

Abstract
Aims
The aim of this study was to elucidate the effects of regulator of G-protein signaling 5 (Rgs5), a negative regulator of G protein-mediated signaling, on atrial repolarization and tachyarrhythmia (ATA) in mice.
Methods and Results
In present study, the incidence of ATA were increased in Rgs5−/− Langendorff-perfused mouse hearts during program electrical stimulation (PES) (46.7%, 7 of 15) and burst pacing (26.7%, 4 of 15) compared with wild-type (WT) mice (PES: 7.1%,1 of 14; burst:7.1%,1 of 14) (P<0.05). And the duration of ATA also shown longer in Rgs5−/− heart than that in WT, 2 out of 15 hearts exhibited sustained ATA (>30 s) but none of them observed in WT mice. Atrial prolonged repolarization was observed in Rgs5−/− hearts including widened P wave in surface ECG recording, increased action potential duration (APD) and atrial effective refractory periods (AERP), all of them showed significant difference with WT mice (P<0.05). At the cellular level, whole-cell patch clamp recorded markedly decreased densities of repolarizing K+ currents including IKur (at +60 mV: 14.0±2.2 pF/pA) and Ito (at +60 mV: 16.7±1.3 pA/pF) in Rgs5−/− atrial cardiomyocytes, compared to those of WT mice (at +60 mV Ito: 20.4±2.0 pA/pF; Ikur: 17.9±2.0 pF/pA) (P<0.05).
Conclusion
These results suggest that Rgs5 is an important regulator of arrhythmogenesis in the mouse atrium and that the enhanced susceptibility to atrial tachyarrhythmias in Rgs5−/− mice may contribute to abnormalities of atrial repolarization.
Introduction
Atrial tachyarrhythmia (ATA), characterized by abnormal, disorganized and rapid atrial electrical activity, is a major public health problem. In particular, atrial fibrillation, which affects 0.5% of people over the age of 50 and 10% of people over 80 [2], is a significant burden on health care resources and increases the risk of serious complications including embolic disease, heart failure and sudden cardiac death. Despite the development of diverse animal models and the application of a multitude of pharmacological and non-pharmacological therapies, understanding of the mechanism underlying ATA remained incomplete. The recent discovery of DNA variants associated with ATA provides new insights into the mechanisms underlying arrhythmias. Changes in the expression of multiple genes including KCNQ1, KCNE1 and KCNE2 have been shown predisposed to atrial fibrillation [1], [4]. Thus, the upstream molecular-regulated targets of pro-arrhythmic substrate need to be better understood.
Rgs5 is a GTPase-activating protein (GAP) for Gα subunits and has an important role in the negative regulation of G-protein-coupled receptor (GPCR)-mediated signaling. Several studies have demonstrated that Rgs5 interacts with Gα(q) and Gα(i) in cardiovascular tissue and inhibits those acting via special GPCRs including AngII type 1 receptor (AT1R) and endothilin-1 receptors [5]. These GPCRs have been implicated in the development of cardiac remodeling during AF, and induced left atrial remodeling and fibrosis are dependent on the extracellular signal-regulated kinase (ERK) pathway [7]. Our previous work showed that Rgs5−/− mice promoted cardiac hypertrophy and fibrosis due to the activation of the MEK-ERK1/2 signaling pathway [15]. As an important regulator of atrial GPCR-dependent signaling, RGS5 protein may potentially exhibit negative way in pro-arrhythmic substrate.
We report here that mice with a knockout of Rgs5 displayed susceptibility of ATA by electrical stimuli and had associated repolarization abnormalities. These observations may provide a new insight into understanding the molecular pathways underlying atrial arrhythmogenesis and for developing strategies for the treatment of atrial tachyarrhythmia.
Materials and Methods
1. Mouse experiments
All animal procedures were performed in accordance with the Guide for the Care and Use of Laboratory Animals, published by the US National Institutes of Health (NIH Publication No. 85-23, revised 1996) and were approved by the Institutional Animal Care and Use Committee at Renmin Hospital of Wuhan University, China.
The generation and genotyping of Rgs5−/− mice (C57BL/6 background) have been described previously. Mice were provided with food and water and held on standard 12 hours light and dark cycles in temperature and humidity controlled house. Male Wild-type and Rgs5−/− mice aged 8 to 10 weeks were used in the studies.
2. ECG recordings
Telemetry ECG (Lead II) was continuously recorded in consciously moving mice by Telemetry ECG (DSI, US). The P-wave duration and amplitude (Pdur and Pamp), PR interval and QRS duration was measured.
3. Echocardiography and histological analysis
Echocardiography was performed to assess left atrial diameter (LAD), left ventricular end-diastolic diameter (LVEDD), left ventricular end-systolic diameter (LVESD), ejection fraction (EF) and fractional shortening (FS). For histological analysis, hearts were excised and cut transversely close to the apex to visualize the left and right ventricles and atrial appendages. Sections of heart (4–5 µm thick) were prepared and stained with Picro Sirius Red (PSR) for collagen deposition [15].
4. Preparation of Langendorff-perfused hearts
The isolated heart was perfused with HEPES-buffered Tyrode's solution. Perfusion was commenced in a retrograde manner through the aorta at 2–2.5 ml/min. The hearts that did not recover to the regular spontaneous rhythm or had inreversible myocardial ischemia were discarded.
5. MAP and BEG recording
To examine the atrial electrical activity, the bipolar electrogram (BEG) was recorded from the epicardial surface of atrial appendage using a silver chloride (2 mm tip diameter) recording electrode. The paired platinum stimulating electrodes paced the epicardial surface of right atrial appendage and the stimulation used a 1 ms square-wave stimuli at three times excitation threshold.
6. Electrical stimulated protocol
The Programmed electrical stimulation (PES) protocol was used for atrial effective refractory period (AERP) and atrial-ventricular effective refractory period (AVERP) examinations. Sinus node recovery time (SNRT) was measured after a 2 s pacing train at a cycle length of 100 ms [16]. Inducibility of atrial tachyarrhythmia was tested by using both PES and burst pacing (2 ms pulses at 50 Hz, 2 s burst duration) [24], burst pacing used up to 3 minutes of pacing in both atrial locations.
7. Transmembrane action potential (TAP) recording
The samples of atrial myocardium were removed from heart and superfused with HEPES-buffered Tyrode's solution. TAP were recorded with borosilicate glass microelectrodes during regular pacing frequency of 1 Hz, 2 Hz, 3.3 Hz, 5 Hz and 6.7 Hz.
8. Isolation of atrial cardiac myocytes
Atrial myocytes were isolated by four steps perfused digestion. At the end of the perfusion, the whole ventricle was dissected from the heart, and the atria were placed in ice-cold KB solution. The temperature of the perfusion was maintained at 37°C. Isolated cardiac myocytes were stored in KB solution at 4°C until needed [28].
9. Cellular electrophysiology recording
All experiments were carried out at room temperature (20–22°C). The whole cell clamp recording protocols, data acquisition and analysis were described in Methods S1.
10. Real-time PCR analysis
RNA levels of the Tgfβ1, Col1α1 and Col3α1 were determined by real-time PCR in Wild-type and Rgs5−/− atria. The mRNA expression of Rgs5−/− hearts was quantified relative to wild-type.
11. Statistical analysis
All data are expressed as mean ± SEM. Statistical analysis performed with a Fisher exact test and Student's t test were completed by SPSS 16.0. A value of P<0.05 was considered significant. Patch-clamp data were analyzed using Origin 6.0 (Microcal Co. USA) for nonlinear curve fitting.
Results
1. ECG recording and echocardiography determination
To analyze whether Rgs5 deficiency results in atrial electrical abnormalities, telemetry ECG were performed. The spontaneous atrial arrhythmias were not observed during 24 h continuous recording, and there was no difference between Rgs5−/− and wild-type mice in heart rate (HR) (P>0.05). However, the P wave usually showed widened in lead II of Rgs5−/− mice, and its duration (Pdur) was significantly different to that in wild-type mice (11.3±2.3 ms vs 9.4±2.4 ms, P<0.05) (Table 1 and Fig. 1). Echocardiography was applied to confirm whether this prolonged P wave is associated with atrial chamber dilation. The left atrial diameter was similar between WT and Rgs5−/− mice (1.43±0.03 mm vs 1.40±0.08 mm, P>0.05). In addition, Rgs5−/− mice did not exhibit ventricular dysfunction by determination of left ventricular ejection fraction (LVEF) and fractional shortening (FS) (Table 2).
Table 1. Measurement of ECG parameters between Rgs5−/− and wild-type mice.
| WT (n = 10) | Rgs5−/− (n = 10) | P | |
| HR(bpm) | 515.8±46.8 | 505.8±52.6 | NS |
| RR(ms) | 118.5±7.7 | 120.6±9.4 | NS |
| PR(ms) | 34.0±1.9 | 36.1±1.5 | <0.05 |
| Pdur(ms) | 7.3±1.1 | 10.6±2.5 | <0.05 |
| Pamp(mV) | 0.19±0.03 | 0.18±0.04 | NS |
| QRS(ms) | 8.0±0.1 | 8.1±0.4 | <0.05 |
Heart rate (HR); RR interval (RR); PR interval (PR); P wave duration (Pdur); P wave amplitude (Pamp); QRS duration (QRS). NS: no statistical significance.
Figure 1. ECG and MAP recordings in Rgs5-/- and WT mice.

Two representative electrocardiograms recorded from telemetry ECG showed the measurements of Pdur and PR intervals (A). Compared with wild-type mice, Rgs5−/− showed widened P wave, and the action potential duration (APD) prolonged in the atria of Rgs5−/− from 50% (APD50) to 90% (APD90) (B,C). The effective refractory periods (ERP) also significantly increased during S1 cycle length of PES at 150 ms, 125 ms and 100 ms.
Table 2. Echocardiography parameters in WT and Rgs5−/− mice.
| WT (n = 8) | Rgs5−/− (n = 8) | P | |
| HR (bpm) | 458.7±39.7 | 449.0±31.9 | NS |
| LVEDD (mm) | 3.60±0.13 | 3.51±0.12 | NS |
| LVESD (mm) | 2.08±0.02 | 2.09±0.04 | NS |
| LAD (mm) | 1.43±0.03 | 1.40±0.08 | NS |
| LVEF (%) | 78.11±1.18 | 80.11±2.04 | NS |
| FS (%) | 43.66±1.45 | 41.35±2.81 | NS |
HR, heart rate; LVEDD, left ventricular end-diastolic diameter; LVESD, left ventricular end-systolic diameter; LAD, left atrial diameter, LVEF, left ventricular ejection fraction; FS, fractional shortening. NS: no statistical significance.
2. Rgs5−/− mice have increased the duration of atrial repolarization
Monophasic action potentials (MAP) were obtained from isolated hearts with amplitudes ranging from 4.8 to 15.1 mV; the mean values did not significantly differ between the two groups. MAP duration (APD) from 30% to 90% was longer in Rgs5−/− than WT mice during regular atrial pacing (CL = 125 ms) (Fig. 1). Similarly, PES revealed longer AERP in Rgs5−/− mice than in wild-type mice, with significant differences during S1 paced CL at 150 ms (43.4±8.8 ms vs 37.6±6.5 ms, P = 0.04), 125 ms (44.9±8.8 ms vs 36.5±6.6 ms, P<0.01) and 100 ms (47.0±11.1 ms vs 36.2±4.7 ms, P<0.01) (Fig. 1 and Table 3).
Table 3. Electrophysiological parameters recorded in isolated mouse heart.
| WT (n = 18) | Rgs5−/− (n = 18) | P | |
| RR | 154.6±28.3 | 167.5±32.1 | NS |
| AERP200 | 38.6±7.6 | 41.9±7.7 | NS |
| AERP150 | 37.6±6.5 | 43.4±8.8 | <0.05 |
| AERP125 | 36.5±6.6 | 44.9±8.8 | <0.01 |
| AERP100 | 36.2±4.7 | 47.0±11.1 | <0.01 |
| AVERP200 | 50.0±12.0 | 58.2±16.3 | NS |
| AVERP150 | 53.0±11.2 | 59.0±17.3 | NS |
| AVERP125 | 51.9±12.8 | 54.9±13.6 | NS |
| AVERP100 | 52.6±14.2 | 46.2±6.5 | NS |
| WCL | 66.8±11.9 | 87.1±20.6 | <0.01 |
| SNRT | 237.5±45.5 | 312.4±67.9 | <0.01 |
| cSNRT | 84.9±11.2 | 144.8±31.7 | <0.01 |
| SNRTi | 1.5±0.1 | 1.8±0.4 | <0.01 |
Values are means±SE. AERP, atrial effective refractory periods; AVERP, atrioventricular effective refractory periods; WCL, Wenkebach cycle length. SNRT, sinus nodal recovery time; cSNRT, corrected sinus nodal recovery time; SNRTi, sinus nodal recovery time index. Subscripts 200, 150, 125 and 100 refer to S1 drive cycle lengths of 200, 150,125 and 100 ms, respectively. NS: no statistical significance.
Transmembrane action potential (TAP) from Rgs5−/− and WT isolated sample of atrium was recorded at different paced CL due to very little spontaneous automaticity. Consistent with MAP findings, TAP recordings at Rgs5−/− samples showed increased APD90 compared to WT group over all paced frequency from 1 Hz to 6.7 Hz. After-depolarization such as EAD was occurred frequently at 1 Hz and 2 Hz in Rgs5−/− group. Moreover, EAD developed in 6 of 11 atrial preparations (54.5%), but none of 10 WT preparations (P<0.05) (Fig. 2).
Figure 2. Transmembrane action potentials (TAP) from WT and Rgs5−/− atrial samples.
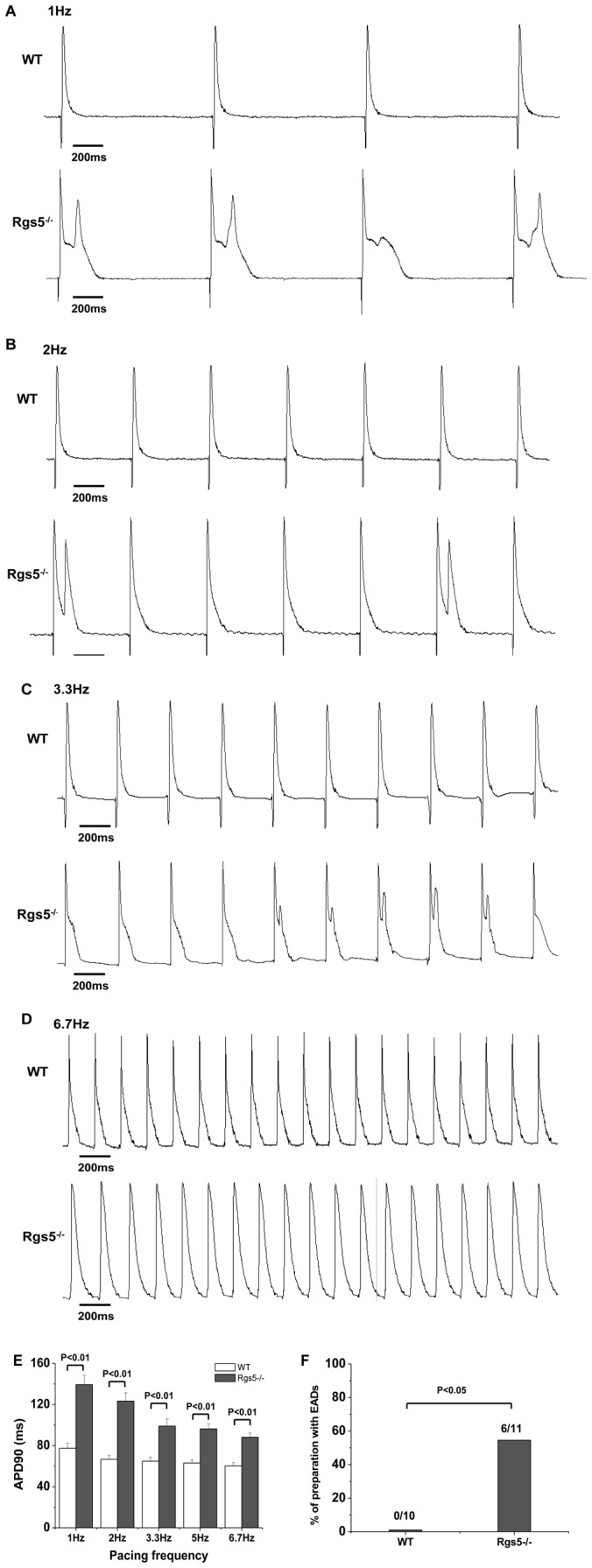
The TAP was recorded at paced frequency from 1 Hz to 6.7 Hz (A–D). Mean±SEM APD values at 90% repolarization (APD90) show APD prolongation at different pacing frequency (E). Early after-depolarizations (EADs) occurred in Rgs5−/− group at 1 Hz, 2 Hz and 3.3 Hz. EADs occurrence was quantified as percentage of preparations (F).
3. Dysfunction of sinus and atrioventricular node in Rgs5−/− mice
Sinus node automaticity was assessed by analyzing the SNRT. Atrial pacing confirmed sinus node dysfunction in Rgs5−/− mice, with SNRT, corrected SNRT and SNRT index being longer in Rgs5−/− hearts than in wild-type hearts (SNRT 312.4±67.9 ms vs 237.5±45.5 ms, P<0.01; cSNRT 144.8±31.7 ms vs 84.9±11.2 ms, P<0.01; 1.8±0.4 vs 1.5±0.1, P<0.01) (Table 3 and Fig. 3). In addition, AVERP was recorded in each S1 paced CL, but there were no significant differences between Rgs5−/− and wild-type mice. However, the wenckebach cycle length (WCL) was significantly longer in Rgs5−/− mice than in wild-type mice (87.1±20.6 ms vs 66.8±11.9 ms, P<0.01) by decremental atrial pacing (Table 3).
Figure 3. Sinus node recovery time (SNRT) in WT (A) and Rgs5−/− (B) heart.

The SNRT was measured by a 2 s pacing train at a cycle length of 100 ms, and defined as the interval between end of the stimuli and recovered sinus rhythm. The bipolar recording electrode was placed on the atrial appendage.
4. Rgs5−/− heart increased the incidence and duration of atrial tachyarrhythmia
Atrial PES and burst pacing were applied to assess the arrhythmic tendency in Rgs5−/− (n = 15) and wild-type (n = 14) hearts. Recordings were obtained during PES at a stimulation frequency from 5 Hz to 10 Hz. The S2 stimulus was continued until atrial refractoriness or arrhythmia occurred. A burst pacing procedure then applied high-frequency trains of S1 stimuli to the right atrium. Each heart reverted to sinus rhythm after arrhythmia. The Rgs5−/− group showed a higher overall incidence of atrial tachyarrhythmia than the WT group through the procedures (P<0.05) (Fig. 4C).
Figure 4. Atrial tachyarrhythmia (ATA) inducibility in Rgs5−/− and WT hearts.

PES (A) and Burst stimuli (50 Hz, 2 s) (B) were applied to induce atrial tachyarrhythmia (ATA). The sustained rapid atrial activities (duration>30 s) were elicited in Rgs5−/− hearts, but Wild-type hearts showed non-sustained rapid atrial activities during both procedures. Overall incidence of ATA induced by PES and burst pacing in WT and Rgs5−/− hearts (C), and the incidence was sorted by the ATA duration (<10 s, 10–30 s and >30 s) during PES (D) and burst pacing (E).
The individual episodes of atrial arrhythmia were sorted by duration within the ranges <10 s, 10–30 s, and >30 s. Most ATA lasted <10 s in both groups, but the incidence of <10 s ATA was significantly higher in the Rgs5−/− group than in the WT (P<0.05). Sustained ATA (>30 s) was induced in one Rgs5−/− heart in both stimulation procedures. In contrast, arrhythmia lasting >30 s was not recorded in WT hearts during either PES or burst pacing (Fig. 4D,E and Table 4).
Table 4. Incidence of atrial tachyarrhythmia induced by PES and burst pacing.
| WT (n = 14) | Rgs5−/− (n = 15) | |
| Inducibility | ||
| PES200 | 7.1% (1/14) | 6.7% (1/15) |
| PES150 | 0 | 6.7% (1/15) |
| PES125 | 0 | 26.7% (4/15)* |
| PES100 | 7.1% (1/14) | 20.0% (3/15)* |
| Burst | 7.1% (1/14) | 26.7% (4/15)* |
| PES Duration | ||
| <10 s | 7.1% (1/14) | 33.3% (5/15)* |
| 10–30 s | 0 | 6.7% (1/15) |
| >30 s | 0 | 6.7% (1/15) |
| Burst Duration | ||
| <10 s | 7.1% (1/14) | 40.0% (6/15) * |
| 10–30 s | 7.1% (1/14) | 13.3% (2/15) * |
| >30 s | 0 | 6.7% (1/15) |
Subscripts 200, 150, 125 and 100 refer to S1 drive cycle lengths of 200, 150,125 and 100 ms, respectively. * P<0.05 Rgs5−/− vs WT.
5. K+ current remodeling in Rgs5−/− atrial cardiomyocytes
Whole-cell recordings were performed with a mean resting membrane potential of-77.6±2.1 mV and 78.3±1.8 mV in WT and Rgs5−/− group, respectively (P>0.05). And the mean capacitance of each group was similar (WT: 130.6±23.9 pF; Rgs5−/−: 114.2±26.8 pF, P>0.05). The results revealed that the density of outward K+ current was markedly changed in Rgs5−/− cardiomyocytes at room temperature (22°C). The I–V curves of total outward current (IPeak) showed the current density at +60 mV was decreased in Rgs5−/− atrial cells compared to wild-type cells (53.5±5.4 pA/pF vs 40.1±4.0 pA/pF, P<0.05) (Fig. 5A). The two components of the IPeak, the transient outward (Ito) and the ultra-rapid delayed rectifier (Ikur), revealed significantly lower density in Rgs5−/− cardiomyocytes than in the wild type (Ito at +60 mV: 16.7±1.3 pA/pF vs 20.4±2.0 pA/pF, P<0.05; Ikur at +60 mV: 14.0±2.2 pF/pA vs 17.9±2.0 pF/pA, P<0.05) (Fig. 5C,E). In addition, the I–V curves of inwardly rectified K+ current (IK1) showed similar densities at −120 mV in Rgs5−/− and wild-type cardiomyocytes (−16.6±2.5 pA/pF vs −18.6±2.1 pA/pF, P>0.05) (Fig. 5G). To determine whether this results are also convinced when data are obtained at more physiological temperatures, we performed the measurement at 37°C. As expected, the mean maximal density of Ipeak, Ito and IKur was significantly larger at 37°C than at room temperature (P<0.05) (Fig. 6). The difference of density between Rgs5−/− and WT also revealed significantly at 37°C in Ipeak, (at +60 mV: 44.3±4.2 pA/pF vs 59.2±4.7 pA/pF, P<0.05), Ito (at +60 mV: 20.4±2.0 pA/pF vs 23.2±2.5 pA/pF, P<0.05) and IKur (at +60 mV: 17.1±1.5 pF/pA vs 19.2±1.9 pF/pA, P<0.05).
Figure 5. Alterations in K+ current were recorded in atrial myocytes of Rgs5−/− and WT mice.
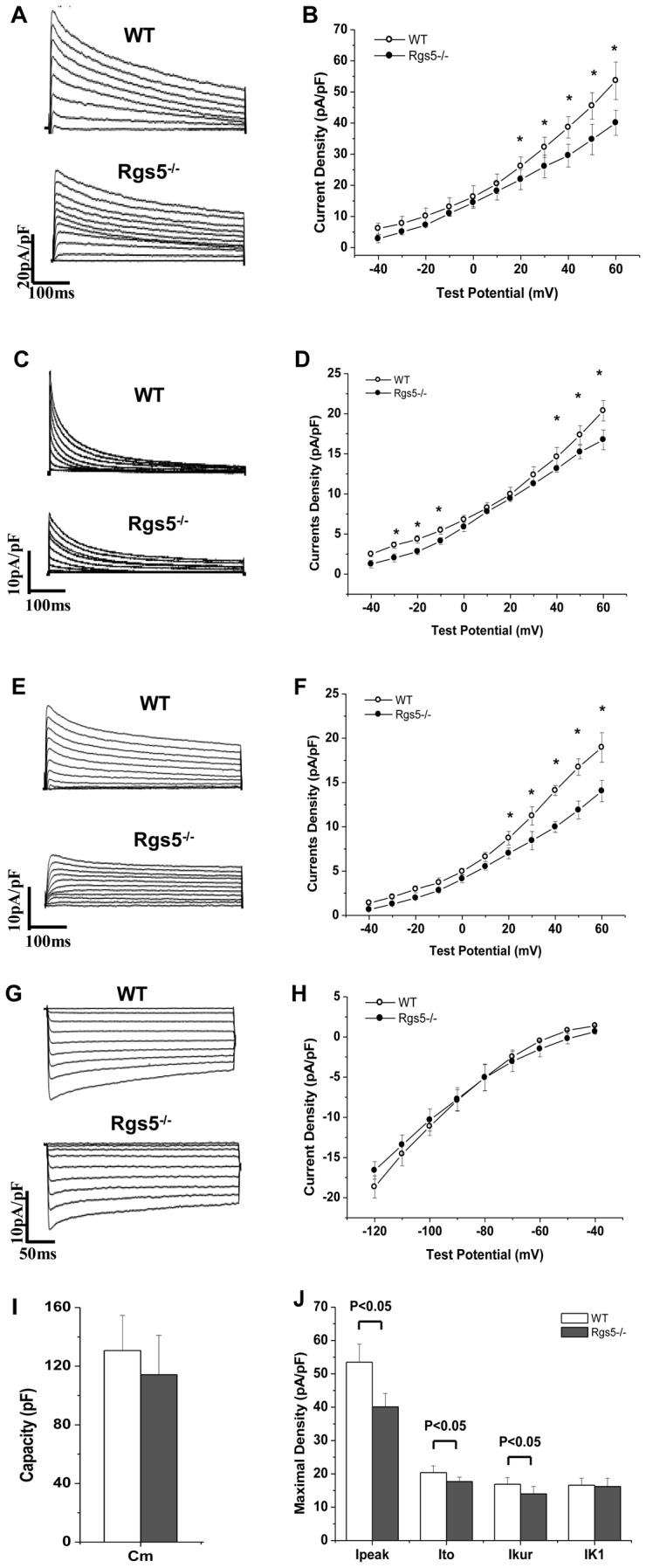
Representative whole-cell total outward K+ currents (Ipeak) (A,B), transient outward currents (Ito) (C, D) and ultra-rapid delayed rectifier currents (Ikur) (E, F) evoked during 500 ms test potential steps from −40 mV to +60 mV from a holding potential (HP) of −80 mV. The inwardly rectified K+ currents (IK1) (G, H), evoked during 350 ms voltage steps to potentials between −120 and −40 mV (HP = −80 mV). The current-voltage relationship (I–V) of Ipeak and IK1 were plotted (B, D). Membrane capacity (Cm) (I) and maximal densities of currents (J) were compared between wild-type and Rgs5−/− mice. *P<0.05 WT vs Rgs5−/−.
Figure 6. Comparison of magnitude of Ipeak, Ito, IKur and IK1 in atrial myocytes of WT and Rgs5−/− mice recorded at 22°C and 37°C (A).

Bar graph represents mean (±S.E.M) maximal current density of each of the four outward K+ current recorded at 22°C and 37°C in WT (B) and Rgs5−/− (C) group.
To analyze the kinetics of Ito and Ikur, the properties of inactivation and recovery were assayed in each group (Fig. 7). For the voltage dependence of steady-state inactivation of Ito, the half-inactivated voltage (V1/2) and slope factor (k) showed no significant difference between Rgs5−/− and wild-type cells (V1/2: −63.1±1.0 mV vs −62.2±1.0 mV, P = 0.13; k: 13.0±0.8 mV vs 13.2±0.7 mV, P P>0.05). However, the recovery from inactivation of Ito was significantly slower in Rgs5−/− myocytes than in the wild type (time constant: 107.5±13.6 ms vs 95.1±13.2 ms, P>0.05). For Ikur, the V1/2 (−51.8±0.9 mV vs −55.8±0.9 mV, P<0.01) and the recovery time (356.9±20.6 ms vs 577.0±38.3 ms, P<0.01) were significantly different between WT and Rgs5−/− cardiomyocytes.
Figure 7. Channel kinetics of Ito and IKur were analyzed in Rgs5−/− and WT cardiomyocytes, respectively.

The steady-state inactivation (A) and reactivation (B) of curves in Ito plotted similar tendency between Rgs5−/− and wild-type atrial myocytes. However, the property of IKur in Rgs5−/− cardiomyocytes showed markedly modification with significant difference of half-inactivated voltage (V1/2) (C) and reactivated time constant (τ) (D). Histogram summarized the statistical significance of V1/2 and τ in WT and Rgs5−/− groups (E, F).
6. Effect of Rgs5−/− on atrial fibrosis
The analysis of mRNA expression levels of mediators of fibrosis including Tgfβ1, procollagen, type Iα1 (Col1α1) and procollagen, type IIIα1 (Col3α1) revealed a similar tendency between WT and Rgs5−/− mouse atrium (P>0.05) (Fig. 8A,B), suggesting that the Rgs5−/− did not promote the formation of fibrosis in atrium. PSR staining section confirms that morphologically through calculated the relative area of fibrosis from atria of WT (n = 8) and Rgs5−/− (n = 8), the area occupied by fibrosis was 12.1%±3.4% in WT and 10.9%±2.7% in Rgs5−/−, it showed not significantly difference between two groups (P>0.05) (Fig. 8C).
Figure 8. Fibrosis assay in Rgs5−/− and WT atrium.

mRNA expression levels of mediators of fibrosis including Tgfβ1, procollagen, type Iα1 (Col1α1) and procollagen, type IIIα1 (Col3α1) were analysis in WT and Rgs5−/− mouse atrium (A, B). Relative abundance was calculated with the value of wild-type as a reference of 100%. PSR staining on histological sections of the atrial appendage and ventricle was performed in WT and Rgs5−/− mice (C).
Discussion
1. Main findings
The present study provided an important original insight into the atrial electrical remodeling of Rgs5−/− mice, As the consequence, we found that (i) Rgs5−/− significantly prolonged atrial repolarization associated with attenuated K+ currents; (ii) the increased incidence and duration of atrial arrhythmia in Rgs5−/− mice may related to the prolonged repolarization.
2. Rgs5 and atrial repolarization
G proteins are key mediators of cardiac signal transduction, particularly following activation of G protein coupled receptor (GPCR). G proteins include α (Gα) and βγ (Gβγ) subunits. Different types of Gα can be activated by special receptors [18]. For example, Gαi can be activated by muscarinic cholinergic receptors, Gαs by β-adrenergic receptors, and Gαq by angiotensin II type 1 receptor (AT1R) and endothelin-1 receptor A (ETA). RGS5, a member of the regulator of G-protein signaling protein super-family, acts as a GTPase-activating factor for a number of Gαi- and Gαq-mediated pathways and as a negative regulator of GPCR signaling [5]. Our previous study demonstrated that Rgs5 transgenic mice were resistant to cardiac hypertrophy and fibrosis through inhibition of MEK-ERK1/2 signaling, whereas Rgs5−/− mice displayed the opposite phenotype in response to pressure overload [15]. Knockdown of RGS5 results in the selective up-regulation of AT1R-mediated activation of ERK1/2 [17]. Previous studies have demonstrated a relationship between AT1R and K+ ion in several tissues. AngII can inhibit several K+ currents, including Kv, KATP, and BKca, and its effects on K+ currents can be reversed by blocking AT1R [9], [11], [23], [25]–[26]. Importantly, AT1R over-expressing mouse hearts had delayed repolarization and reduced density of several repolarizing K+ currents, together with an increased incidence of spontaneous arrhythmia [14]. Thus, as a negative mediator of GPCR signaling pathways, Rgs5 may directly regulate atrial repolarization through K+ currents.
As the primary repolarizing component in mouse heart, the voltage dependent K+ current exhibits more powerful than other mammals [8]. Decreased K+ currents may seriously prolong repolarizing phase. In present study, we observed the atrial action potential duration was prolonged in Rgs5−/− mice, which associated with greatly decreased K+ currents including IPeak, Ito and IKur, but the inwardly rectified K+ current (IK1) did not showed significant difference between Rgs5−/− and WT group. Rivard et al. demonstrated that prolongation of APD in AT1R-overexpressing mouse hearts was due to decreased Ito and IKur densities and alteration of their reactivation kinetics by chronic stimulation of the AT1R signaling pathway. Notably, these alterations preceded anatomic disturbances and were not a consequence of cardiac remodeling [20]. However, the reduced density of the IK1 current is more common in mouse heart with cardiac dysfunction and increased myocytes size [3]. In present study, Rgs5−/− heart did not show increased atrial size and cellular membrane capacitance compared to WT mice, and the function of Rgs5−/− heart was normal. Based on these observations, it has been concluded that reductions of IK1 are closely related to cardiomyocyte hypertrophy and may not be directly due to the increased stimulation of AT1R. Consistent with these notions, the markedly decreased densities of IKur and Ito in Rgs5−/− atria possibly contributed to the effect on the AT1R/Gq signaling pathway.
3. Rgs5 and atrial tachyarrhythmia
Toumi et al reported that Rgs2, another member of the RGS protein super-family, is expressed at high levels in mouse atrium and plays a critical role in atrial arrhythmias. Rgs2−/− mice were more susceptible to PES-induced atrial tachycardia and fibrillation (AT/AF), and a higher percentage of them were accompanied by heterogeneities in refractory periods and regional conduction block. This phenotype was likely due to the reentry mechanism [27]. Atrial tachyarrhythmia can be initialized by rapidly firing ectopic foci (e.g., from the pulmonary veins) and maintained by functional reentrant circuit (mother rotor) [12], [19]. As a major risk factor for atrial arrhythmogenesis, prolonged repolarization forms the substrate for ectopic stimulation, which is generated from pulmonary and caval vein, cardiac ganglion or other atrial special tissues. Several studies have examined predisposition to atrial arrhythmia in mouse models of congenital long QT syndrome (LQT). Dautova et al. showed that in mice with a ΔKPQ knock-in LQT3 mutation, Langendorff-perfused hearts are more likely to develop atrial arrhythmia [13]. Lemoine et al. demonstrated that atrial arrhythmias induced by rapid rate alteration occurred significantly more often in the LQT3 mouse model, suggestive of after-depolarization-dependent arrhythmogenic mechanisms [21]. In present study, although we did not observe the spontaneous atrial arrhythmia during 24 h-ECG recording, the trigger activity including EAD was observed in Rgs5−/− atrial tissue, and the electrical stimulation (PES and burst pacing), which simulate a physiological ectopic excitation and is thought to more readily induce reentry than triggered activity [27], induced atrial arrhythmias were facilitated in Rgs5−/− heart. It suggested that Rgs5−/− induced prolonged repolarizaiton provided a potential substrate and the ectopic activity facilitated this effect for arrhythmogenesis in atria.
As the substrate for atrial arrhythmogenesis, interstitial fibrosis and atrial enlargement have been implicated in theoretical models. In present study, we did not observed significant sign of atrial enlarged or fibrosis. However, the prolonged P waves were recorded in Rgs5−/− conscious mice. As a sign of atrial enlargement, prolonged P wave was also seemed to be related to regional atrial conduction disturbances, which have been suggested to favour the development of AF [29]. Thus, the result suggested that the Rgs5−/− induced ATA was mainly dependent on the atrial electrophysiological modifications.
In addition, SNRT was measured to assess the function of the sinus node, which maintains the normal sinus rate. Several RGS mRNAs are expressed at high levels in the sinus node and atrioventricular nodal region and regulate parasympathetic tone of pacemaker myocytes. Rgs4 and Rgs6 are highly expressed in the sinus node, and hearts from mice deficient in these genes showed marked bradycardia with a sustained lower HR and prolonged RR intervals [6], [10], [22]. Alterations in RGS protein expression or activity could potentially contribute to diseases such as sick sinus syndrome. The Rgs5−/− hearts, which have significantly longer SNRT and cSNRT, had abnormal sinus function and thus promoted the maintenance of atrial ectopic rhythm.
Conclusion
In conclusion, the present study confirmed the relationship between the Rgs5 and atrial arrhythmogenesis, the Rgs5−/− facilitated ATA was due to the prolonged repolarization, and this electrophysiological alteration could be accounted for attenuated repolarizing K+ currents.
Supporting Information
(DOC)
Funding Statement
This research was supported by National Key Basic Research Development Program (The “973” Program) (Grant number: 2012CB518604), National Natural Science Foundation of China Grants (Grant number: 81070142 and 81100129) and Fundamental Research Funds for the Central Universities, China (grant number: 201130202020023 and 201130202020003). The funder has designed this study and decided to publish this manuscript.
References
- 1. Yang Y, Xia M, Jin Q, Bendahhou S, Shi J, et al. (2004) Identification of a KCNE2 Gain-of-function mutation in patients with familial atrial fibrillation. Am J Hum Genet 75: 899–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Benjamin EJ, Wolf PA, D'Agostino RB, Silbershatz H, Kannel WB, et al. (1998) Impact of atrial fibrillation on the risk of death:the Framingham Heart Study. Circulation 98: 946–952. [DOI] [PubMed] [Google Scholar]
- 3. Domenighetti AA, Boixel C, Cefai D, Abriel H, Pedrazzini T (2007) Chronic angiotensin II stimulation in the heart produces an acquired long QT syndrome associated with IK1 potassium current downregulation. J Mol Cell Cardiol 42: 63–70. [DOI] [PubMed] [Google Scholar]
- 4. Chen YH, Xu SJ, Bendahhou S, Wang XL, Wang Y, et al. (2003) KCNQ1 gain-of-function mutation in familial atrial fibrillation. Science 299: 251–254. [DOI] [PubMed] [Google Scholar]
- 5. Xiao B, Zhang Y, Niu WQ, Gao PJ, Zhu DL (2009) Haplotype-based association of regulator of G-protein signaling 5 gene polymorphisms with essential hypertension and metabolic parameters. Clin Chem Lab Med 47: 1483–1488. [DOI] [PubMed] [Google Scholar]
- 6. Cifelli C, Rose RA, Zhang HJ, Bolz JV, Bolz SS, et al. (2008) RGS4 Regulates parasympathetic signaling and heart rate control in the sinoatrial node. Circ Res 103: 527–535. [DOI] [PubMed] [Google Scholar]
- 7. De Boer RA, Pokharel S, Flesch M, van Kampen DA, Suurmeijer AJ, et al. (2004) Extracellular signal regulated kinase and SMAD signaling both mediate the angiotensin II driven progression towards overt heart failure in homozygous TGR (mRen2)27. J Mol Med 82: 678–687. [DOI] [PubMed] [Google Scholar]
- 8. Nerbonne JM Molecular basis of functional voltage gated K+ channel diversity in the mammalian myocardium (2000). J Physiol 525: 285–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Fischer R, Dechend R, Gapelyuk A, Shagdarsuren E, Gruner K, et al. (2007) Angiotensin II-induced sudden arrhythmic death and electrical remodeling. Am J Physiol 293: H1242–H1253. [DOI] [PubMed] [Google Scholar]
- 10. Fu Y, Huang XY, Piao L, Lopatin AN, Neubig RN (2007) Endogenous RGS proteins modulate SA and AV nodal functions in isolated heart: implications for sick sinus syndrome and AV block. Am J Physiol Heart Circ Physiol 292: H2532–H2539. [DOI] [PubMed] [Google Scholar]
- 11. Goette A, Lendeckel U, Klein HU (2002) Signal transduction systems and atrial fibrillation. Cardiovasc Res 54: 247–258. [DOI] [PubMed] [Google Scholar]
- 12. Nattel S, Takeshita AS, Brundel BJ, Rivard L (2005) Mechanisms of Atrial Fibrillation: Lessons From Animal Models. Prog Cardiovasc Dis 48: 9–28. [DOI] [PubMed] [Google Scholar]
- 13. Dautova Y, Zhang Y, Sabir I, Grace AA, Huang CL (2009) Atrial arrhythmogenesis in wild–type and SCN5a+/delta murine hearts modelling LQTS3 syndrome. Pflugers Arch 458: 443–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Katy R, Pierre P, Mona N, Celine F (2008) Cardiac-specific overexpression of the human type 1 angiotensin II receptor causes delayed repolarization. Cardiovasc Res 78: 53–62. [DOI] [PubMed] [Google Scholar]
- 15. Li HL, He CW, Feng JH, Zhang Y, Tang QZ, et al. (2010) Regulator of G protein signaling 5 protects against cardiac hypertrophy and fibrosis during biomechanical stress of pressure overload. Proc. Natl. Acad. Sci. U.S.A 107: 13818–13823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Mangoni ME, Traboulsie A, Leoni AL, Couette B, Marger L, et al. (2006) Bradycardia and slowing of the atrioventricular conduction in mice lacking CaV3.1/α1G T-Type Calcium Channels. Circ Res 98: 1422–1430. [DOI] [PubMed] [Google Scholar]
- 17. Manzur M, Ganss R (2009) Regulator of G protein signaling 5: A new player in vascular remodeling. Trends Cardiovasc Med 19: 26–30. [DOI] [PubMed] [Google Scholar]
- 18. Nattel S (2009) G-protein signaling and arrhythmogenic atrial remodeling: Relevance to novel therapeutic targets in atrial fibrillation. Heart Rhythm 6: 85–86. [DOI] [PubMed] [Google Scholar]
- 19. Nattel S, Burstein B, Dobrev D (2008) Atrial remodeling and atrial fibrillation: mechanisms and implication. Circ Arrhythmia Electrophysiol 1: 62–73. [DOI] [PubMed] [Google Scholar]
- 20. Katy R, Pierre P, Mona N, Celine F (2008) Cardiac-specific overexpression of the human type 1 angiotensin II receptor causes delayed repolarization. Cardiovasc Res 78: 53–62. [DOI] [PubMed] [Google Scholar]
- 21. Lemoine MD, Duverger JE, Naud P, Chartier D, Qi XY, et al. (2011) electrophysiology in a murine geneticlong QT syndrome model. Cardiovasc Res 92: 67–74. [DOI] [PubMed] [Google Scholar]
- 22. Posokhova E, Wydeven N, Allen KL, Wickman K, Martemyanov KA (2010) RGS6/Gβ5 complex accelerates IKAch gating kinetics in atrial myocytes and modulates parasympathetic regulation of heart rate. Circ Res 107: 00–00. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hayabuchi Y, Davies NW, Standen NB (2001) Angiotensin II inhibits rat arterial KATP channels by inhibiting steady-state protein kinase A activity and activating protein kinase C1. J Physiol 530: 193–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Reil JC, Hohl M, Oberhofer M, Kazakov A, Kaestner L, et al. (2010) Cardiac Rac1 overexpression in mice creates a substrate for atrial arrhythmias characterized by structural remodeling. Cardiovasc Res 87: 485–93. [DOI] [PubMed] [Google Scholar]
- 25. Zhang TT, Takimoto K, Stewart AFR, Zhu C, Levitan ES (2001) Independent regulation of cardiac Kv4.3 potassium channel expression by angiotensin II and phenylephrine. Circ Res 88: 476–482. [DOI] [PubMed] [Google Scholar]
- 26. Shimoni Y (2001) Inhibition of the formation or action of angiotensin II reverses attenuated Kt currents in type 1 and type 2 diabetes. J Physiol 537: 83–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Tuomi JM, Chidiac P, Jones DL (2010) Evidence for enhanced M3 muscarinic receptor function and sensitivity to atrial arrhythmia in the RGS2-deficient mouse. Am J Physiol Heart Circ Physiol 298: H554–H561. [DOI] [PubMed] [Google Scholar]
- 28. Brouillette J, Clark RB, Giles WR, Fiset C (2004) Functional properties of K+ currents in adult mouse ventricular myocytes. J Physiol 559: 777–798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Thile'n U, Carlson J, Platonov PG, Havmoller R, Olsson SB (2007) Prolonged P wave duration in adults with secundum atrial septal defect: a marker of delayed conduction rather than increased atrial size? Europace 9: vi105–vi108. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(DOC)