mice were primed in vitro using a mixture of irradiated antigen-presenting cells (APCs) and peptides. Five days later, the splenocytes were tested for their ability to proliferate in the presence of GLAST peptides. Lymphocytes from immunized mice proliferated significantly more than lymphocytes from vehicle-receiving mice. The ability of peptides 2, 3, and 4 to induce robust proliferation correlated well with their in vivo immunogenicity (Fig. 3C).
The lack of autoimmune reactions and demyelination supports the safety of GLAST-peptide administration
GLAST is not a glioma-specific antigen, and the risk of vaccinating with this antigen must be weighed carefully owing to the possibility of inducing autoimmune reactions targeting the CNS. To test whether the administration of GLAST-derived peptides might damage healthy areas of the CNS, we evaluated brain sections from immunized mice by hematoxylin and eosin (H&E) and Luxol Fast Blue (LFB) staining. We examined brain samples collected at different time points after immunization and observed that highly myelinated areas, such as the corpus callosum and the internal capsule, were densely stained with LFB, demonstrating that demyelination had not occurred in these mice. We also observed the integrity of the SVZ, where GLAST is expressed by radial glia-derived neural stem cells (Fig. 4A and B) and of the cerebellum (Bergmann glia). There was no evidence of immune cell infiltration, and GLAST positivity was comparable in immunized and control mice (Fig. 4C and D).
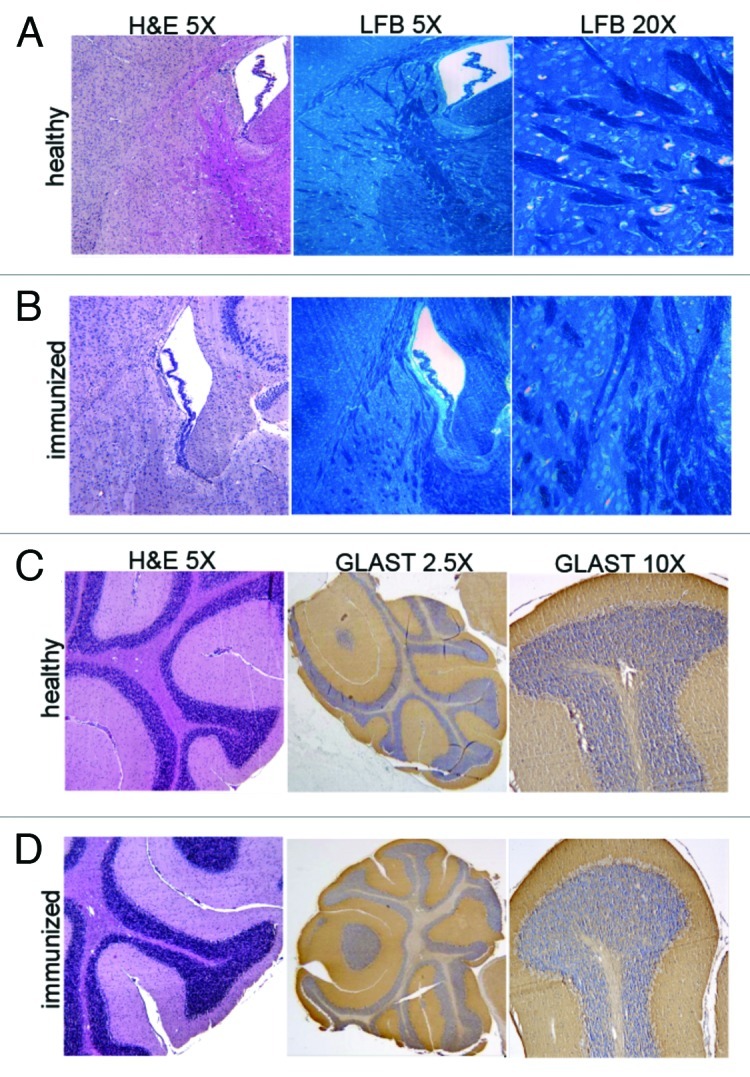
Figure 4. (A,B) H&E and LFB staining of the subventricular zone revealed the total absence of demyelination in the brains of immunized when compared with healthy brains. Highly myelinated areas appear densely stained with LFB (magnification 5X and 40X). (C,D) H&E staining of cerebellar tissue showed a structural integrity (magnification 5X) that was also confirmed by strongly positive GLAST staining at the level of Bergmann glial processes in immunized mice when compared with healthy mice (magnification 2.5X and 20X). Representative images from three mice are shown.
Increased local recruitment and tumor tropism of antigen-specific T cells in immunized mice appear to be related to enhanced expression of chemoattractants
The absence of autoimmune signs supports the idea that activated immune cells selectively travel to the tumor site. We isolated infiltrating lymphocytes from freshly harvested tissues (n = 4 for each group) and evaluated the percentage of CD4+ and CD8+ T cells within the CD3+ T cell population by flow cytometry. Representative plots show that CD4+ T and CD8+ T cell infiltration of tumor tissues was significantly increased in immunized mice (p < 0.001 and p = 0.01, respectively) (Fig. 5A). In addition, the percentage of CD8+ T cells expressing VLA-4 was higher in immunized than in control mice (p = 0.004), supporting the efficient CNS homing of CTLs activated upon peptide vaccination.23 The frequency of NKp46+CD3- NK cells was very low in tumors from vehicle-treated mice and appeared to be slightly, but significantly, increased in tumors from immunized mice (p = 0.002).

Figure 5. (A) CNS-infiltrating CD4+ and CD8+ T cells and NK cells isolated from the glioma-bearing hemisphere on day 15 were more abundant in immunized mice than in control mice. The numbers reported in the dot plots represent the frequency of the investigated subpopulations. RT-PCR analysis showed the expression profile of different chemokines. (B) CCL2 expression was measurable in vehicle control mice and decreased significantly in immunized mice. CXCL10 and VLA-4 expression levels were higher in immunized mice when compared with control mice. (C and D) CCL3, CCL4, CCL5 and NKG2DL were expressed at higher levels in immunized than in control mice. mRNA levels from the hemisphere where the tumor cells were implanted and from the contralateral hemisphere were normalized to the relative quantity of β2-microglobulin. The relative expression of chemokines was compared with that detected in normal brain tissue.
We next hypothesized that the attraction and recruitment of immune cells may be directed by the presence of chemoattractant factors. We thus examined the expression of CCL2 (which is involved in attracting immunosuppressive regulatory T cells, Tregs), CXCL10 (which is responsible for specific CTL recruitment), CCL3, CCL4, and CCL5 in immunized and control mice 15 d after tumor implantation. We performed RT-PCR and compared the results from the left hemisphere, where tumor cells were implanted, to those from the contralateral hemisphere and from tumor-free brain tissue.
We found that CCL2 expression was 5.80 ± 0.10-fold lower and CXCL10 expression was 11.70 ± 0.01-fold higher in immunized mice, compared with their control counterparts (p < 0.0001). Similarly, we found a significant increase in VLA-4 (CD49d) expression (26.60 ± 0.20-fold higher in immunized mice vs. controls, p < 0.001) (Fig. 5A) and in CCL3 and CCL4 expression (2.20 ± 0.10- and 2.00 ± 0.30-fold higher, respectively, p < 0.001) (Fig. 5B). CCL5 was expressed at lower levels than the other chemokines in control mice and was increased significantly in immunized mice (15.00 ± 0.70-fold higher, p < 0.0001) (Fig. 5C).
These data suggest that the CCL3/CCL4/CCL5 axis may play a role in the accumulation of immune cells (and particularly of NK cells) at the sites of tumor formation. To test this hypothesis, we investigated the expression of the NKG2D ligand (NKG2DL), and found that its was expressed at very low levels or was absent in control mice, while it was upregulated in immunized mice, suggesting that the GLAST peptide-induced anti-tumor immune response involves NKG2D-mediated NK cell recognition of tumor cells (Fig. 5C). In further support of the hypothesis that infiltrating and local cells cooperate to create a chemotactic gradient, we found that the expression levels of these chemokines were increased in immunized mice after a single injection of GLAST peptides (data not shown).
Discussion
Our studies focus on increasing the therapeutic efficacy of GB immunotherapy by targeting GSCs, the fraction of GB tumor cells endowed with characteristics of stem cells. GLAST, the radial glia marker used in this study as a target for immunotherapy, has not previously been reported to be expressed in GSCs. By characterizing the expression profiles of GL261 GSCs, we detected the upregulation of five genes that are also expressed in radial glia.17 We then investigated the possibility of isolating GSCs using GLAST as a membrane marker. GLAST-derived peptides emulsified with the adjuvant Montanide ISA-51 and administered in conjunction with GM-CSF were effective in prolonging survival, enhancing systemic and local immunity, and modifying the tumor microenvironment. Loss of GLAST expression in gliomas from immunized mice provided evidence of the induction of a specific antitumor immune response against GLAST-expressing tumor cells that resulted in subsequent immunoediting and tumor escape.24,25 A recent example of immunoediting was provided by studies involving immunotherapy targeting the tumor-specific EGFRvIII mutation, which is highly expressed in GB.26
In spite of immunoediting, however, we found that targeting the radial glia marker GLAST significantly increased the survival of mice bearing GL261 gliomas. We also observed a remarkable modulation of the glioma microenvironment and an increase in peripheral NK cells in immunized mice. In addition, the NK cell-specific transcription factor E4BP427 was expressed at higher levels in gliomas from immunized mice than in those from control mice, confirming the contribution of NK cells to the antitumor response. NK-cell infiltration led to increased IFNγ expression, and its local accumulation exacerbated CXCL10 production in the glioma microenvironment. CXCL10, in turn, was responsible for the recruitment of antigen-specific CTLs, defined as Tc1, to GL261 gliomas. The production of IFNγ and the expression of VLA-4 by antigen-specific Tc1 cells were found to be critical for efficient infiltration into the glioma mass.11,28
NK cells are able to recognize the loss of MHC I molecule, an important event in tumor immunoediting. Recent studies also show the potential of an NK-cell response in fighting GSCs.29,30 In agreement with these findings, we detected upregulation of NKG2DL expression in gliomas from immunized mice. NKG2DL is weakly expressed or absent in GSCs,31 and TGFβ seems to be responsible for both this decreased expression of NKG2DL32,33 and the downregulation of NKG2D receptor on NK cells.34 Thus, the downregulation of TGFβ observed in mice immunized with GLAST peptides could influence NKG2DL expression and reverse the suppression of NK cells.
The efficacy of our therapeutic strategy could be hampered by immune tolerance, as GLAST is expressed by the normal CNS tissue; conversely, the absence of tolerance could result in autoimmune reactions. Our data suggest that the absence of toxicity following GLAST-derived peptide immunization may be attributed to the recruitment of activated immune cells to the tumor site by a chemotactic gradient. In particular, gliomas in GLAST peptide-treated mice displayed upregulation of CXCL10 concomitant with downregulation of CCL2, an important chemokine involved in Treg migration.35 Furthermore, we found that CCL5 was highly expressed in gliomas of immunized mice. This chemokine may play a dual role depending on its source. CCL5/RANTES (regulated upon activation, expressed and secreted by normal T cells) induces chemotaxis in T cells, monocytes, dendritic cells, NK cells, eosinophils and basophils.36-41 When produced by CD8+ T cells, CCL5 is associated with effector functions, together with CCL3 and CCL4,42-44 and antigen-specific T cells are activated in response to CCL5.45 In contrast, tumor-secreted CCL5 is deleterious and acts as an immunosuppressor, and its inhibition improves the efficacy of immunochemotherapy.46 GL261 gliomas treated with vehicle express low levels of CCL5, whereas gliomas from mice immunized with GLAST peptides express much higher levels of CCL5. These findings support the idea that, in our experimental system, CCL5 expression is related to the presence of infiltrating immune cells. In conclusion, both NK cells and CD8+ T cells can play an important role in tumor rejection: NK cells may be one important source of IFNγ, a crucial cytokine that mediates CD8+ T-cell activation.47
GLAST peptide immunotherapy delayed and, at least in some cases, abolished tumor growth, resulting in the survival of 40% of the immunized mice. Recent studies suggest that some chemotherapeutic agents possess immune stimulatory effects.48,49 We thus plan to design immunochemotherapy combinations in an attempt to potentiate the specific antitumor immunity achieved following GLAST peptide immunization. In addition, a more complete appraisal of the safety of GLAST immunotherapy is essential to assess the translational potential of these findings.
Materials and Methods
Cell culture
GL261 glioma cells were cultured in DMEM-F12 Glutamax™-1 (Life Technologies), B-27 supplement 1X (Life Technologies), penicillin/streptomycin 1X, human recombinant epidermal growth factor (EGF; 20 ng/ml; Peprotech), and human recombinant fibroblast growth factor-2 (FGF-2; 20 ng/ml; Peprotech). The spleens and draining lymph nodes of immunized and control mice were surgically removed and mechanically processed in RPMI 1640 basal medium (LONZA) containing 5% fetal bovine serum, 20 mM HEPES, 2% L-glutamine and penicillin/streptomycin. Erythrocytes were lysed with ice-cold ACK buffer, and lymphocytes were resuspended in RPMI 1640 supplemented with 10% fetal bovine serum, 2% penicillin/streptomycin, 2% L-glutamine, 50 μM β-mercaptoethanol, 100 mM sodium pyruvate and 100X nonessential amino acids. We also added 10 U/ml human recombinant IL-2 (Roche) to the medium for all in vitro experiments.
Peptide prediction
To determine whether GLAST peptide-based immunotherapy is protective against GL261 gliomas, we used the SYFPEITHI (http://www.syfpeithi.de/) and BIMAS (http://www.bimas.cit.nih.gov/molbio/hla_bind/) binding-motif algorithms to identify high-scoring target peptides containing both the murine major histocompatibility complex (MHC) H-2Db and the human HLA-A*0201. We selected four peptides: Pep1(44–52) YLFRNAFVL, score 23; Pep2(103–11) SLVTGMAAL, score 18; Pep3(368–376) TLPITFKCL, score 16; and Pep4(403–411) ALYEALAAI, score 15. The complete sequence of GLAST is available in the UniProt database (http://www.uniprot.org/). Lyophilized peptides were purchased from Primm (Primm S.r.l) and resuspended at a final concentration of 5 mg/ml/peptide using SEPICLEAR™ 01 PPI (SEPPIC), a lipoamino acid compound designed to dissolve poorly soluble peptides.
In vivo experiments
A total of 38 C57BL/6N mice (Charles River Laboratory), 19 immunized and 19 controls (10 mice per group for survival studies and 9 mice per group for immunological and histological studies), were injected with 1x105 GL261 cells/mouse into the nucleus caudatum using a stereotactic frame. The stereotactic coordinates with respect to the bregma were as follows: 0.7 mm posterior, 3 mm left lateral, and 3.5 mm deep into the nucleus caudatum. The immunized mice received subcutaneous (sc) injections of all four peptides (15 μg/peptide) into different areas of the flank on days 4, 11 and 18 after tumor implantation (day 0). The peptides were emulsified with Montanide™ ISA 51 VG (1:1) (SEPPIC) using two 2-ml sterile Luer Lock glass syringes (Artiglass S.r.l) and a three-way stopcock connector (MOVI S.p.A.) as recommended by the manufacturer. The immunized mice received a total of 3 μg of recombinant murine granulocyte macrophage colony-stimulating factor (GM-CSF) (Miltenyi Biotec) spread out over three injections into the same area of the peptide injections, beginning one day before the first vaccination. The control mice were treated with vehicle only (Montanide) on the same days as the immunized mice were vaccinated and injected with GM-CSF according to the experimental schema (Fig. 1A).
Cytotoxicity assay
Lymphocytes from immunized and control mice were isolated on day 15 after tumor implantation. We tested their ability to recognize and lyse GL261 cells in vitro. A total of 2x106 splenocytes were pre-stimulated for 5 d in the presence of 5x105 3-Gy-irradiated naïve splenocytes as antigen presenting cells and 5 μg/mL of each peptide. The cells were cultured in 24-well tissue culture plates in a final volume of 2 mL/well of complete RPMI 1640 supplemented with 10 U/mL IL-2. Splenocytes from healthy mice were irradiated (43855F Cabinet X-ray System, Faxitron®X-ray Corporation) and used as antigen presenting cells. Pre-stimulated lymphocytes were tested for GL261-specific cytotoxicity using different effector:target (E:T) ratios (10:1, 20:1, and 40:1). NIH 3T3 cells were used as negative controls. To quantify cell lysis, a non-radioactive cytotoxic assay (Cytotoxicity detection kitplus LDH, Roche) was performed according to the manufacturer’s instructions. Absorbances for the various cell groups were used to calculate the percentage of specific cytotoxicity according to the following equation: (effector:target cell mix - effector cell control) - low control / (high control - low control) x 100, where the high and low controls correspond to the background target absorbance and the maximum target absorbance, respectively.
Proliferation assay
Immunized and control mice were sacrificed 15 d after tumor implantation, and a total of 2x106 splenocytes were primed for 5 d in the presence of 5x105 3-Gy-irradiated naïve splenocytes as antigen presenting cells, 5 μg/mL of all peptides and 10 U/ml of IL-2. After pre-stimulation, 5x105 splenocytes were incubated for 24 h or 48 h in the presence of single peptides (5 μg/ml) and IL-2 and tested for their ability to proliferate. The number of viable cells was assessed using MTT Reagent (Millipore). The data are expressed as the percentage of proliferation, which is calculated according to the following equation: (OD stimulated splenocytes - OD splenocytes without peptide) / OD stimulated splenocytes x 100.
Isolation of CNS-infiltrating lymphocytes
CNS-infiltrating lymphocytes were isolated using a tumor dissociation kit (mouse, Miltenyi Biotec) on day 15 after tumor implantation. Briefly, glioma-bearing hemispheres (where GL261 cells were injected) from immunized and control mice (n = 4/group) were explanted, cut into small pieces of 2–4 mm, and dissociated using GentleMACS (Miltenyi Biotec) according to manufacturer’s instruction. The cells were then suspended in PEB buffer (PBS/0.5% bovine serum albumin/2 mM EDTA) for labeling and flow cytometry evaluation.
Flow cytometry
Lymphocytes from spleens, cervical draining lymph nodes and explanted gliomas of immunized and control mice were used for immune monitoring. Briefly, 1.5x106 cells were stained in PBS for 30 min at 4°C with the following antibodies: anti-CD4 PE-Cy5 (BD Bioscience), anti-CD3 FITC (Biolegend), anti-CD8-FITC (BD Bioscience), anti-CD8-PE (Biolegend), and anti-CD49d-APC (Miltenyi Biotec). For NK cell detection, an anti-NKp46-PE antibody (Miltenyi Biotec) was used according to the manufacturer’s instructions. Flow cytometry acquisition was performed on a MACSQuant instrument, and the data were analyzed with the MACSQuantify Software (Miltenyi Biotec).
Real-time PCR (RT-PCR)
Total RNA was isolated from freshly harvested GL261 gliomas, lymphocytes and paraffin-embedded samples from immunized and control mice and used for gene expression analysis. Similar studies were also performed on cells from in vitro experiments. RNA was extracted with TRIzol reagent (Life Technologies) using the RNeasy MINI KIT (Qiagen) and the RNase-Free DNase Set (Qiagen). For paraffin-embedded samples, the Absolutely RNA FFPE kit (Stratagene) was used according to the manufacturer’s instructions. cDNA was synthesized from total RNA using oligo (dT) and M-MLV Reverse Transcriptase (Life Technologies). Specific primers for target genes were designed for Fast SYBR Green chemistry (Applied Biosystems) and purchased from Primm S.r.l. The relative mRNA levels were measured using a 7500 Real-Time PCR System (Applied Biosystems) and calculated using the ΔΔCt method. The expression levels of the target genes were normalized to the expression level of β2-microglobulin. The sequences of the primers are reported in the supplementary data.
Histology and immunohistochemistry
Immunohistochemical analysis of GLAST (Santa Cruz), Ki67 (BD Bioscience), CD8 (R&D Systems), CD4 (R&D Systems), and CD11b (BD Bioscience) was performed on paraffin-embedded sections. For double immunofluorescence, the tumor sections were incubated with anti-GLAST and anti-nestin antibodies overnight at 4°C and then incubated with Alexa Fluor 488-conjugated anti-rabbit secondary antibody. Quantitative analyses were performed on three to five independent fields per tumor by counting the number of cells in the photographed fields using the 40X objective of a Leica DM-LB microscope.
Statistical analysis
The differences between groups were analyzed using two-tailed Student’s t-tests and were considered statistically significant when p < 0.05.
Supplementary Material
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
This work has been supported by fund from Associazione Italiana per la Ricerca sul Cancro (AIRC) to GF and by a grant from Il Fondo di Gio, a charity affiliated to the International Brain Tumor Alliance. The manuscript was edited by American Journal Experts (AJE).
Supplemental Materials
Supplemental materials can be found at: www.landesbioscience.com/journals/oncoimmunology/article/20637
Footnotes
Previously published online: www.landesbioscience.com/journals/oncoimmunology/article/20637
References
- 1.Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ, et al. European Organisation for Research and Treatment of Cancer Brain Tumor and Radiotherapy Groups. National Cancer Institute of Canada Clinical Trials Group Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005;352:987–96. doi: 10.1056/NEJMoa043330. [DOI] [PubMed] [Google Scholar]
- 2.Stupp R, Hegi ME, Mason WP, van den Bent MJ, Taphoorn MJ, Janzer RC, et al. European Organisation for Research and Treatment of Cancer Brain Tumour and Radiation Oncology Groups. National Cancer Institute of Canada Clinical Trials Group Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 2009;10:459–66. doi: 10.1016/S1470-2045(09)70025-7. [DOI] [PubMed] [Google Scholar]
- 3.Liau LM, Prins RM, Kiertscher SM, Odesa SK, Kremen TJ, Giovannone AJ, et al. Dendritic cell vaccination in glioblastoma patients induces systemic and intracranial T-cell responses modulated by the local central nervous system tumor microenvironment. Clin Cancer Res. 2005;11:5515–25. doi: 10.1158/1078-0432.CCR-05-0464. [DOI] [PubMed] [Google Scholar]
- 4.Wheeler CJ, Black KL, Liu G, Mazer M, Zhang XX, Pepkowitz S, et al. Vaccination elicits correlated immune and clinical responses in glioblastoma multiforme patients. Cancer Res. 2008;68:5955–64. doi: 10.1158/0008-5472.CAN-07-5973. [DOI] [PubMed] [Google Scholar]
- 5.De Vleeschouwer S, Fieuws S, Rutkowski S, Van Calenbergh F, Van Loon J, Goffin J, et al. Postoperative adjuvant dendritic cell-based immunotherapy in patients with relapsed glioblastoma multiforme. Clin Cancer Res. 2008;14:3098–104. doi: 10.1158/1078-0432.CCR-07-4875. [DOI] [PubMed] [Google Scholar]
- 6.Okada H, Lieberman FS, Walter KA, Lunsford LD, Kondziolka DS, Bejjani GK, et al. Autologous glioma cell vaccine admixed with interleukin-4 gene transfected fibroblasts in the treatment of patients with malignant gliomas. J Transl Med. 2007;5:67. doi: 10.1186/1479-5876-5-67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Okada H, Lieberman FS, Edington HD, Witham TF, Wargo MJ, Cai Q, et al. Autologous glioma cell vaccine admixed with interleukin-4 gene transfected fibroblasts in the treatment of recurrent glioblastoma: preliminary observations in a patient with a favorable response to therapy. J Neurooncol. 2003;64:13–20. doi: 10.1007/BF02700016. [DOI] [PubMed] [Google Scholar]
- 8.Kikuchi T, Akasaki Y, Abe T, Fukuda T, Saotome H, Ryan JL, et al. Vaccination of glioma patients with fusions of dendritic and glioma cells and recombinant human interleukin 12. J Immunother. 2004;27:452–9. doi: 10.1097/00002371-200411000-00005. [DOI] [PubMed] [Google Scholar]
- 9.Yamanaka R, Homma J, Yajima N, Tsuchiya N, Sano M, Kobayashi T, et al. Clinical evaluation of dendritic cell vaccination for patients with recurrent glioma: results of a clinical phase I/II trial. Clin Cancer Res. 2005;11:4160–7. doi: 10.1158/1078-0432.CCR-05-0120. [DOI] [PubMed] [Google Scholar]
- 10.Sampson JH, Archer GE, Mitchell DA, Heimberger AB, Herndon JE, 2nd, Lally-Goss D, et al. An epidermal growth factor receptor variant III-targeted vaccine is safe and immunogenic in patients with glioblastoma multiforme. Mol Cancer Ther. 2009;8:2773–9. doi: 10.1158/1535-7163.MCT-09-0124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhu X, Nishimura F, Sasaki K, Fujita M, Dusak JE, Eguchi J, et al. Toll like receptor-3 ligand poly-ICLC promotes the efficacy of peripheral vaccinations with tumor antigen-derived peptide epitopes in murine CNS tumor models. J Transl Med. 2007;5:10. doi: 10.1186/1479-5876-5-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Okada H, Kalinski P, Ueda R, Hoji A, Kohanbash G, Donegan TE, et al. Induction of CD8+ T-cell responses against novel glioma-associated antigen peptides and clinical activity by vaccinations with alpha-type 1 polarized dendritic cells and polyinosinic-polycytidylic acid stabilized by lysine and carboxymethylcellulose in patients with recurrent malignant glioma. J Clin Oncol. 2011;29:330–6. doi: 10.1200/JCO.2010.30.7744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Singh SK, Clarke ID, Terasaki M, Bonn VE, Hawkins C, Squire J, et al. Identification of a cancer stem cell in human brain tumors. Cancer Res. 2003;63:5821–8. [PubMed] [Google Scholar]
- 14.Singh SK, Hawkins C, Clarke ID, Squire JA, Bayani J, Hide T, et al. Identification of human brain tumour initiating cells. Nature. 2004;432:396–401. doi: 10.1038/nature03128. [DOI] [PubMed] [Google Scholar]
- 15.Tunici P, Bissola L, Lualdi E, Pollo B, Cajola L, Broggi G, et al. Genetic alterations and in vivo tumorigenicity of neurospheres derived from an adult glioblastoma. Mol Cancer. 2004;3:25. doi: 10.1186/1476-4598-3-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Galli R, Binda E, Orfanelli U, Cipelletti B, Gritti A, De Vitis S, et al. Isolation and characterization of tumorigenic, stem-like neural precursors from human glioblastoma. Cancer Res. 2004;64:7011–21. doi: 10.1158/0008-5472.CAN-04-1364. [DOI] [PubMed] [Google Scholar]
- 17.Pellegatta S, Poliani PL, Corno D, Menghi F, Ghielmetti F, Suarez-Merino B, et al. Neurospheres enriched in cancer stem-like cells are highly effective in eliciting a dendritic cell-mediated immune response against malignant gliomas. Cancer Res. 2006;66:10247–52. doi: 10.1158/0008-5472.CAN-06-2048. [DOI] [PubMed] [Google Scholar]
- 18.Merkle FT, Tramontin AD, García-Verdugo JM, Alvarez-Buylla A. Radial glia give rise to adult neural stem cells in the subventricular zone. Proc Natl Acad Sci U S A. 2004;101:17528–32. doi: 10.1073/pnas.0407893101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shashidharan P, Plaitakis A. Cloning and characterization of a glutamate transporter cDNA from human cerebellum. Biochim Biophys Acta. 1993;1216:161–4. doi: 10.1016/0167-4781(93)90057-k. [DOI] [PubMed] [Google Scholar]
- 20.Danbolt NC, Storm-Mathisen J, Kanner BI. An [Na+ + K+]coupled L-glutamate transporter purified from rat brain is located in glial cell processes. Neuroscience. 1992;51:295–310. doi: 10.1016/0306-4522(92)90316-T. [DOI] [PubMed] [Google Scholar]
- 21.Ehlers S, Benini J, Held HD, Roeck C, Alber G, Uhlig S. Alphabeta T cell receptor-positive cells and interferon-gamma, but not inducible nitric oxide synthase, are critical for granuloma necrosis in a mouse model of mycobacteria-induced pulmonary immunopathology. J Exp Med. 2001;194:1847–59. doi: 10.1084/jem.194.12.1847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Flórido M, Cooper AM, Appelberg R. Immunological basis of the development of necrotic lesions following Mycobacterium avium infection. Immunology. 2002;106:590–601. doi: 10.1046/j.1365-2567.2002.01459.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Calzascia T, Masson F, Di Berardino-Besson W, Contassot E, Wilmotte R, Aurrand-Lions M, et al. Homing phenotypes of tumor-specific CD8 T cells are predetermined at the tumor site by crosspresenting APCs. Immunity. 2005;22:175–84. doi: 10.1016/j.immuni.2004.12.008. [DOI] [PubMed] [Google Scholar]
- 24.Schreiber RD, Old LJ, Smyth MJ. Cancer immunoediting: integrating immunity’s roles in cancer suppression and promotion. Science. 2011;331:1565–70. doi: 10.1126/science.1203486. [DOI] [PubMed] [Google Scholar]
- 25.Pellegatta S, Cuppini L, Finocchiaro G. Brain cancer immunoediting: novel examples provided by immunotherapy of malignant gliomas. Expert Rev Anticancer Ther. 2011;11:1759–74. doi: 10.1586/era.11.102. [DOI] [PubMed] [Google Scholar]
- 26.Sampson JH, Heimberger AB, Archer GE, Aldape KD, Friedman AH, Friedman HS, et al. Immunologic escape after prolonged progression-free survival with epidermal growth factor receptor variant III peptide vaccination in patients with newly diagnosed glioblastoma. J Clin Oncol. 2010;28:4722–9. doi: 10.1200/JCO.2010.28.6963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gascoyne DM, Long E, Veiga-Fernandes H, de Boer J, Williams O, Seddon B, et al. The basic leucine zipper transcription factor E4BP4 is essential for natural killer cell development. Nat Immunol. 2009;10:1118–24. doi: 10.1038/ni.1787. [DOI] [PubMed] [Google Scholar]
- 28.Sasaki K, Zhu X, Vasquez C, Nishimura F, Dusak JE, Huang J, et al. Preferential expression of very late antigen-4 on type 1 CTL cells plays a critical role in trafficking into central nervous system tumors. Cancer Res. 2007;67:6451–8. doi: 10.1158/0008-5472.CAN-06-3280. [DOI] [PubMed] [Google Scholar]
- 29.Castriconi R, Daga A, Dondero A, Zona G, Poliani PL, Melotti A, et al. NK cells recognize and kill human glioblastoma cells with stem cell-like properties. J Immunol. 2009;182:3530–9. doi: 10.4049/jimmunol.0802845. [DOI] [PubMed] [Google Scholar]
- 30.Avril T, Vauleon E, Hamlat A, Saikali S, Etcheverry A, Delmas C, et al. Human glioblastoma stem-like cells are more sensitive to allogeneic NK and T cell-mediated killing compared with serum-cultured glioblastoma cells. Brain Pathol 2012; 22:159-74; PMID: 20493562; DOI: 10.1111/j.1750-3639.2011.00515.x; 10.1111/j.1750-3639.2011.00515.x. [DOI] [PMC free article] [PubMed]
- 31.Di Tomaso T, Mazzoleni S, Wang E, Sovena G, Clavenna D, Franzin A, et al. Immunobiological characterization of cancer stem cells isolated from glioblastoma patients. Clin Cancer Res. 2010;16:800–13. doi: 10.1158/1078-0432.CCR-09-2730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Friese MA, Wischhusen J, Wick W, Weiler M, Eisele G, Steinle A, et al. RNA interference targeting transforming growth factor-beta enhances NKG2D-mediated antiglioma immune response, inhibits glioma cell migration and invasiveness, and abrogates tumorigenicity in vivo. Cancer Res. 2004;64:7596–603. doi: 10.1158/0008-5472.CAN-04-1627. [DOI] [PubMed] [Google Scholar]
- 33.Eisele G, Wischhusen J, Mittelbronn M, Meyermann R, Waldhauer I, Steinle A, et al. TGF-beta and metalloproteinases differentially suppress NKG2D ligand surface expression on malignant glioma cells. Brain. 2006;129:2416–25. doi: 10.1093/brain/awl205. [DOI] [PubMed] [Google Scholar]
- 34.Crane CA, Han SJ, Barry JJ, Ahn BJ, Lanier LL, Parsa AT. TGF-beta downregulates the activating receptor NKG2D on NK cells and CD8+ T cells in glioma patients. Neuro Oncol. 2010;12:7–13. doi: 10.1093/neuonc/nop009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jordan JT, Sun W, Hussain SF, DeAngulo G, Prabhu SS, Heimberger AB. Preferential migration of regulatory T cells mediated by glioma-secreted chemokines can be blocked with chemotherapy. Cancer Immunol Immunother. 2008;57:123–31. doi: 10.1007/s00262-007-0336-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schall TJ, Bacon K, Toy KJ, Goeddel DV. Selective attraction of monocytes and T lymphocytes of the memory phenotype by cytokine RANTES. Nature. 1990;347:669–71. doi: 10.1038/347669a0. [DOI] [PubMed] [Google Scholar]
- 37.Roth SJ, Carr MW, Springer TA. C-C chemokines, but not the C-X-C chemokines interleukin-8 and interferon-gamma inducible protein-10, stimulate transendothelial chemotaxis of T lymphocytes. Eur J Immunol. 1995;25:3482–8. doi: 10.1002/eji.1830251241. [DOI] [PubMed] [Google Scholar]
- 38.de la Rosa G, Longo N, Rodríguez-Fernández JL, Puig-Kroger A, Pineda A, Corbí AL, et al. Migration of human blood dendritic cells across endothelial cell monolayers: adhesion molecules and chemokines involved in subset-specific transmigration. J Leukoc Biol. 2003;73:639–49. doi: 10.1189/jlb.1002516. [DOI] [PubMed] [Google Scholar]
- 39.Taub DD, Sayers TJ, Carter CR, Ortaldo JR. Alpha and beta chemokines induce NK cell migration and enhance NK-mediated cytolysis. J Immunol. 1995;155:3877–88. [PubMed] [Google Scholar]
- 40.Kameyoshi Y, Dörschner A, Mallet AI, Christophers E, Schröder JM. Cytokine RANTES released by thrombin-stimulated platelets is a potent attractant for human eosinophils. J Exp Med. 1992;176:587–92. doi: 10.1084/jem.176.2.587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Conti P, Pang X, Boucher W, Letourneau R, Reale M, Barbacane RC, et al. RANTES is a pro-inflammatory chemokine and chemoattracts basophil cells to extravascular sites. J Pathol. 1997;183:352–8. doi: 10.1002/(SICI)1096-9896(199711)183:3<352::AID-PATH938>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- 42.Taub DD, Ortaldo JR, Turcovski-Corrales SM, Key ML, Longo DL, Murphy WJ. Beta chemokines costimulate lymphocyte cytolysis, proliferation, and lymphokine production. J Leukoc Biol. 1996;59:81–9. doi: 10.1002/jlb.59.1.81. [DOI] [PubMed] [Google Scholar]
- 43.Hadida F, Vieillard V, Mollet L, Clark-Lewis I, Baggiolini M, Debré P. Cutting edge: RANTES regulates Fas ligand expression and killing by HIV-specific CD8 cytotoxic T cells. J Immunol. 1999;163:1105–9. [PubMed] [Google Scholar]
- 44.Cocchi F, DeVico AL, Garzino-Demo A, Arya SK, Gallo RC, Lusso P. Identification of RANTES, MIP-1 alpha, and MIP-1 beta as the major HIV-suppressive factors produced by CD8+ T cells. Science. 1995;270:1811–5. doi: 10.1126/science.270.5243.1811. [DOI] [PubMed] [Google Scholar]
- 45.Appay V, Dunbar PR, Cerundolo V, McMichael A, Czaplewski L, Rowland-Jones S. RANTES activates antigen-specific cytotoxic T lymphocytes in a mitogen-like manner through cell surface aggregation. Int Immunol. 2000;12:1173–82. doi: 10.1093/intimm/12.8.1173. [DOI] [PubMed] [Google Scholar]
- 46.Conforti R, Ma Y, Morel Y, Paturel C, Terme M, Viaud S, et al. Opposing effects of toll-like receptor (TLR3) signaling in tumors can be therapeutically uncoupled to optimize the anticancer efficacy of TLR3 ligands. Cancer Res. 2010;70:490–500. doi: 10.1158/0008-5472.CAN-09-1890. [DOI] [PubMed] [Google Scholar]
- 47.Salem ML, El-Naggar SA, Kadima A, Gillanders WE, Cole DJ. The adjuvant effects of the toll-like receptor 3 ligand polyinosinic-cytidylic acid poly (I:C) on antigen-specific CD8+ T cell responses are partially dependent on NK cells with the induction of a beneficial cytokine milieu. Vaccine. 2006;24:5119–32. doi: 10.1016/j.vaccine.2006.04.010. [DOI] [PubMed] [Google Scholar]
- 48.Zitvogel L, Apetoh L, Ghiringhelli F, Kroemer G. Immunological aspects of cancer chemotherapy. Nat Rev Immunol. 2008;8:59–73. doi: 10.1038/nri2216. [DOI] [PubMed] [Google Scholar]
- 49.Galluzzi L, Senovilla L, Zitvogel L, Kroemer G. The secret ally: Immunostimulation by anticancer drugs. Nat Rev Drug Discov 2012; 11:215-33; PMID: 22252132; DOI: 10.1038/nrd3626; 10.1038/nrd3626. [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.