

Abstract
Described herein are preparations of synthetic models for the deactivated Ni(II)Fe(II) states of the [NiFe]-hydrogenases. Iodination of the S = ½ species [(dppe)Ni(pdt)Fe(CO)3]+ afforded the diamagnetic iodo complex [(dppe)Ni(pdt)IFe(CO)3]+. Crystallographic analysis of this species confirmed the presence of square-pyramidal Ni linked to an octahedral Fe centre. The Ni···Fe separation of 3.018 Å indicated the absence of metal-metal bonding. This complex could be reduced to give (dppe)Ni(pdt)Fe(CO)3 and, in the presence of iodide, decarbonylated to afford (dppe)Ni(pdt)FeI2. Derivatives of the type [(diphosphine)Ni(dithiolate)XFe(CO)2L]+ (X = Cl, Br, I) were prepared by halogenation of mixed-valence precursors [(diphosphine)Ni(dithiolate)Fe(CO)2L]+ (diphosphine = dppe, dcpe; L = tertiary phosphine or CO). The Fe(CO)2(PR3)-containing derivatives are more robust than the related tricarbonyl derivatives. Exploiting this greater stability, we characterised examples of chloride and bromide derivatives. Related fluorides could be prepared by F− abstraction from BF4−. Spectroscopic evidence is presented for the hydroperoxide [(diphosphine)Ni(dithiolate)(OOH)Fe(CO)2L]+, which was prepared by oxidation of a model for Ni-SU and Ni-SIr states.
Introduction
Hydrogen processing in Nature is mediated by the hydrogenases (H2ases), a class of enzyme prevalent in anaerobic bacteria.1 A major subclass of these enzymes, the nickel-iron hydrogenases, is expressed to catalyse the redox reaction 2H+ + 2e− ⇌H2, which is central to the metabolism of certain bacteria and archaea.2 The [NiFe]-H2ase active site features a Ni(cysteinato)4 centre, two thiolate ligands of which bridge to a Fe(CN)2(CO) fragment (Figure 1). The states of the enzyme are classified according to the identity (or absence) of the bridging ligand X, the oxidation states of the metal centres, and the protonation of two of the four thiolate ligands. Of relevance to this paper, aerobic deactivation of the enzyme affords states in which oxygenic ligands bridge the two metal ions.3 For example, a Ni(II)(OH)Fe(II) core is observed for (Ni-SIr)I,4 whereas in the Ni-SU state the divalent metal ions are thought to be bridged by a hydroperoxo ligand.5 Our attention was thus drawn to the rarity of model compounds featuring Fe(μ-SR)2(μ-X)Ni cores.6
Figure 1.
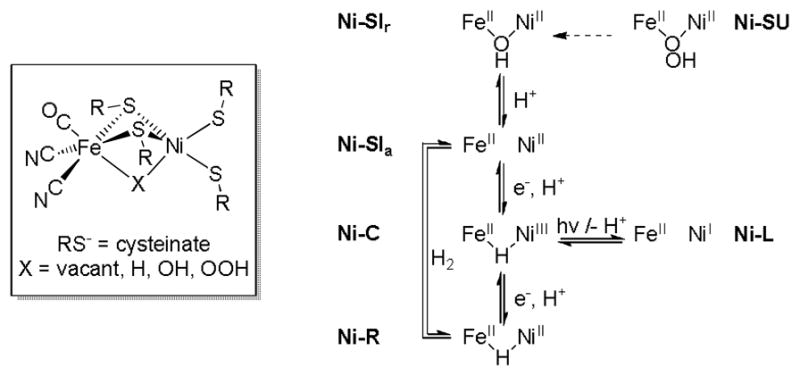
Schematic of the [NiFe]-H2ase active site (left) and some of the enzyme states (right).
Much recent work has focused on synthetic modelling of the [NiFe]-H2ase active sites,7 no doubt motivated by the increased understanding of the natural systems and hydrogen processing catalysts that such work promises.8 The Ni(I)Fe(I) models of type (diphosphine)Ni(dithiolate)Fe(CO)3 are of particular interest,9 as their conjugate acids, the only synthetic nickel-iron dithiolate hydrides, represent structural and functional models for Ni-R.10 We have reported a promising procedure for the neutral complexes from the precursors (diphosphine)Ni(dithiolate) and cis-[FeI2(CO)4].11 The Ni(II)Fe(II) intermediate thus generated, tentatively formulated as [(diphosphine)Ni(dithiolate)IFe(CO)3]I, is reduced with CoCp2 to afford the targeted Ni(I)Fe(I) complex. Attempts to isolate the metastable intermediate have been unfruitful, as exemplified by the crystallization of the CO-free species (dcpe)Ni(2,2-dimethyl-1,3-propanedithiolate)FeI2 (dcpe = 1,2-bis(dicyclohexylphosphino)ethane), thought to arise from nucleophilic attack of I− and concomitant decarbonylation.11 According to IR spectroscopic data, solutions of the putative iodide salt can also be prepared by treatment of (diphosphine)Ni(dithiolate)Fe(CO)3 with I2, the ensuing heterolysis resulting in the bridging iodo species as its iodide salt. Presented here are syntheses and characterization of bridging halide complexes of type [(diphosphine)Ni(pdt)XFe(CO)3]+ (pdt2− = 1,3-propanedithiolate) as well as substituted derivatives [(diphosphine)Ni(pdt)XFe(CO)2(monophosphine)]+. The work illustrates the radical character of [(diphosphine)Ni(pdt)Fe(CO)3]+ and its participation in atom transfer reactions.
Results
Tricarbonyl Halides [(diphosphine)Ni(pdt)XFe(CO)3]+
As mentioned in the Introduction, halide salts of type [(diphosphine)Ni(pdt)XFe(CO)3]X are unstable towards decarbonylation. In CH2Cl2 solution, the tricarbonyls [(dppe)Ni(pdt)IFe(CO)3]Br([1I]I, dppe = 1,2- bis(diphenylphosphino)ethane) and [(dppe)Ni(pdt)BrFe(CO)3]Br [1Br]Br were found to convert to (dppe)Ni(pdt)FeI2 and (dppe)Ni(pdt)FeBr2, respectively. IR analysis of these paramagnetic materials indicated the absence of νCO bands, with elemental analyses also being consistent with these formulations (see Experimental section). Consequently, our focus shifted to salts of [(diphosphine)Ni(pdt)XFe(CO)3]+ with non-nucleophilic anions. Oxidation of (diphosphine)Ni(pdt)Fe(CO)3 with ferrocenium tetrafluoroborate (FcBF4) furnishes the paramagnetic Ni(II)Fe(I) derivatives [(diphosphine)Ni(pdt)Fe(CO)3]BF4.12 The salt [(dppe)Ni(pdt)Fe(CO)3]BF4 ([1]BF4) reacts with I2 (0.5 equiv.) in CH2Cl2, resulting in a colour change from dark brown to red. The solutions thus formed have νCO bands identical to those of [(diphosphine)Ni(dithiolate)Fe(CO)3]/I2 mixtures. Notably, the stretching frequencies (νCO = 2097, 2055, 2026 cm−1) are higher than those for the hydride [1H]BF4 (νCO = 2082, 2024 cm−1), reflecting the poorer σ-donating ability of the iodo ligand vs the hydride ligand (Figure 2). Furthermore, three νCO bands are easily resolved in the IR spectra for these halides, whereas the hydrides exhibit an A-E pattern in the νCO region typical of a more symmetrical coordination environment.
Figure 2.

FT-IR spectrum (νCO region, CH2Cl2) of [1I]BF4 (top) and [2I]BF4 (bottom).
The new compound was obtained as an orange powder, analytical data for which were consistent with its formulation as the diamagnetic salt [(dppe)Ni(pdt)IFe(CO)3]BF4 ([1I]BF4). The complex cation could be identified by ESI-MS (m/z 828.9) and 31P NMR spectroscopy (δ 49.2), the latter confirming its high symmetry. Analogously, iodination of the dcpe-containing species [(dcpe)Ni(pdt)Fe(CO)3]BF4 ([2]BF4)13 afforded the salt [(dcpe)Ni(pdt)IFe(CO)3]BF4 ([2I]BF4). The transformations are summarised in Scheme 1.
Scheme 1.
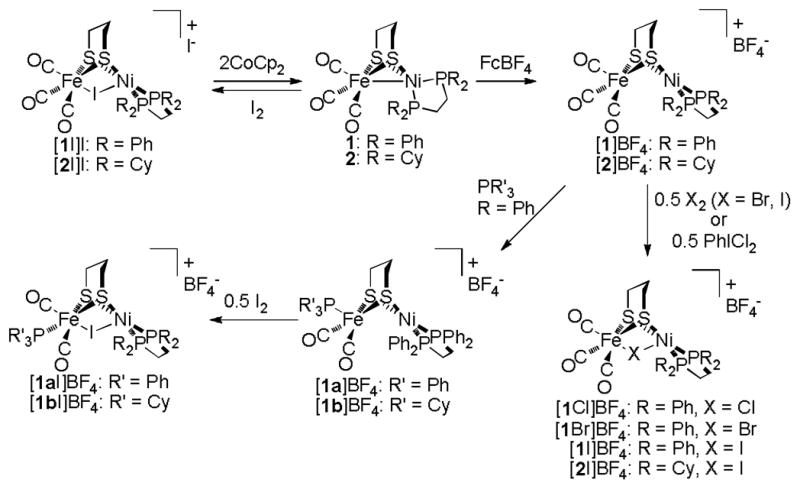
Reactions of Ni(I)Fe(I), Ni(II)Fe(I) and Ni(II)XFe(II) complexes.
Structure of (diphosphine)Ni(pdt)IFe(CO)3]+
The solid state structure of [1I]BF4 was determined by X-ray crystallography; an ORTEP of the cation is presented in Figure 3. As expected, the complex features Fe and Ni centres bridged by dithiolate and iodo ligands. The intermetallic separation (3.018 Å) is consistent with the absence of metal-metal bonding, which is expected for a Ni(II)Fe(II) system. The Fe coordination environment is approximately octahedral, with the Ni centre being best described as square-pyramidal, although the long Ni···I distance (2.931 Å) suggests that this apical ligand interacts only weakly with Ni. The iodo ligand is indeed nearer to the Fe centre (2.682 Å) and the situation is similar in the hydride [1H]+ (Fe–H, 1.460 Å; Ni–H, 1.637 Å), with which [1I]+ is almost isostructural. The related chloro complex [(dppe)Ni(pdt)ClFe(CO)(dppe)]+ has a similar Ni–Fe distance (3.076 Å), with the Cl− ligand being much closer to the metal centres (Fe–Cl, 2.362 Å; Ni–Cl, 2.668Å) than I− is in [1I]+.10 Notably, [(dppe)Ni(pdt)ClFe(CO)(dppe)]+ is prepared from the divalent metal precursors (dppe)NiCl2 and (pdt)Fe(CO)2(dppe), whereas [1I]+ is formed by a pathway probably involving I2 homolysis.
Figure 3.

ORTEP of [1I]BF4·2CH2Cl2 with ellipsoids drawn at the 50% probability level. The H atoms, disordered CH2Cl2 solvate molecules and BF4− anion are omitted for clarity. Selected distances (Å): Ni1–Fe1, 3.018; Ni1–P1, 2.189; Ni1–P2, 2.187; Ni1–S1, 2.240; Ni1–S2, 2.227; Ni1–I1, 2.931; Fe1–S1, 2.318; Fe1–S2, 2.311; Fe1–I1, 2.682; Fe1–C30, 1.815; Fe1–C31, 1.775; Fe1–C32, 1.836. Selected angles (°): Ni1–I1–Fe1, 64.9.
Reactivity of [(diphosphine)Ni(pdt)XFe(CO)3]+
Solid [1I]BF4 is air-stable, although in CH2Cl2 solution it decomposes over the course of hours even in the absence of air. The νCO bands for [1I]BF4 rapidly disappear when CH2Cl2 solutions of the compound are treated with NBu4I (1 equiv.), consistent with the formation of the CO-free product (dppe)Ni(pdt)FeI2. This halide-induced decarbonylation is consistent with the instability of the iodide salt [1I]I (vide supra) and is depicted in Scheme 2. Bromination of [1]BF4 using Br2 (0.5 equiv.) or N-bromosuccinimide (1 equiv.) affords the bromide derivative [(dppe)Ni(pdt)BrFe(CO)3]BF4 ([1Br]BF4). However, in comparison to the iodide complex, the bromide is less stable, decomposition to insoluble CO-free materials being apparent within minutes in CH2Cl2. Also unstable is the chloro species [(dppe)Ni(pdt)ClFe(CO)3]BF4 ([1Cl]BF4), which was prepared from [1]BF4 and PhICl2 (0.5 equiv.). This trend in the stability of the halide complexes mirrors that observed for the FeX2(CO)4 compounds (X = I, Br, Cl).14
Scheme 2.
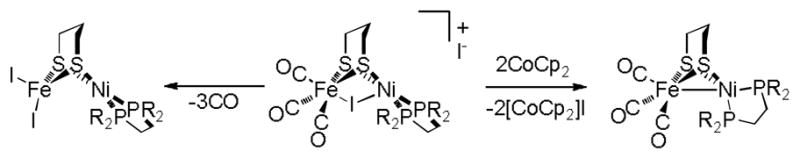
Reactions of the μ-iodo cation [1I]+.
Conversion of [1Br]BF4 to [1I]BF4 (as well as the decomposition product) proceeds in the presence of I2, reflecting the better donor ability of I− versus Br−. Similarly, the hydride [1H]BF4 is completely consumed upon treatment with excess I2, [1I]BF4 being the only CO-containing product. A CO-free byproduct is also likely given the lower intensity of the CO bands and the detection of free CO in the headspace. Like [(dppe)Ni(pdt)ClFe(CO)(dppe)]+, [1Br]+ and [1I]+ are unreactive toward Ag+ salts, behaviour that is often characteristic of bridging halide ligands.15 The bromo complex [1Br]+ also forms from [1]+ upon abstraction of Br· from benzyl bromide.
Dicarbonyl-Phosphine Iodides [(diphosphine)Ni(pdt)IFe(CO)2(PR3)]+
Given the Fe(CN)2(CO) fragment present in the enzyme, it was of interest to prepare derivatives in which of one or more of the carbonyl ligands was replaced by stronger σ-donor ligands. Similar approaches were previously used by us to obtain the mixed-valence species [(diphosphine)Ni(pdt)Fe(CO)2(PR3)]+, and the hydrides [(diphosphine)Ni(pdt)HFe(CO)2(PR3)]+, models for the Ni-L12 and Ni-R states,16 respectively. The tricarbonyl iodide [1I]+ reacts only slowly with tertiary phosphines. A more efficient route to the derivatives [(diphosphine)Ni(pdt)IFe(CO)2(PR3)]+ involves radical iodination of the substituted Ni-L models. For example, treatment of [(dppe)Ni(pdt)Fe(CO)2(PPh3)]BF4 (1a]BF4)12 with I2 (0.5 equiv.) in CH2Cl2 afforded [(dppe)Ni(pdt)IFe(CO)2(PPh3)]BF4 ([1aI]BF4). The phosphine-substituted iodide exhibits enhanced stability relative to its unsubstituted parent [1I]BF4. The product was characterised according to FT-IR, NMR, ESI-MS and analytical data. In particular, 31P NMR spectra of [1aI]BF4 in CD2Cl2 feature three resonances, consistent with a basal orientation of the PPh3 ligand. This stereochemistry is also found in the hydride [1aH]BF4,16 but it contrasts that of the precursor [1a]BF4, in which the monophosphine is apical.12 The PCy3-iodo complex salt [(dppe)Ni(pdt)IFe(CO)2(PCy3)]BF4 ([1bI]BF4) could be prepared in a similar fashion; selected data for new compounds are presented below (Table 1). Cationic diiron dithiolato halides, exemplified by [Fe2(pdt)Cl(CO)6]+,15 are also stabilised by phosphine ligands.17
Table 1.
IR data for complexes of type [(dxpe)Ni(pdt)XFe(CO)2L]BF4 in CH2Cl2 solution.
| Species | dxpe | X | L | νCO/cm−1 | Ered |
|---|---|---|---|---|---|
| [1I]BF4 | dppe | I | CO | 2097, 2055, 2026 | −0.99 (irrev.) |
| [1Br]BF4 | dppe | Br | CO | 2106, 2065, 2028 | |
| [1Cl]BF4 | dppe | Cl | CO | 2111, 2072, 2031 | −1.00 (irrev.) |
| [1aI]BF4 | dppe | I | PPh3 | 2031, 1978 | |
| [1bI]BF4 | dppe | I | PCy3 | 2024, 1965 | −1.43 (irrev.) |
| [1aCl]BF4 | dppe | Cl | PPh3 | 2042, 1981 | −1.34 (irrev.) |
| [1bCl]BF4 | dppe | Cl | Pcy3 | 2033, 2068 | −1.44 (irrev.) |
| [2I]BF4 | dcpe | I | CO | 2096, 2054, 2023 | −0.86 (ipc/ipa = 1.87) |
The well-resolved 31P NMR spectrum indicates that [1aI]BF4 is stereochemically rigid. In contrast, the corresponding hydride is dynamic, resulting from rotation at the Ni(diphosphine) centre. This difference is consistent with Fe-I forming a stronger bond to Ni than does Fe-H.
Cyclic Voltammetry Studies
The salts [1I]BF4, [1bI]BF4 and [2I]BF4 were studied by cyclic voltammetry. Whereas the oxidation waves for the compounds are complicated, the reductions are more obvious. In the case of [1I]BF4, irreversible reduction occurs at −0.99 V, whereas for [2I]BF4 the process is partially reversible and occurs at −0.86 V. The hydride analogues [1H]BF4 and [2H]BF4 are irreversibly reduced at −1.34 and −1.56 V, respectively.11 Overall, trends in reduction potentials are consistent with the stronger donor ability of H− versus I−. The ease of reduction of the tricarbonyls [1I]+ and [2I]+ is consistent with their reduction by cobaltocene (CoCp2), exploited by our group to prepare the neutral species 1 and 2 (Scheme 2).
The PCy3-substituted derivative [1bI]BF4 is reduced at −1.43 V, considerably more cathodic that the tricarbonyl parent [1I]BF4. Similar shifts (~ −0.4 V) are observed for the mixed-valence salt [1b]BF4 relative to [1]BF4.13 As evidenced by 31P NMR spectroscopy, chemical reduction of [1I]BF4 and [2I]BF4 with CoCp2 (2 equiv.) affords the expected crude mixtures containing the neutral species 1 (δ 63.0) and 2 (δ 80.9 ppm) respectively.
Dicarbonyl-Phosphine Fluorides [(diphosphine)Ni(pdt)FFe(CO)2(PR3)]+
Attempts were made to oxidise the substituted mixed-valence derivatives [1a]+ and [1b]+ to the 32e− clusters [1a]2+ and [1b]2+, these latter species representing models for the highly electrophilic Ni-SIa state responsible for H2 heterolysis. However, treatment of [1a]BF4 with FcBF4 in CH2Cl2 instead afforded the μ-fluorido species [(dppe)Ni(pdt)FFe(CO)2(PPh3)]BF4 ([1aF]BF4), presumably from the abstraction of F− from BF4−. FT-IR analysis revealed two sets of overlapping νCO bands (2063, 2040, 1984, 1977 cm−1), consistent with the presence of two isomers (not uncommon for such pdt2−-bridged systems).12 The mean νCO frequency (2016 cm−1) is slightly higher than that for [1aCl]BF4 (2011 cm−1), which is in line with the poorer donor ability of the lighter halido ligands. The identity of [1aF]BF4 was further confirmed by ESI-MS (m/z 955.3) and analytical data. The PCy3 congener [1bF]BF4 could be prepared in a similar fashion from [1b]BF4.
Attempted Synthesis of Hydroxo- and Hydroperoxo-Bridged Complexes
Several sets of experiments were conducted in an effort to install bridging oxygenic ligands, as seen in the Ni-SI and Ni-SU states of the enzyme. The mixed-valence Ni(II)Fe(I) complexes, e.g. [1]+ react with water but not in a rationalizable manner, and no hydroxides could be detected. Thus treatment of a CH2Cl2 solution of [1]BF4 with water afforded 1 in ~50% yield according to IR analysis. Sensitive qualitative tests failed to indicate the formation of hydrogen peroxide in the reaction solutions.
Attempts were also made to install peroxo bridging groups. Treatment of a CH2Cl2 solution of the hydride [1aH]BF4 with aqueous H2O2 resulting in a colour change from brown to red. The proposed stoichiometry is shown in Scheme 3.
Scheme 3.
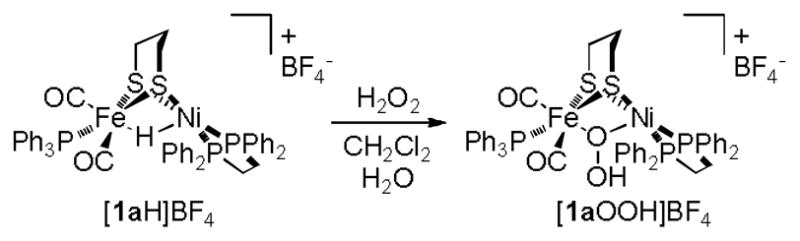
Formation of a hydroperoxo complex from a hydride precursor.
Although we were unable to purify the product, ESI-MS analysis of the red solution allowed for the detection of [(dppe)Ni(pdt)(OOH)Fe(CO)2PPh3]BF4 ([1aOOH]+, m/z 971.3). The IR spectrum exhibited two νCO bands (2042, 1981 cm−1) at energies similar to those of the iodo salt [1aI]BF4 (2031, 1978 cm−1).
Discussion
Complexes of the type (diphosphine)Ni(dithiolate)Fe(CO)2L represent the most versatile models for the active site of the [NiFe]-hydrogenases, despite their abiological terminal ligation. In previous reports, we have described models for the hydride-containing state Ni-R9 and the S = ½ state Ni-L.12 This paper summarises efforts to prepare synthetic models for the Ni-SU and Ni-SIr states. These enzyme states feature Ni(II)Fe(II) centres bridged by hydroxide and hydroperoxide.18
As for other complexes in this series of models, substitution of one CO ligand on Fe by a phosphine ligand enhances the stability of these compounds considerably. The halide-bridged species are not only important precursors to functional models19 but also represent the closest approach to synthetic analogues of these inactive states. Halide complexes incorporating NiFe nuclei have also been prepared by Bouwman and co-workers.20 Our attempts to purify hydroxide-, hydroperoxide- and alkoxide-bridged NiFe models have been unsuccessful, although we did detect a hydroperoxide derivative in one case. Derivatives incorporating Ni(II)(OR)(OH)Fe(II), Ni(II)(O)2Fe(II)21 and Ni(II)(OH)Fe(II) cores22 have been reported by the Driess and Holm groups, respectively. Metal carbonyl complexes containing hydroperoxide ligands are rare, probably owing to the facility of intramolecular reactions involving CO and the peroxide. As highlighted by Stiebritz and Reiher,23 the importance of studying O2-induced hydrogenase inhibition cannot be understated, as robust enzyme variants (and their models) represent promising catalysts for hydrogen processing.
Experimental
Unless otherwise stated, all chemicals were purchased from commercial sources and used as received. N-Bromosuccinimide was recrystallized from H2O. CD2Cl2 was distilled from CaH2. PhICl2,24 125 and 211 were prepared according to the literature methods. All reactions were conducted in an MBraun glovebox equipped with a solvent purification system; the concentrations of O2 and H2O in the N2 atmosphere were typically 1 and 0.2 ppm, respectively. IR spectra of complexes (in CH2Cl2) were recorded on a Perkin-Elmer Spectrum 100 FTIR spectrometer. A Micromass Quattro II spectrometer was used to acquire ESI-MS data for analytes as dilute CH2Cl2 solutions. Analytical data were acquired using an Exeter Analytical CE-440 elemental analyser. NMR spectra were recorded at room temperature on a Varian Unity 500 spectrometer. 1H and 31P{1H} spectra were collected at 500 and 202 MHz, respectively, and chemical shifts are referenced to residual CHDCl2 (5.32 ppm vs TMS for 1H) and external 85% H3PO4 (for 31P). Cyclic voltammetry experiments were carried out in a single compartment glass cell using a CH Instruments CHI630D electrochemical analyser. The working, counter and pseudo-reference electrodes were glassy carbon, platinum and silver, respectively. The analyte (1 mM) and NBu4PF6 (100 mM) were dissolved in CH2Cl2, and potentials (reported here relative to internal Fc/Fc+) were swept at 0.1 Vs−1. Crystallographic data were collected using a Bruker X8 diffractometer equipped with a Cu Kα source (λ = 1.54178 Å) and an Apex II detector.
(dppe)Ni(pdt)FeI2
Compound 1 (21.1 mg, 30 μmol) was partially dissolved in CH2Cl2 (0.5 mL) and treated with I2 (3.8 mg, 15 μmol) in CH2Cl2 (1 mL). The solution was stirred for a further 3 days, after which IR analysis indicated complete decarbonylation. Pentane (−28 °C, 15 mL) was added and the mixture allowed to stand at −28 °C for 1 h. The solids were isolated by filtration, washed with pentane (−28 °C, 2 × 2 mL) and dried briefly to afford the title compound as a red-brown powder (24.6 mg, 28 μmol, 94%).
Anal. calcd for C29H30FeI2NiP2S2: C, 39.90; H, 3.46; N, 0.00. Found: C, 40.11; H, 3.47; N, 0.50.
(dppe)Ni(pdt)FeBr2
This compound was prepared analogously to the iodo derivative, using Br2 in place of I2. Yield: 91%, red-brown powder.
Anal. calcd for C29H30FeBr2NiP2S2: C, 44.71; H, 3.88; N, 0.00. Found: C, 44.68; H, 3.98; N, 0.49.
[(dppe)Ni(pdt)IFe(CO)3]BF4 ([1I]BF4)
Compound 1 (21.1 mg, 30 μmol) and FcBF4 (8.2 mg, 30 μmol) were dissolved in CH2Cl2 (1 mL) with rapid stirring. After 1 min the solution was treated with I2 (3.8 mg, 15 μmol) in CH2Cl2 (1 mL). The solution was stirred for a further 0.5 min and pentane (−28 °C, 15 mL) was added and the mixture allowed to stand at −28 °C for 1 h. The solids were isolated by filtration, washed with pentane (−28 °C, 2 × 2 mL) and dried briefly to afford the title compound as an orange powder (26.9 mg, 29 μmol, 98%). 31P NMR (CD2Cl2, 202 MHz): δ 49.2. Anal. calcd for C32H30O3S2P2NiFeIBF4·CH2Cl2: C, 39.56; H, 3.22; N, 0.00. Found: C, 39.71; H, 3.09; N, 0.00. ESI-MS: m/z 828.9 [M – BF4−]+. Orange prismatic single crystals of [1I]BF4·2CH2Cl2 formed upon slow diffusion of pentane vapour into a concentrated CH2Cl2 solution of the title compound at −28 °C. One crystal (0.366 × 0.102 × 0.098 mm3) was subjected to X-ray diffraction at 193 K. Its space group was determined to be monoclinic C2/c with cell parameters: a 22.916Å, b 17.958 Å, c 21.500 Å, α 90.00°, β 107.87°, γ 90.00°. Integration of 5113 reflections and solution by direct methods using SHELXTL V6.1226 afforded a model with R1 = 0.0548 and wR2 = 0.0920.
[(dppe)Ni(pdt)BrFe(CO)3]BF4 ([1Br]BF4)
This compound was prepared analogously to [1I]BF4, using Br2 in place of I2. Yield: 98%, orange powder. 31P{1H} NMR (CD2Cl2, 202 MHz): δ 59.6 (~18% impurity), 53.4. Anal. calcd for C32H30O3S2P2NiFeBrBF4: C, 44.18; H, 3.48; N, 0.00. Found: C, 44.45; H, 3.40; N, 0.00. ESI-MS: m/z 782.8 [M – BF4−]+. N-Bromosuccinimide (5.3 mg, 30 μmol) can be used in place of Br2. Treatment with CoCp2 (2 equiv.) in CH2Cl2 gave a green solution and IR bands at 2028 and 1952 cm−1, consistent with formation of the neutral species.
[(dppe)Ni(pdt)ClFe(CO)3]BF4 ([1Cl]BF4)
This compound was prepared analogously to [1I]BF4, using PhICl2 in place of I2. Yield: 93%, orange powder.
31P{1H} NMR (CD2Cl2, 202 MHz): δ 54.5. Anal. calcd for C32H30O3S2P2NiFeClBF4: C, 46.56; H, 3.66; N, 0.00. Found: C, 46.81; H, 3.52; N, 0.00. ESI-MS: m/z 736.8 [M – BF4−]+.
[(dppe)Ni(pdt)IFe(CO)2(PR3)]BF4
Compound 1 (14.1 mg, 20 μmol) and FcBF4 (5.5 mg, 20 μmol) were dissolved in CH2Cl2 (1 mL) with rapid stirring. After 1 min the solution was added dropwise to PR3 (25 μmol) in CH2Cl2 (0.5 mL). The solution was stirred for a further 5 min before being treated with I2 (2.5 mg, 10 μmol) in CH2Cl2 (1 mL) with rapid stirring. After 5 min, pentane (−28 °C, 15 mL) was added and the mixture allowed to stand at −28 °C for 1 h. The solids were isolated by filtration, washed with pentane (−28 °C, 2 × 2 mL) and dried briefly to afford the product.
[(dppe)Ni(pdt)IFe(CO)2(PPh3)]BF4 ([1aI]BF4)
Yield: 72%, orange powder. 31P NMR (CD2Cl2, 202 MHz): δ 48.6 (dd, JPP = 36, 30 Hz), 45.1 (d, JPP = 36 Hz), 42.6 (d, JPP = 30 Hz). Anal. calcd for C49H45BF4FeINiO2P3S2·0.3CH2Cl2: C, 50.32; H, 3.91; N, 0.00. Found: C, 50.22; H, 3.91; N, 0.00. ESI-MS: m/z 1063.0 [M – BF4−]+.
[(dppe)Ni(pdt)IFe(CO)2(PCy3)]BF4 ([1bI]BF4)
Yield: 68%, orange powder. 31P{1H} NMR (CD2Cl2, 202 MHz): δ 44.9 (dd, JPP = 36, 24 Hz), 44.2 (d, JPP = 36 Hz), 42.8 (d, JPP = 24Hz). Anal. calcd for C49H63BF4FeINiO2P3S2: C, 50.33; H, 5.43; N, 0.00. Found: C, 50.09; H, 5.54; N, 0.00. ESI-MS: m/z 1081.2 [M – BF4−]+.
[(dppe)Ni(pdt)ClFe(CO)2(PR3)]BF4
Compound 1 (14.1 mg, 20 μmol) and FcBF4 (5.5 mg, 20 μmol) were dissolved in CH2Cl2 (1 mL) with rapid stirring. After 1 min the solution was added dropwise to PR3 (25 μmol) in CH2Cl2 (0.5 mL). The solution was stirred for a further 5 min before being treated with PhICl2 (4.1 mg, 15 μmol) in CH2Cl2 (1 mL) with rapid stirring. After 20 min, pentane (−28 °C, 15 mL) was added and the mixture allowed to stand at −28 °C for 1 h. The solids were isolated by filtration, washed with pentane (−28 °C, 2 × 2 mL) and dried briefly to afford the products.
[(dppe)Ni(pdt)ClFe(CO)2(PPh3)]BF4 ([1aCl]BF4)
Yield: 95%, orange powder. 31P NMR (CD2Cl2, 202 MHz): δ 53.1 (m), 44.7 (m) ppm. Anal. calcd for C49H45BF4FeClNiO2P3S2: C, 55.54; H, 4.34; N, 0.00. Found: C, 55.59; H, 4.49; N, 0.00. ESI-MS: m/z 1054.9 [M + CH2Cl2 –BF4−]+, 970.9 [M – BF4−]+.
[(dppe)Ni(pdt)ClFe(CO)2(PCy3)]BF4 ([1bCl]BF4)
Yield: 92%, orange powder. 31P NMR (CD2Cl2, 202 MHz): δ 53.7 (m), 51.6 (m) ppm. Anal. calcd for C49H63BF4FeClNiO2P3S2: C, 54.60; H, 5.89; N, 0.00. Found: C, 54.48; H, 5.99; N, 0.00. ESI-MS: m/z 989.2 [M – BF4−]+.
[(dppe)Ni(pdt)FFe(CO)2(PPh3)]BF4 ([1aF]BF4)
[1a]BF4 (21.2 mg, 20 μmol) in CH2Cl2 (1 mL) was treated with FcBF4 (5.5 mg, 20 μmol) in CH2Cl2 (1 mL) with stirring. After 5 min, pentane (−28 °C, 15 mL) was added and the mixture allowed to stand at −28 °C for 1 h. The solids were isolated by filtration, washed with pentane (2 × 2 mL) and dried briefly to afford the title compound as an orange powder (18.9 mg, 18 μmol, 90%). FT-IR: νCO = 2063, 2040, 1984, 1977 cm−1. Anal. calcd for C49H45BF5FeNiO2P3S2·CH2Cl2: C, 53.23; H, 4.20; N, 0.00. Found: C, 53.27; H, 4.20; N, 0.09. ESI-MS: m/z 955.3 [M –BF4−]+.
[(dppe)Ni(pdt)FFe(CO)2(PCy3)]BF4 ([1bF]BF4)
This compound was prepared analogously to [1aF]BF4, instead using the precursor [1b]BF4. Yield = 86%, orange powder. FT-IR: νCO = 2051, 2030, 1975, 1961 cm−1. Anal. calcd for C49H63BF5FeNiO2P3S2·1.2CH2Cl2: C, 51.83; H, 5.67; N, 0.00. Found: C, 51.90; H, 5.76; N, 0.09. ESI-MS: m/z 972.6 [M –BF4−]+.
Generation of [(dppe)Ni(pdt)OOHFe(CO)2(PPh3)]BF4 ([1aOOH]BF4)
[1aH]BF4 (20.5 mg, 20 μmol) in CH2Cl2 (2 mL) was treated with H2O2 (30% aqueous solution, 100 μL, ~900 μmol) and stirred in the absence of light. The dark brown solution became pale red, with ESI-MS and IR analyses indicating conversion to the new salt. ESI-MS: m/z 971.3 [M – BF4−]+.
[(dcpe)Ni(pdt)IFe(CO)3]BF4 ([2I]BF4)
This compound was prepared analogously to [1I]BF4, instead using 2 as the precursor. Yield: 72%, orange powder. 31P NMR (CD2Cl2, 202 MHz) : δ 63.6. Anal. calcd for C32H54O3S2P2NiFeIBF4: C, 40.84; H, 5.78; N, 0.00. Found: C, p40.78; H, 5.96; N, 0.00. ESI-MS: m/z 853.0 [M – BF4−]+.
Supplementary Material
Figure 4.

Positive ion ESI mass spectrum (top) and FT-IR spectrum (νCO region, CH2Cl2) of [1aOOH]BF4 (bottom).
Acknowledgments
The authors wish to thank Drs Danielle L. Gray and Amy L. Fuller for X-ray crystallography. This work was supported by the National Institutes of Health (GM61153).
Footnotes
Electronic Supplementary Information (ESI) available: Spectroscopic, electrochemical, and crystallographic details. See DOI: 10.1039/b000000x/
References
- 1.Frey M. ChemBioChem. 2002;3:153–60. doi: 10.1002/1439-7633(20020301)3:2/3<153::AID-CBIC153>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- 2.Hatchikian EC, Bruschi M, Le Gall J. Biochem Biophys Res Commun. 1978;82:451–61. doi: 10.1016/0006-291x(78)90896-3. [DOI] [PubMed] [Google Scholar]
- 3.Lacey ALD, Fernández VM, Rousset M, Cammack R. Chem Rev. 2007;107:4304–30. doi: 10.1021/cr0501947. [DOI] [PubMed] [Google Scholar]
- 4.Pandelia ME, Ogata H, Lubitz W. Chem Phys Chem. 2010;11:1127–40. doi: 10.1002/cphc.200900950. [DOI] [PubMed] [Google Scholar]
- 5.Ogata H, Lübitz W, Higuchi Y. Dalton Trans. 2009:7577–87. doi: 10.1039/b903840j. [DOI] [PubMed] [Google Scholar]
- 6.Ohki Y, Yasumura K, Ando M, Shimokata S, Tatsumi K. Proc Natl Acad Sci. 2010;107:3994–97. doi: 10.1073/pnas.0913399107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tard C, Pickett CJ. Chem Rev. 2009;109:2245–74. doi: 10.1021/cr800542q. [DOI] [PubMed] [Google Scholar]; Ohki Y, Tatsumi K. Eur J Inorg Chem. 2011:973–85. [Google Scholar]; Ichikawa K, Matsumoto T, Ogo S. Dalton Trans. 2009:4304–09. doi: 10.1039/b819395a. [DOI] [PubMed] [Google Scholar]
- 8.Lewis NS, Nocera DG. Proc Natl Acad Sci USA. 2006;103:15729–35. doi: 10.1073/pnas.0603395103. [DOI] [PMC free article] [PubMed] [Google Scholar]; Helm ML, Stewart MP, Bullock RM, Rakowski DuBois M, DuBois DL. Science. 2011;333:863–66. doi: 10.1126/science.1205864. [DOI] [PubMed] [Google Scholar]
- 9.Zhu W, Marr AC, Wang Q, Neese F, Spencer DJE, Blake AJ, Cooke PA, Wilson C, Schröder M. Proc Natl Acad Sci. 2005;102:18280–85. doi: 10.1073/pnas.0505779102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Barton BE, Whaley CM, Rauchfuss TB, Gray DL. J Am Chem Soc. 2009;131:6942–43. doi: 10.1021/ja902570u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Carroll ME, Barton BE, Gray DL, Mack AE, Rauchfuss TB. Inorg Chem. 2011;50:9554–63. doi: 10.1021/ic2012759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schilter D, Nilges MJ, Chakrabarti M, Lindahl PA, Rauchfuss TB, Stein M. Inorg Chem. 2012;51:2338–48. doi: 10.1021/ic202329y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schilter D, Rauchfuss TB, Stein M. Inorg Chem. 2012;51:0000. doi: 10.1021/ic300910r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.King RB. Organometallic Syntheses: Transition-Metal Compounds. Academic Press; New York: 1965. [Google Scholar]
- 15.Matthews SL, Heinekey DM. Inorg Chem. 2011;50:7925–27. doi: 10.1021/ic2009573. [DOI] [PubMed] [Google Scholar]
- 16.Barton BE, Rauchfuss TB. J Am Chem Soc. 2010;132:14877–85. doi: 10.1021/ja105312p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Haines RJ, de Beer JA, Greatrex R. J Chem Soc, Dalton Trans. 1976:1749–57. [Google Scholar]
- 18.Fontecilla-Camps JC, Volbeda A, Cavazza C, Nicolet Y. Chem Rev. 2007;107:4273–303. doi: 10.1021/cr050195z. [DOI] [PubMed] [Google Scholar]
- 19.Carroll ME, Barton BE, Gray DL, Mack AE, Rauchfuss TB. Inorg Chem. 2011;50:9554–63. doi: 10.1021/ic2012759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Verhagen JAW, Lutz M, Spek AL, Bouwman E. Eur J Inorg Chem. 2003;2003:3968–74. [Google Scholar]
- 21.Yao S, Driess M. Acc Chem Res. 2012;45:276–87. doi: 10.1021/ar200156r. [DOI] [PubMed] [Google Scholar]
- 22.Huang D, Holm RH. J Am Chem Soc. 2010;132:4693–701. doi: 10.1021/ja1003125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Stiebritz MT, Reiher M. Chem Sci. 2012;3:1739. [Google Scholar]
- 24.Zhao XF, Zhang C. Synthesis. 2007;4:551–57. [Google Scholar]
- 25.Barton BE, Rauchfuss TB. J Am Chem Soc. 2010;132:14877–85. doi: 10.1021/ja105312p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sheldrick GM. Acta Cryst. 2008;A64:112–22. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.