

Abstract
Kaposi’s sarcoma (KS) herpesvirus (KSHV) is the etiological agent of several immunodeficiency-linked cancers, including KS. Our previous work showed that the proto-oncogene c-kit is upregulated in KSHV-infected endothelial cells (EC), as well as in KS lesions. We show here that KSHV-dependent induction of both c-kit mRNA and protein requires the establishment of a latent infection and that this upregulation occurs in primary DMVEC as well as in immortalized DMVEC (eDMVEC). Interestingly, we find that while the lymphatic EC (LEC) subpopulation exhibits KSHV-induced c-Kit upregulation, the blood EC (BEC) subpopulation does not. Despite this upregulation of c-Kit, receptor activation and phosphorylation of downstream effectors such as MAP Kinase Erk 1/2 and GSK-3 still requires the addition of exogenous c-Kit ligand, stem cell factor (SCF). These data indicate that KSHV does not induce constitutive c-Kit signaling, but instead upregulates c-Kit receptor levels, thus allowing infected EC to respond to endogenous and exogenous SCF. Nonetheless, inhibition of either c-Kit activation or its downstream effectors reverses the characteristic spindle phenotype of infected eDMVEC. Together, these results contribute to our overall understanding of the role that the c-kit proto-oncogene plays in KS pathogenesis.
Keywords: Kaposi’s Sarcoma, KSHV, herpesvirus 8, viral transformation, c-kit, endothelial cells
Introduction
KSHV/HHV8 is an oncogenic γ-herpesvirus from the Rhadinovirus genus and is associated with several immunodeficiency-linked cancers, including Kaposi’s sarcoma (KS). KS is the most prevalent malignancy affecting AIDS patients, and it has become the most common cancer in sub-Saharan Africa, where KSHV is endemic and HIV is widespread. Unlike most tumors that arise from the expansion of a single transformed cell, KS tumors are most often polyclonal (Gill et al., 1998). The majority of cells that comprise a KS lesion have a distinctive spindle morphology and express endothelial markers such as CD31 and CD34 (Nickoloff, 1991; Nickoloff, 1993). However, cells expressing macrophage, fibroblast and smooth muscle cell markers have also been identified within KS lesions (Uccini et al., 1994; Uccini et al., 1997). Over the past 15 years many studies have been published in an attempt to pinpoint the exact origin of these cells (reviewed in (Ganem, 2006)). Initial studies based on histology and tumor location suggested that the spindle cells (SC) were of lymphatic EC (LEC) origin (Beckstead, Wood, and Fletcher, 1985). The majority of SC express lymphatic specific markers such as lymphatic endothelial hyaluronan receptor-1 (LYVE-1), D2-40, VEGFR-3 and podoplanin, but factor VIII and PAL-E, which are markers for blood vascular EC (BEC), have also been identified (Jussila et al., 1998; Kahn, Bailey, and Marks, 2002; Nadimi et al., 1988; Weninger et al., 1999; Xu et al., 2004). In addition, KS lesions are not found in areas devoid of lymphatics, such as the brain (Herndier and Ganem, 2001). Recently, two groups have used microarray analysis to further address this question. Using comparative transcriptomics (Wang et al., 2004), one group found that the gene expression profile of KS tissue was more similar to EC than that of any other cell type, and that the KS profile more closely matched that of LEC than BEC, thus confirming the majority of the histology data. The most intriguing results were obtained from the comparison of the transcriptomes of both infected and uninfected LEC and BEC. It appears that KSHV can efficiently infect both cell types, resulting in a reprogramming that makes each cell type more closely resemble the other. A similar transcriptomic analysis of KSHV-infected BEC also concluded that infected BEC are reprogrammed towards a more lymphatic expression profile, as evidenced by the upregulation of the lymphatic-specific marker prospero-related homeobox-1 (PROX-1) (Hong et al., 2004). To a large extent, the determination of the mechanisms of KSHV-induced tumorigenesis has been hampered by the inability to identify truly relevant KSHV target cells and to culture KS cells as explant cultures that maintain the viral genome. The inability to grow infected KS-derived cells led us to develop an in vitro endothelial cell infection model to facilitate KSHV pathogenesis studies. We have previously described a model system that utilizes HPV E6/E7-immortalized dermal microvascular EC (eDMVEC) that are chronically infected (~ 4 weeks) with KSHV (Moses et al., 1999). In this system, KSHV-infected eDMVEC develop a KS-like spindle phenotype and exhibit features of transformation, particularly the capacity to grow post confluence to form multi-layered foci. A subsequent cDNA microarray analysis of cellular genes upregulated by KSHV infection identified several proto-oncogenes including c-kit, the receptor for stem cell factor (SCF, also mast cell growth factor, steel factor, Kit ligand) (Moses et al., 2002a). Further study revealed that the KSHV-induced upregulation of c-kit was necessary for the transformed phenotype that develops in eDMVEC with time post-infection, suggesting that c-kit may play a role in KS development. It was subsequently shown that c-kit is strongly expressed in KS biopsies representing all epidemiologic and histologic forms of KS (Pantanowitz et al., 2004; Pantanowitz et al., 2005). These results led to a clinical study that demonstrated the efficacy of the tyrosine kinase inhibitor Gleevec for KS tumor regression (Koon et al., 2005).
c-Kit is a member of the PDGF family of receptor tyrosine kinases and is normally expressed on hematopoietic stem cells, mast cells, melanocytes, and germ cells. The receptor is activated by oligomerization, which is triggered by the binding of its ligand SCF (reviewed in (Lennartsson et al., 2005b)). Ligand-dependent auto-phosphorylation of c-Kit results in a series of downstream signaling events that include the activation of various kinase pathways such as Ras/Erk, phosphoinositide 3’-kinase, JAK/STAT, Src family kinase, and phospholipase Cγ (Lennartsson et al., 2005a). SCF stimulation of c-Kit and synergy with other cytokines results in cell survival and proliferation. However, oncogenesis can occur when SCF is secreted constitutively, or when c-Kit is either overexpressed or mutated to allow ligand-independent activation (Lennartsson et al., 2005b).
To further characterize the transformation of EC by KSHV, we have undertaken an analysis of c-Kit expression and signaling. Here we show that virus-induced over-expression of c-Kit rather than aberrant c-Kit activation is responsible for KSHV’s transforming capability. Moreover, we show that this c-kit upregulation is cell type dependent and only occurs in lymphatic, but not blood EC.
Results
c-kit mRNA is significantly induced only in latently KSHV-infected eDMVECs
We have previously described a microarray analysis to characterize the transcriptome of eDMVEC latently infected with KSHV (Moses et al., 2002a). This was done in an effort to identify cellular genes involved in KSHV-dependent transformation, and minimize the contribution of genes involved in acute responses to viral infection. One of the KSHV-induced genes identified in this screen was c-kit. In order to determine the kinetics of c-kit upregulation during KSHV infection, we infected eDMVEC with virus purified from PMA-stimulated BCBL cells and then cultured these cells without passage for 3 weeks. RNA was isolated at regular intervals from both KSHV and mock-infected eDMVEC and real-time quantitative PCR (qPCR) was performed to measure the levels of c-kit message. Gapdh mRNA levels were used for normalization. Figure 1A shows that c-kit levels began to increase in the KSHV-infected eDMVEC by day 8 post infection (PI), but were not significantly upregulated until day 12. c-Kit message levels in mock-infected eDMVEC remained low throughout the timecourse. Interestingly, these mRNA levels correlate with the degree of spindle cells in the culture (data not shown). To confirm that viral latency had been established, expression of both a latent (ORF73) and an early lytic (ORF59) protein were measured via immunofluorescent staining of infected monolayers and counting of positive cells. As shown in Figure 1B, latent protein expression was detectable at day 4 PI in approximately 25% of the cells compared to <1% expressing ORF59. Lytic protein expression remained at a similar low level throughout the timecourse, while latent protein expression increased to an average of 55-60%. To determine if c-kit message levels correlated with an increase in viral copy number, DNA was isolated at each of the timecourse intervals and viral copy number was measured by qPCR of the ORF26 gene. As shown in Figure 1C, the viral copy number increased between day 4 and day 8 PI and then did not significantly change throughout the remainder of the timecourse. Therefore the viral copy number peaked several days before the observed c-kit upregulation. These data suggest that the KSHV-induced increase in c-kit occurs after the establishment of latency and requires a certain viral load threshold and/or accumulation of a viral or virally-induced factor.
Figure 1. c-kit mRNA levels, late and lytic viral protein expression and viral copy number during a KSHV infection.
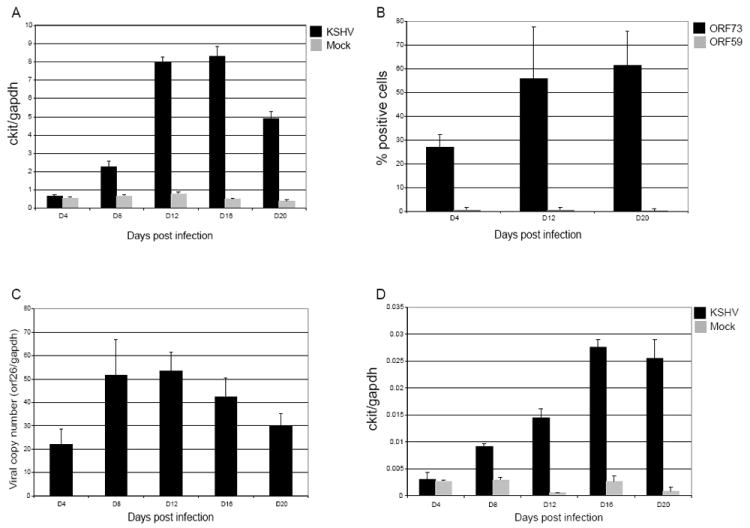
To assess the kinetics of c-kit upregulation during a long-term KSHV infection, eDMVEC (A) and primary DMVEC (D) were either mock (grey bars) or KSHV-infected (black bars) as described in materials and methods. RNA was isolated at the indicated times, qPCR for c-kit and gapdh was performed and c-kit levels were then normalized to gapdh levels. The data shown is representative of at least two independent infection timecourses. To analyze late and lytic viral protein expression, KSHV-infected eDMVEC were fixed and stained for ORF73 (late) and ORF59 (lytic) viral proteins (B). The percentages of positive cells in 5 imaged fields are depicted in the graph. To quantitate the viral copy number for the eDMVEC infection, DNA was isolated from the indicated times and qPCR for ORF26 and gapdh was performed (C). The ORF26 copy number relative to the copies of gapdh is shown in the graph. Error bars represent the standard deviation from duplicate qPCR reactions.
c-Kit upregulation also occurs in latently infected primary DMVEC
The eDMVEC system that we previously developed to study transformation by KSHV depends upon the prior immortalization of primary DMVECs with the papillomavirus E6 and E7 gene products to create a life-extended cell line (Moses et al., 1999). To confirm that c-kit upregulation was not an artifact of this immortalization process, we repeated the KSHV infection timecourse experiments in primary DMVEC. As seen in Figure 1D, qPCR revealed a similar latent expression pattern for KSHV-induced c-kit upregulation in primary DMVEC compared to eDMVEC. However, the overall levels of c-kit were significantly lower in the primary DMVEC (compare scale of Fig. 1A to Fig. 1D). This was not due to a lower infection efficiency, as the viral copy number in the primary DMVEC was equivalent to the viral copy number in the eDMVEC (data not shown). The efficient KSHV-induction of c-Kit in E6/E7-expressing cells may recapitulate the tumor microenvironment where additional factors likely exacerbate KSHV-associated KS lesion growth.
c-Kit receptor expression is upregulated in KSHV-infected eDMVECs
To confirm that the increased c-kit message levels in KSHV-infected cells correlated with an increase in c-Kit protein expression and proper cell surface localization, immunoprecipitation/immunoblot analysis of latently infected eDMVEC and flow cytometric analysis of a KSHV infection timecourse were performed. Figure 2A shows that only KSHV-infected eDMVEC and the positive Ad-cKit control express an appreciable level of c-Kit compared to mock-infected eDMVEC. Immunoblot analysis alone was not sufficient to consistently visualize c-Kit expression unless it was overexpressed by adenovirus (data not shown). The flow cytometric analysis in Figure 2B indicates that cell surface expression of c-Kit in KSHV-infected eDMVEC follows the same pattern of upregulation seen with the c-kit message (compare Figure 2B to Figure 1A). However, while the increase in c-kit message is detectable at 8 days, the protein expression is not evident until 13 days post infection. Despite this delay, the kinetics of c-Kit protein induction in infected eDMVEC cultures is consistent with KSHV-induced c-kit mRNA levels in latently-infected cells.
Figure 2. c-Kit protein expression and cell surface localization.
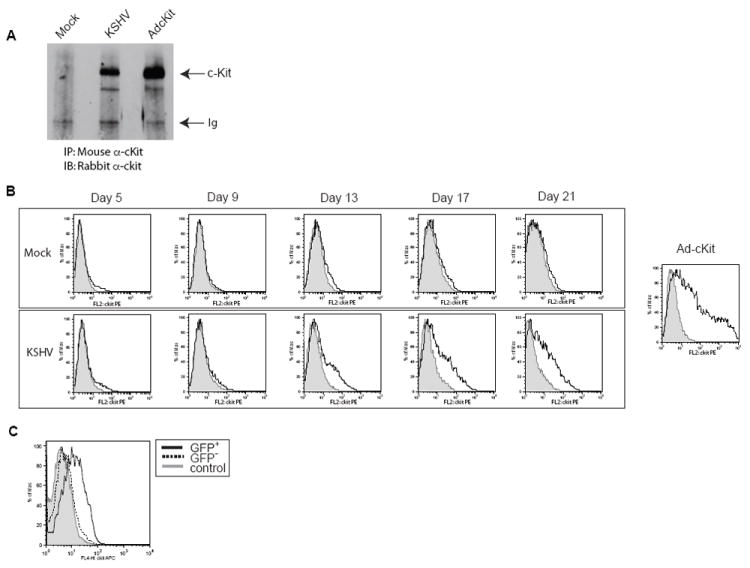
To verify that the upregulation of the c-kit message by KSHV correlated with an increase in c-Kit receptor that was properly localized to the cell surface, immunoprecipitation/immunoblots (A) and flow cytometric analysis (B) were performed on Mock-, latent KSHV-, and Ad-c-Kit-infected eDMVEC cells. (A) Total c-Kit protein levels were determined by a combination of c-Kit immunopreciptations (IP) from the indicated cells followed by c-Kit immunoblots (IB). (B, upper panel) Flow cytometric analysis for cell surface c-Kit was performed on Mock-infected eDMVEC at the indicated days post infection. The solid lines represent cells stained with a c-Kit antibody conjugated to PE and the shaded areas represent cells stained with a PE-conjugated isotype control. (B, bottom panel) Flow cytometric analysis for cell surface c-Kit was performed on KSHV-infected eDMVEC at the indicated days post infection. The solid lines represents cells stained with a c-Kit antibody conjugated to PE and the shaded areas represent cells stained with a PE-conjugated isotype control. Ad-c-Kit infected eDMVEC are shown for comparison (far right). The Y axis represents cell number as the percent maximum and the X-axis represents PE fluorescence intensity (C) Flow cytometric analysis for cell surface c-Kit was performed on rKSHV.219-infected eDMVEC at three weeks post-infection. The solid line represents GFP positive cells stained with a c-Kit antibody conjugated to APC, the dashed line represents GFP negative cells stained with the same c-Kit antibody and the shaded histogram represent cells stained with an APC-conjugated isotype control antibody.
To determine whether the induction of c-Kit only occurs in those cells directly infected with KSHV and is not due to a bystander effect, we utilized the recombinant GFP-expressing KSHV, rKSHV.219 (Vieira and O’Hearn, 2004) (kindly provided by Jeff Vieira). Cells infected with this recombinant KSHV express GFP allowing us to use GFP expression as a marker for viral infection. As shown in Figure 2C only the GFP positive population of cells exhibited an increase in surface c-Kit expression, suggesting a direct correlation between c-Kit induction and KSHV infection. The GFP negative population of cells expressed no detectable c-Kit when compared to the isotype control, indicating that c-Kit is not induced merely by proximity to infected cells.
This recombinant virus also allowed us to determine whether the induction of c-Kit was a consequence of viral spread through the culture. Accordingly, eDMVEC were infected with rKSHV.219 and cultured for 20 days. Samples were collected at 4 day intervals and analyzed by flow cytometry for both GFP (as a measure of viral infection) and c-Kit expression. As shown in Figure 3 (top panels), virus was detected in 17% of the cells at day 4 and increased only 2 fold to a final level of 32% by day 20. In contrast (bottom panels), c-Kit expression was not detectable at day 4 in the GFP+ KSHV-infected cells, but was evident on day 8. From day 8 to day 20 the levels of c-Kit remained consistent and only present in the GFP+ population of cells. We conclude that the significant increase in c-Kit levels at day 8 is not due simply to an increase in the quantity of virally infected cells, as the percentage of KSHV-infected cells did not significantly change between day 4 and day 8. However, both the viral copy number (Figure 1C) and latent gene expression (Figure 1B) increased between day 4 and day 12, coincident with the increase in c-Kit levels. Overall, c-Kit expression in a KSHV-infected culture is influenced by both the net level of infected cells and the viral load per infected cell. In vivo, these parameters could change independently with time and/or due to exogenous factors.
Figure 3. Viral spread and c-Kit expression during a KSHV infection.
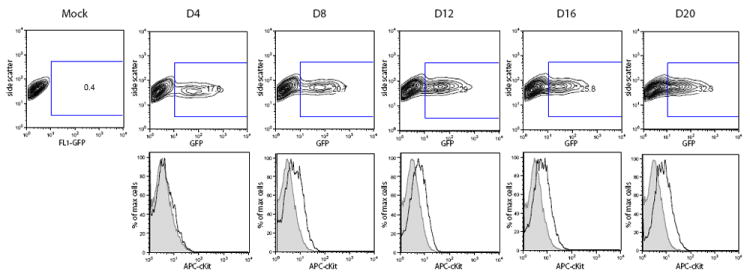
To determine whether c-Kit induction correlated with an increase in KSHV-infected cells, flow cytometric analysis of GFP expression in both mock and rKSHV.219 infected eDMVEC were performed at the indicated days post infection. The contour plots in the top panel show the percentage of GFP positive, KSHV-infected cells at each timepoint. The corresponding histograms in the bottom panel show c-Kit expression of the mock (shaded) or GFP-positive/rHSKV.219-infected (black) cells. The inset rectangles in the top panel are the gates used to denote GFP-positive cells.
SCF stimulation induces phosphorylation of c-Kit in KSHV-infected eDMVECs
To determine if KSHV affects c-Kit receptor activation, the level of phosphorylation of the c-Kit receptor was measured in mock- and latently-infected eDMVEC in the presence and absence of exogenous SCF after 24 hours of serum starvation. Immunoprecipitations of infected cell lysates were performed with a c-Kit antibody, and the resulting immune complexes were then subjected to a phospho-c-Kit immunoblot (Figure 4, top panel). To determine total c-Kit protein levels, the blot was stripped and re-probed with a c-Kit antibody (Figure 4, bottom panel). Significant phosphorylation of c-Kit only occurred in the presence of SCF. The phosphorylation is greatest following 5 to 20 minutes of SCF stimulation and diminishes by 40 minutes. This decrease in phosphorylation is consistent with ligand-induced receptor internalization, which is also evidenced by a decrease in total c-Kit levels. In addition, this pattern of c-Kit phosphorylation is consistent with SCF stimulation in hematopoietic stem cells (Wandzioch et al., 2004). These results suggest that while KSHV infection upregulates c-Kit receptor expression, ligand-independent activation of the receptor does not occur. Exogenous SCF is still required for c-Kit phosphorylation, at least under conditions of in vitro culture.
Figure 4. c-Kit phosphorylation in the presence or absence of SCF.

To determine if KSHV affects c-Kit activation, mock and latent KSHV-infected cells were cultured in the presence or absence of SCF ligand after 24 hours of starvation in minimal medium without serum. Cell lysates were then prepared, total c-Kit protein was recovered by immunoprecipitation, and the presence of phosphorylated c-Kit (Y568) was detected via immunoblot analysis (top panels). The blot was striped and re-probed for total c-Kit (bottom blots). Infection status and incubation times of SCF stimulation in minutes are indicated above the blots. Densitometric measurements obtained using Image J software are shown below each blot. Density values were calculated as background subtracted mean grey values normalized to the 0 minute samples set to 1.
SCF-induced activation of the Map Kinase Erk 1/2 and Akt pathways is enhanced in KSHV-infected eDMVECs
Activation of the c-Kit receptor via SCF binding results in a signaling cascade that involves several kinase pathways, including MAP Kinase, Erk 1/2, and Akt. To determine whether latent KSHV infection affected signaling events downstream of c-Kit, the phosphorylation states of Erk 1/2 and glycogen synthase kinase 3 (GSK-3), two effectors in these signaling pathways, were analyzed in the presence or absence of SCF stimulation. GSK-3 phosphorylation was used to measure the activation of the Akt pathway because the phosphorylation of Akt was not reliably detected. Both Erk 1/2 (Figure 5A, top panel) and GSK-3 (Figure 5B, top panel) phosphorylation were upregulated in SCF-stimulated cells, however this activation was enhanced in KSHV-infected cells, consistent with our previous data showing increased c-Kit levels in KSHV-infected cells. In the absence of SCF, phosphorylation levels of both effectors were slightly increased in KSHV-infected cells. This was likely due to minimal activation of various other upstream receptors that signal through these effectors. Total effector protein levels were not significantly affected by SCF or KSHV (Figure 5A & 5B, middle panels).
Figure 5. Activation of c-Kit signaling pathways.
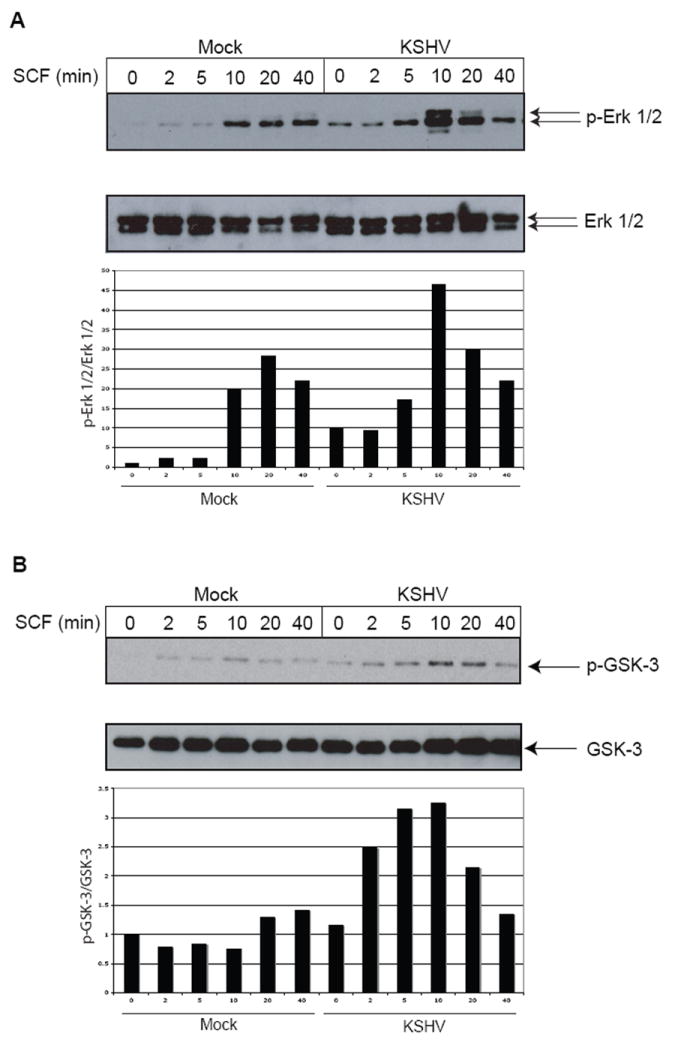
To ascertain whether signaling downstream of c-Kit was affected by KSHV infection, immunoblots for phosphorylated Erk 1/2 (p-Erk 1/2) (A, top blot) and phosphorylated GSK-3 (p-GSK-3) (B, top blot) were performed on mock or KSHV-infected cells (3 weeks post-infection) that had been treated with SCF as described in Figure 4. To confirm that total protein levels were unchanged, the blots were stripped and reprobed with antibodies to Erk 1/2 (A, bottom blot) and GSK-3 (B, bottom blot). Quantitation of the phosphorylated protein levels compared to total protein levels were obtained using Image J software and are shown in the graphs below each of the blots. All values are normalized to the mock-infected, No SCF sample, which was set to 1.
Various kinase inhibitors block SCF-induced activation of Map Kinase Erk 1/2
To further confirm that c-Kit signaling in KSHV-infected cells was dependent upon exogenous SCF, the phosphorylation of ERK 1/2 was analyzed in the presence of kinase inhibitors that act at various stages of the c-Kit signaling cascade (diagramed in Figure 6A). As expected, the tyrosine kinase inhibitors PP1 and Gleevec both prevented Erk 1/2 activation in the presence of SCF (Figure 6B). Similarly, PD98059 and the Raf1 inhibitor, which are inhibitors of the Erk1/2 pathway, blocked Erk 1/2 phosphorylation in a dose dependent manner. Interestingly, the PI-3 kinase inhibitor LY294002 also showed a decrease in Erk 1/2 activation, which suggests that PI-3 kinase may be involved in the Erk 1/2 pathway as well as the Akt pathway. This observation is consistent with a study showing that PI-3 kinase may act downstream of Ras in the Erk 1/2 pathway in SCF-stimulated hematopoietic stem cells (Wandzioch et al., 2004). None of the kinase inhibitors affected total Erk 1/2 levels. Our results support KSHV-induced c-Kit expression, but normal SCF-dependent activation.
Figure 6. Inhibition of c-Kit signaling.
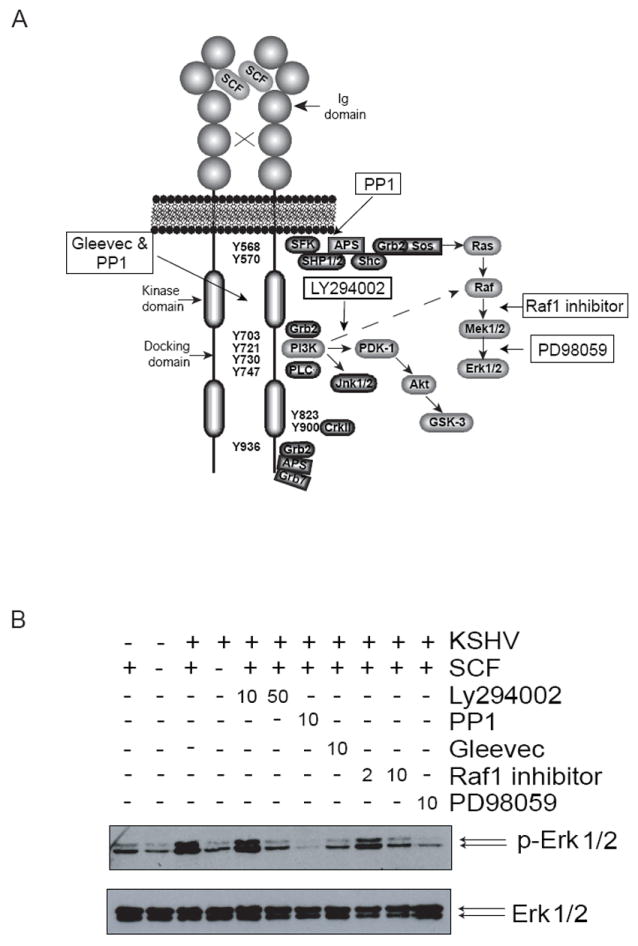
To confirm the activation of c-Kit signaling by KSHV, a series of kinase inhibitors were utilized. The diagram (A) depicts the c-Kit kinase-signaling pathway and where each inhibitor acts (modified from (Lennartsson et al., 2005a)). A p-Erk 1/2 immunoblot was performed on KSHV-infected cells pre-incubated with the indicated kinase inhibitors prior to 10 minutes of SCF stimulation (B, top blot). The blot was stripped and reprobed with antibodies to Erk 1/2 to measure total protein levels (B, bottom blot). The concentration (μM) of inhibitor is indicated above the blots.
Inhibition of kinases in the c-Kit signaling pathway reverses the spindle phenotype of KSHV-infected eDMVECs
Previous data demonstrated the importance of c-Kit in KSHV-induced transformation of EC, as evidenced by prominent spindling of infected cells and the growth of infected monolayers post-confluence into multi-layered foci (Moses et al., 2002b). To determine if the inhibition of various kinases in the c-Kit pathway would affect the virus-induced transformation phenotype, latently infected, spindling eDMVECS were treated with two concentrations (0.5μM and 2μM) of the kinase inhibitiors previously described. As shown in Figure 7A, PP1 (tyrosine kinase inhibitor), PD98059 (MEK inhibitor), and LY294002 (PI-3 kinase inhibitor) all significantly inhibited the spindle-cell phenotype (compare panels 1-3 to panel 5), with the most robust effect occurring at the higher inhibitor concentration. The Raf1 inhibitor only partially reversed the phenotype at the 2μM concentration (compare panel 4 to panel 5). Restoration of the normal cell morphology in the presence of inhibitors was not due to a loss of KSHV-infected cells, as the number of ORF73-expressing cells (60-80%) was not significantly affected (Figure 6B). These data indicate that KSHV-induced transformation of EC is dependent on the continued activation of Erk 1/2.
Figure 7. Inhibition of KSHV-induced transformation.

To investigate whether the kinases in the c-Kit signaling pathway are involved in KSHV-induced transformation of EC, inhibitors at either 0.5μM (A, top panels) or 2μM (A, bottom panels and B) were added to mock and KSHV-infected eDMVECs (21 days post infection). Media plus inhibitor was changed every 2-3 days. After 8 days the cells were (A) photographed live to record any changes in the spindle cell morphology and then (B) fixed and stained with ORF73 antibodies to assess any alterations in the level of viral infection. (B, top panels) ORF73 immunofluorescence (B, bottom panels) corresponding phase images.
ckit mRNA does not appear to be stabilized in a latent KSHV culture
To address the mechanism of KSHV-induced c-kit upregulation, we first evaluated the half-life of c-kit mRNA in mock or latently KSHV-infected eDMVEC. The cells were treated with Actinomycin D and RNA was then isolated at regular intervals for 8 hours. Figure 8A depicts the mRNA levels of both a stable control gene (gapdh) and an unstable control gene (c-myc) as measured by qPCR. Gapdh had a half-life of >8 hours in both mock and KSHV-infected samples, while c-myc had a half-life of 2 hours regardless of infection. As shown in Figure 8B, the c-kit mRNA levels for KSHV-infected cells was 10 fold higher than mock-infected cells, however the half-lives of c-kit mRNA in both mock and KSHV-infected cells was ~7 hours. These results indicate that the KSHV-induced increase in c-kit mRNA levels is not a result of increased mRNA stability.
Figure 8. c-kit mRNA half-life determination.
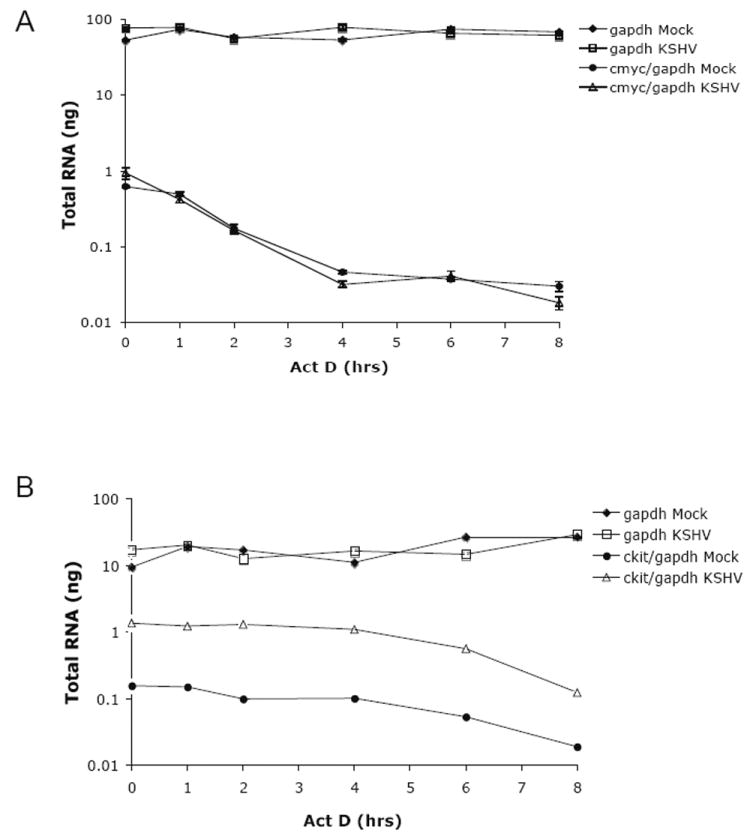
To determine if KSHV influences the stability of c-kit mRNA, latently infected eDMVEC were treated with Actinomycin D and qPCR was performed on RNA isolated from various times post-treatment. Values are plotted as total RNA (ng) determined by relative quantitation based on standard curves. c-myc (A) and c-kit (B) values are normalized to gapdh values. Error bars represent the standard deviation of duplicate samples and the data shown is representative of three independent experiments.
c-kit upregulation occurs in the LEC, but not the BEC subpopulation of DMVEC
Because DMVEC obtained from commercial sources are a mixture of lymphatic and blood EC, we wanted to know if the upregulation of c-kit could be induced in both subpopulations of cells. We immortalized the purified LEC and BEC with papillomavirus E6 and E7 gene products as previously described for DMVEC (Moses et al., 1999). These cells were then infected with KSHV and c-kit mRNA levels were measured at various timepoints by qPCR. Notably, only the infected LEC showed an upregulation of c-kit (Figure 9). There was no increase in c-kit observed in infected BEC although the level of infection was similar as measured by ORF73 expression (data not shown). Interestingly, the endogenous levels of c-kit in mock-infected BEC were comparable to the induced c-kit levels in KSHV-infected LEC (compare Figure 9A to 9B). These results suggest that the c-kit upregulation is not only virus dependent, but also cell-type dependent. KSHV-induction of c-Kit in LEC correlates with data showing that KSHV infection of EC sub-types drives the cells to a more convergent phenotype such that LEC acquire features of BEC and vice versa (Hong et al., 2004; Wang et al., 2004). Alternately, but not exclusively, c-Kit induction may reflect a transition towards a more pluripotent, dedifferentiated phenotype.
Figure 9. c-kit expression in immortalized LEC and BEC.
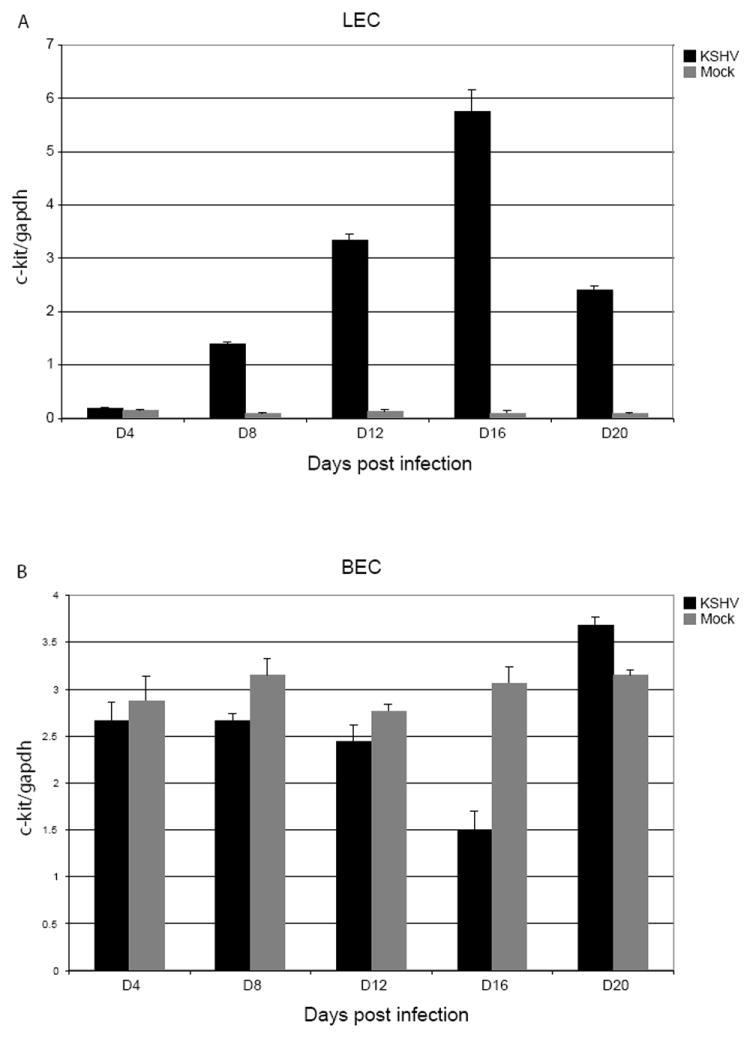
To ascertain whether both subpopulations of DMVEC can exhibit KSHV-induced c-kit upregulation, RNA samples from mock (grey bars) and KSHV-infected (black bars) LEC (A) and BEC (B) were isolated at the indicated times, qPCR for c-kit and gapdh was performed and c-kit levels were then normalized to gapdh levels. The data shown is representative of at least two independent infection timecourses.
Discussion
The goal of this study was to understand how c-Kit expression contributes to the KSHV-induced transformation of EC, an in vitro surrogate of KS tumorigenesis. We first investigated the kinetics of c-Kit expression and discovered that infected EC exhibited significant increases in both c-Kit message and protein levels only after 12 days in culture, which is well after the establishment of viral latency. The lack of c-Kit expression at early times after infection suggests a requirement for the accumulation of a viral or virally induced factor(s). However, our data does not rule out the possibility that a certain viral load threshold is also required. The latent kinetics of c-kit upregulation seen with infected eDMVEC was reiterated in primary DMVEC. However, the levels of c-kit were significantly higher in both mock and infected eDMVEC. This overall increase in c-kit could be due to the papillomavirus E6/E7 proteins used to immortalize the DMVEC. It should be noted that E6/E7 alone cannot fully transform cells. Instead, E6/E7 immortalize cells via the degradation of p53 (Scheffner et al., 1990) and the inactivation of pRb (Dyson et al., 1989). A synergistic affect between E6/E7 and v-HA-ras has been implicated in papillomavirus’s oncogenic phenotype (Phelps et al., 1988; Riou et al., 1988). The eDMVEC transformation model was developed to overcome KSHV’s inability to transform primary EC, so it is not surprising that E6/E7 might enhance KSHV-induced expression of c-kit. We also discovered that the KSHV-induced upregulation of c-Kit is a direct effect of viral infection and not due to a bystander effect from infected neighbor cells (Figure 2C).
While c-Kit plays a crucial role in hematopoietic cell development, abnormal c-Kit expression or activation leads to the development of various cancers. This occurs via temporal dysregulation of receptor expression (e.g. acute myelongenous leukemia), constitutive activation of the receptor by mutations in the juxtamembrane and kinase domains (e.g. gastrointestinal stromal cell tumors), or autocrine secretion of c-Kit’s ligand, SCF (e.g. small lung carcinomas). We found that the phosphorylation of c-Kit in latent KSHV-infected cells still required exogenous SCF. In addition, two of the downstream kinase pathways for c-Kit, Erk 1/2 and Akt were significantly activated only in latently infected cells treated with SCF. Previous data has shown that KSHV does not significantly increase SCF expression in eDMVEC (Moses et al., 2002b). Therefore, these data suggest that KSHV’s ability to transform eDMVEC occurs via the over-expression of the c-Kit receptor rather than constitutive receptor activation or induction of SCF secretion. Both endogenous SCF produced from the eDMVEC and exogenous SCF from the culture medium (human serum contains several nanograms per milliliter of SCF (Hsu et al., 1997)) would likely provide the stimuli to maintain c-Kit activation. In KS lesions, the inflammatory infiltrate comprised of mast cells and macrophages could provide a continual source of elevated SCF.
The importance of Erk 1/2 activation for the establishment of KSHV infection in EC has been described (Sharma-Walia et al., 2005). Our data suggests that this activation is also important for the KSHV-induced transformed spindle phenotype. Using various inhibitors that act on kinases in the Erk 1/2 pathway we were able to reverse the spindle phenotype. However, we cannot rule out a role for other kinases such as Akt and JNK 1/2, because the most effective reversion occurred with PP1 and LY294002, which also inhibit these pathways. In light of the data suggesting that Erk 1/2 kinase inhibitors prevented KSHV infection and reduced viral transcription of both lytic and late viral genes such as ORF50 and ORF73 (Sharma-Walia et al., 2005), it was surprising that the percentage of infected cells and the level of ORF73 appeared unaffected by the inhibitor treatment. Perhaps the profound dependence on Erk 1/2 signaling for KSHV replication only occurs during the lytic cycle, and once latency has been established, Erk 1/2 is no longer required for genome maintenance.
Due to the possibility that the latently expressed Kaposin B protein stabilizes certain mRNAs (McCormick and Ganem, 2005), we investigated the ability of KSHV to stabilize the c-kit message. Using actinomycin D to block transcription, we found that there was no significant difference in the c-kit mRNA half-lives between mock and KSHV-infected DMVEC. This data suggests that the KSHV-induced increase in c-kit mRNA levels was not due to enhanced message stability. Therefore, promoter activation is the most likely mechanism for the observed upregulation of c-kit, although further analysis will be necessary to confirm this hypothesis.
In an attempt to determine whether any of the latent gene products are responsible for the c-Kit induction we established stable eDMVEC lines expressing the individual latent proteins, including v-FLIP, v-cyclin, Lana-1 and KapA (data not shown). Unfortunately, none of the individual latent genes induced any significant levels of c-kit compared to control cells. In addition, none of the individual latent genes induced the spindle-cell phenotype normally associated with a latent KSHV infection. This was in contradiction to a previously published report that v-FLIP could induce endothelial cell spindling when expressed alone (Grossmann et al., 2006). Immunoblot analysis confirmed that our v-FLIP cell line did indeed express v-FLIP protein, so it is not clear what could account for this discrepancy. Our data suggests that no single latent gene is responsible for the KSHV-induced c-kit upregulation or the spindle-cell phenotype associated with transformation. This implies that either multiple latent genes and/or low lytic gene expression are responsible.
It has not been conclusively established whether KSHV infects and reprograms EC committed to the LEC or BEC lineages, or infects endothelial cell precursors and influences their lineage commitment. Interestingly, when we looked at KSHV’s ability to induce c-kit in either the LEC or BEC subpopulations of primary DMVEC, the levels of c-kit were upregulated in LEC over time, while the amount of c-kit message slightly decreased in BEC. These results are intriguing in light of the potential for KSHV to reprogram EC, such that infected LEC mimic untreated BEC and vice versa (Hong et al., 2004; Wang et al., 2004). c-kit is a developmentally regulated gene that is highly expressed in hematopoietic and endothelial progenitor cells, but decreases as the cells differentiate. Exceptions include mast cells, melanocytes and umbilical vein EC (HUVEC) (reviewed in (Lennartsson et al., 2005a)). We show here that normal DMVEC and LEC do not express any significant levels of c-kit message or protein until they are latently infected with KSHV. Conversely, the endogenous levels of c-kit in BEC were much higher than in mock-infected LEC. The differentiation pathway of progenitor EC leading to either the LEC or BEC phenotype is not clear; however one possibility is that LEC may be derived from vascular origins (Adams and Alitalo, 2007). Perhaps the loss of c-kit expression is part of the normal lymphatic differentiation pathway, while vascular EC maintain a higher level of c-kit expression. The significant levels of c-Kit observed in HUVEC would support this hypothesis (Broudy et al., 1994).
Previously we have shown that the induction of c-kit by KSHV is necessary for the transformed spindle cell phenotype observed in infected eDMVEC. Here we have expanded on these observations and demonstrate that the KSHV-induced increase in c-kit is dependent on the establishment of viral latency in an appropriate endothelial cell-type. However, despite this c-kit induction, KSHV does not appear to directly affect the phosphorylation of the c-Kit receptor, which remains SCF-dependent. Further characterization of the mechanism(s) underlying KSHV-induced activation of the c-kit promoter in EC will advance our understanding of KSHV-associated tumorigenesis and may lead to the identification of new cellular targets for KS.
Materials and Methods
Cells, viral production, titering and infections
Primary adult DMVEC, neonatal LEC and BEC (Lonza, Walkersville, MD) were infected with a retroviral vector encoding E6 and E7 from HPV type 16 to generate life-extended DMVEC (eDMVEC) as previously described (Moses et al., 1999). All endothelial cells were maintained in human endothelial serum-free medium (SFM) (Invitrogen) supplemented with 37ug/ml endothelial cell growth supplement (ECGS) (BD, Franklin Lakes, NJ), 100UI/ml penicillin, 100ug/ml streptomycin, 3mg/ml glutamine (PSG) (Invitrogen), and 10% human serum (Sigma, St. Louis, MO). KSHV used for all infections was obtained from phorbol 12-myristate 13-acetate (PMA, Sigma) stimulated body cavity based lymphoma (BCBL) cells (NIH AIDS Research & Reference Reagent Program, Germantown, MD). To produce cell-free virus, BCBL cells were cultured in RPMI supplemented with 1mM HEPES, 1mM sodium pyruvate, PSG and 10% fetal bovine serum (FBS). Cells were stimulated with 20ng/ml PMA for 4 days and harvested via centrifugation for 10min at 400 × g. The resulting supernatant was layered onto a 5% sucrose cushion in TNE (150mM NaCl, 10mM Tris pH 8, 2mM EDTA pH 8) and centrifuged for 2 hours at 21,000 × g. Viral pellets were resuspended in 150th the supernatant volume of TNE. Virus titers were determined via a modification of a previously described replication and transcription activator (RTA) assay (Inoue et al., 2003). Briefly, T1H6 cells (a 293T-based cell line harboring the lacZ (β-galactosidase) gene expressed from the RTA-dependent PAN promoter) obtained from Dr. Margaret Offermann (Emory University, Atlanta, GA) were maintained in DMEM (high glucose) plus 10% FBS, PSG, and 50ug/ml hygromycin B. T1H6 cells were seeded at 4 × 104 cells/well of a 96 well plate without hygromycin B. Various dilutions of KSHV preparations were made in 50uL of serum free DMEM plus 8ug/ml polybrene. These were then added to the cells, followed by a 2hr incubation at 37°C. After infection, 50ul of complete media was added to each well and the cells were incubated at 37°C for 3 days. To assay for β-galactosidase activity, 100ul of Beta-Glo reagent (Promega, Madison, WI) was added directly to each well. The plate was incubated at room temperature for 30min and then read in a luminometer (Turner BioSystems). Viral titers were then calculated as β-galactosidase units/ml. To convert β-galactosidase units into infectious units, the amount of virus needed to infect approximately 50-80% eDMVEC was determined. For infections, EC were seeded at 3 × 105 cells/well in 6-well plates. To each well, KSHV was added at 40 β-galactosidase units/per cell in 1 mL of serum-free medium plus 8ug/ml polybrene, and the plates were then centrifuged at 400 × g for 30min (spinoculation). The cells were then incubated at 37°C with 5% CO2 for an additional 1.5 hr. After incubation, 1.5ml of complete medium was added. The media was changed every 3-4 days for the duration of the experiment. The GFP-expressing KSHV, rKSHV.219 was obtained from Jeff Vieira and produced as described previously (Vieira and O’Hearn, 2004).
Nucleic acid isolation and quantitative real-time pcr (qPCR)
RNA was isolated from cells using an RNeasy kit (Qiagen, Valencia, CA) with “on-column” DNAse treatment. RNA was reverse transcribed using a Superscript III First Strand Synthesis Kit with random hexamer priming (Invitrogen). DNA was isolated from cells using a DNeasy tissue kit (Qiagen). qPCR was performed using the Power SYBR Green PCR master mix in an ABI PRISM 7700 Sequence detection system or an ABI 7500 Real-time PCR system (Applied Biosystems, Foster City, CA). Primer sequences for quantitating cDNA were: gapdh forward 5’-GTCCACTGGCGTCTTCACCA-3’, gapdh reverse 5’-GTGGCAGTGATGGCATGGAC-3’, c-kit forward 5’-CTCAACCATCTGTGAGTCCA-3’, c-kit reverse 5’-AAGCCGTGTTTGTTGGTGCA, c-myc forward 5’- CTCCTACGTTGCGGTCACAC-3’, c-myc reverse 5’- CCGGGTCGCAGATGAAACTC. Relative quantitation of gene expression using the standard curve method was performed as outlined by ABI. For determining DNA copy numbers, absolute quantitation was performed using plasmids containing either a portion of an intron from the gapdh gene or the BamHI fragment of the ORF26 gene. Primer sequences for determining DNA copy number were: gapdh forward 5’-TGCCTTCTTGCCTCTTGTCTCT-3’, gapdh reverse 5’-GGCTCACCATGTAGCACTCACC-3’, ORF26 forward 5’-AGCCGAAAGGATTCCACCAT-3’, ORF26 reverse 5’-TCCGTGTTGTCTACGTCC-3’. For RNA stability assays, actinomycin D (10ug/ml, Fisher Scientific) was added to the media of mock and KSHV-infected eDMVEC (3 weeks post infection) for the indicated times. RNA was isolated and qPCR was performed as described above.
Flow cytometric analysis and immunofluorescence
Mock and KSHV-infected eDMVEC were trypsinized and resuspended in flow cytometry buffer (1 × phosphate buffered saline (PBS) plus 3% FBS). The cells were stained for c-Kit with anti-CD117 (104D2) antibody conjugated to phycoerythrin (PE) or the corresponding PE-conjugated isotype control (BD, Franklin Lakes, NJ). Samples were washed 3 times with flow cytometry buffer and then analyzed on a FACScalibur (BD). For experiments using rKSHV.219, the cells were stained for c-Kit with anti-CD117 (104D2) antibody conjugated to APC or the corresponding APC-conjugated isotype control (BioLegend, SanDiego, CA). ORF73 and ORF59 immunofluorescence was performed by first fixing the cells in 5% glacial acetic acid in ethanol for 8 min, washing them 3 times in 1 × PBS, and permeabilizing them in 0.5% triton-X-100 for 8 min. The cells were then washed 3 times in 1 × PBS, blocked in 20% normal goat serum (NGS, Sigma), washed once in 1 × PBS, and incubated in 1% NGS containing rabbit anti-ORF73 or mouse anit-ORF59 antibody (kindly provided by Bala Chandran (Chicago Medical School, North Chicago, IL)). After primary antibody incubation, the cells were washed 3 times in 1 × PBS and incubated in 1% NGS with AlexaFluor 595-conjugated secondary anti-rabbit antibody (Invitrogen), AlexaFluor 488-conjugated secondary anti-mouse antibody (Invitrogen), and 1ug/ml DAPI. Fluorescent images were obtained from an Axiocam MRm camera mounted to a Zeiss Axioskop 2 Plus microscope (Zeiss, Thornwood, NY).
Ligand stimulation, immunoprecipitations, immunoblots and kinase inhibitors
For ligand stimulation, mock and KSHV-infected EC were first starved in M199 medium (basal medium for endo-SFM) (Invitrogen) for 24 hours followed by stimulation with 50ng/ml recombinant human stem cell factor (SCF) (R & D Systems, Minneapolis, MN) for various times. The reactions were stopped by incubation on ice and immediately lysed in either 1% sodium dodecyl sulfate (SDS) buffer (1% SDS, 1mM phenylmethylsulphonyl fluoride (PMSF), 2mM sodium vanidate, 0.05M Tris pH 8.0, 1x SigmaFast protease inhibitors, 1 × phosphatase inhibitor 2 (Sigma)) for immunoblots or 1% Igepal lysis buffer (50mM Tris pH 7.6) plus protease and phosphatase inhibitors (Sigma) for immunoprecipitations. For c-Kit immunoprecipitations, the cell lysates were incubated for 30min on ice followed by centrifugation at 10,000 × g for 10min at 4°C to pellet the cellular debris. Protein concentrations were determined using a BCA protein assay (Pierce, Rockford, IL). 250-500ug total protein was then precleared with 1ug of normal mouse IgG and 20ul of a 25% Protein G PLUS-Agarose slurry (Santa Cruz Biotech., Santa Cruz, CA) at 4°C for 30min. The agarose beads were then pelleted by centrifugation at 1000 × g for 5min at 4°C. Immunoprecipitations were performed on the supernatants by adding 1ug of mouse anti-c-Kit antibody (AB 81, Santa Cruz Biotech.) for 1hr at 4°C, followed by the addition of 20ul of the 25% Protein G-PLUS-Agarose slurry and incubating for 16-20 hours rocking at 4°C. The immune complexes were then centrifuged at 1000 × g for 5min at 4°C and washed with 1ml lysis buffer. The centrifugation and wash steps were repeated 3 times. After the final centrifugation, the supernatant was removed and the pellet was resuspended in 25ul protein loading buffer (10% glycerol, 5% β-mercaptoethanol, 2% SDS, 50uM Tris pH 6.8 and 0.02% bromophenol blue). The samples were boiled, resolved by SDS-PAGE and transferred to nitrocellulose membranes. To identify activated c-Kit the blots were probed with rabbit anti-phospho-cKit (Tyr 568/570) antibodies (Santa Cruz Biotech.) followed by secondary horseradish peroxidase (HRP) conjugated anti-rabbit antibody. Immune complexes were detected using an ECL Advance kit (GE Healthcare, Pittsburgh, PA). To detect total c-Kit the phospho-c-Kit antibodies were removed by soaking the immunoblots in strip buffer (100mM 2-Mercaptoethanol, 2% SDS, 62.5mM Tris pH 6.7) at 50°C for 30min. The blots were re-probed with a rabbit anti-cKit (H300) antibody (Santa Cruz Biotech.). For detection of the phosphorylated c-Kit signaling molecules (phospho-Erk 1/2 and phospho-GSK-3), direct immunoblots with the following antibodies were used; mouse anti-phospho-p44/42 Map kinase (Thr202/Tyr204) (E10) and rabbit anti-phospho-GSK-3 α/β (Ser21/9). For detection of total Erk 1/2 and GSK-3 levels, the phospho immunoblots were striped and re-probed with rabbit anti-p44/42 MAP kinase antibody and rabbit anti-GSK-3β antibody, respectively. All antibodies to signaling molecules were obtained from Cell Signaling Tech. (Danvers, MA).
For kinase inhibition, EC that had been previously starved for 24 hours were incubated in the following inhibitors or the equivalent amount of dimethyl sulfoxide (DMSO) for 30min prior to SCF stimulation; 4-amino-5-(4-methylphenyl)-7-(t-butyl)pyrazolo[3,4-d]pyrimidine (PP1), 2’-amino-3’methoxyflavone (PD98059), 2-(4-morpholinyl)-8-phenyl-4H-1-benzopyran-4-one (LY294002), 5-iodo-3-[(3,5-dibromo-4-hydroxyphenyl)methylene]-2-indolinone (raf1 kinase inhibitor I) (Biosource, Camarillo, CA) and the 2-phenylaminopyrimidine derivative STI 571 (Gleevec) provided by Elisabeth Buchdunger (Novartis, Basel, Switzerland). All compounds were dissolved in DMSO to make 10mM stocks.
Acknowledgments
This research was supported by National Institutes of Health Grants CA099906 and RR00163. This manuscript is the sole responsibility of the authors and does not necessarily represent the official views of the NIH. Funding for this research was also provided by a grant from the Collins Medical Trust (J.D.).
The authors would like to thank Jean Gustin for his critical review of the manuscript. They would also like to thank Margaret Offermann for the T1H6 cell line, Bala Chandran for the ORF73 and ORF59 antibodies and Jeff Vieira for the rKSHV.219 virus.
References
- Adams RH, Alitalo K. Molecular regulation of angiogenesis and lymphangiogenesis. Nat Rev Mol Cell Biol. 2007;8(6):464–78. doi: 10.1038/nrm2183. [DOI] [PubMed] [Google Scholar]
- Beckstead JH, Wood GS, Fletcher V. Evidence for the origin of Kaposi’s sarcoma from lymphatic endothelium. Am J Pathol. 1985;119(2):294–300. [PMC free article] [PubMed] [Google Scholar]
- Broudy VC, Kovach NL, Bennett LG, Lin N, Jacobsen FW, Kidd PG. Human umbilical vein endothelial cells display high-affinity c-kit receptors and produce a soluble form of the c-kit receptor. Blood. 1994;83(8):2145–52. [PubMed] [Google Scholar]
- Dyson N, Howley PM, Munger K, Harlow E. The human papilloma virus-16 E7 oncoprotein is able to bind to the retinoblastoma gene product. Science. 1989;243(4893):934–7. doi: 10.1126/science.2537532. [DOI] [PubMed] [Google Scholar]
- Ganem D. KSHV infection and the pathogenesis of Kaposi’s sarcoma. Annu Rev Pathol. 2006;1:273–96. doi: 10.1146/annurev.pathol.1.110304.100133. [DOI] [PubMed] [Google Scholar]
- Gill PS, Tsai YC, Rao AP, Spruck CH, 3rd, Zheng T, Harrington WA, Jr, Cheung T, Nathwani B, Jones PA. Evidence for multiclonality in multicentric Kaposi’s sarcoma. Proc Natl Acad Sci U S A. 1998;95(14):8257–61. doi: 10.1073/pnas.95.14.8257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grossmann C, Podgrabinska S, Skobe M, Ganem D. Activation of NF-kappaB by the latent vFLIP gene of Kaposi’s sarcoma-associated herpesvirus is required for the spindle shape of virus-infected endothelial cells and contributes to their proinflammatory phenotype. J Virol. 2006;80(14):7179–85. doi: 10.1128/JVI.01603-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herndier B, Ganem D. The biology of Kaposi’s sarcoma. Cancer Treat Res. 2001;104:89–126. doi: 10.1007/978-1-4615-1601-9_4. [DOI] [PubMed] [Google Scholar]
- Hong YK, Foreman K, Shin JW, Hirakawa S, Curry CL, Sage DR, Libermann T, Dezube BJ, Fingeroth JD, Detmar M. Lymphatic reprogramming of blood vascular endothelium by Kaposi sarcoma-associated herpesvirus. Nat Genet. 2004;36(7):683–5. doi: 10.1038/ng1383. [DOI] [PubMed] [Google Scholar]
- Hsu YR, Wu GM, Mendiaz EA, Syed R, Wypych J, Toso R, Mann MB, Boone TC, Narhi LO, Lu HS, Langley KE. The majority of stem cell factor exists as monomer under physiological conditions. Implications for dimerization mediating biological activity. J Biol Chem. 1997;272(10):6406–15. doi: 10.1074/jbc.272.10.6406. [DOI] [PubMed] [Google Scholar]
- Inoue N, Winter J, Lal RB, Offermann MK, Koyano S. Characterization of entry mechanisms of human herpesvirus 8 by using an Rta-dependent reporter cell line. J Virol. 2003;77(14):8147–52. doi: 10.1128/JVI.77.14.8147-8152.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jussila L, Valtola R, Partanen TA, Salven P, Heikkila P, Matikainen MT, Renkonen R, Kaipainen A, Detmar M, Tschachler E, Alitalo R, Alitalo K. Lymphatic endothelium and Kaposi’s sarcoma spindle cells detected by antibodies against the vascular endothelial growth factor receptor-3. Cancer Res. 1998;58(8):1599–604. [PubMed] [Google Scholar]
- Kahn HJ, Bailey D, Marks A. Monoclonal antibody D2-40, a new marker of lymphatic endothelium, reacts with Kaposi’s sarcoma and a subset of angiosarcomas. Mod Pathol. 2002;15(4):434–40. doi: 10.1038/modpathol.3880543. [DOI] [PubMed] [Google Scholar]
- Koon HB, Bubley GJ, Pantanowitz L, Masiello D, Smith B, Crosby K, Proper J, Weeden W, Miller TE, Chatis P, Egorin MJ, Tahan SR, Dezube BJ. Imatinib-induced regression of AIDS-related Kaposi’s sarcoma. J Clin Oncol. 2005;23(5):982–9. doi: 10.1200/JCO.2005.06.079. [DOI] [PubMed] [Google Scholar]
- Lennartsson J, Jelacic T, Linnekin D, Shivakrupa R. Normal and oncogenic forms of the receptor tyrosine kinase kit. Stem Cells. 2005a;23(1):16–43. doi: 10.1634/stemcells.2004-0117. [DOI] [PubMed] [Google Scholar]
- Lennartsson J, Voytyuk O, Heiss E, Sundberg C, Sun J, Ronnstrand L. C-Kit signal transduction and involvement in cancer. Cancer Therapy. 2005b;3:5–28. [Google Scholar]
- McCormick C, Ganem D. The kaposin B protein of KSHV activates the p38/MK2 pathway and stabilizes cytokine mRNAs. Science. 2005;307(5710):739–41. doi: 10.1126/science.1105779. [DOI] [PubMed] [Google Scholar]
- Moses AV, Fish KN, Ruhl R, Smith PP, Strussenberg JG, Zhu L, Chandran B, Nelson JA. Long-term infection and transformation of dermal microvascular endothelial cells by human herpesvirus 8. J Virol. 1999;73(8):6892–902. doi: 10.1128/jvi.73.8.6892-6902.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moses AV, Jarvis MA, Raggo C, Bell YC, Ruhl R, Luukkonen BG, Griffith DJ, Wait CL, Druker BJ, Heinrich MC, Nelson JA, Fruh K. A functional genomics approach to Kaposi’s sarcoma. Ann N Y Acad Sci. 2002a;975:180–91. doi: 10.1111/j.1749-6632.2002.tb05951.x. [DOI] [PubMed] [Google Scholar]
- Moses AV, Jarvis MA, Raggo C, Bell YC, Ruhl R, Luukkonen BG, Griffith DJ, Wait CL, Druker BJ, Heinrich MC, Nelson JA, Fruh K. Kaposi’s sarcoma-associated herpesvirus-induced upregulation of the c-kit proto-oncogene, as identified by gene expression profiling, is essential for the transformation of endothelial cells. J Virol. 2002b;76(16):8383–99. doi: 10.1128/JVI.76.16.8383-8399.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nadimi H, Saatee S, Armin A, Toto PD. Expression of endothelial cell markers PAL-E and EN-4 and Ia-antigens in Kaposi’s sarcoma. J Oral Pathol. 1988;17(8):416–20. doi: 10.1111/j.1600-0714.1988.tb01307.x. [DOI] [PubMed] [Google Scholar]
- Nickoloff BJ. The human progenitor cell antigen (CD34) is localized on endothelial cells, dermal dendritic cells, and perifollicular cells in formalin-fixed normal skin, and on proliferating endothelial cells and stromal spindle-shaped cells in Kaposi’s sarcoma. Arch Dermatol. 1991;127(4):523–9. [PubMed] [Google Scholar]
- Nickoloff BJ. PECAM-1 (CD31) is expressed on proliferating endothelial cells, stromal spindle-shaped cells, and dermal dendrocytes in Kaposi’s sarcoma. Arch Dermatol. 1993;129(2):250–1. [PubMed] [Google Scholar]
- Pantanowitz L, Dezube BJ, Pinkus GS, Tahan SR. Histological characterization of regression in acquired immunodeficiency syndrome-related Kaposi’s sarcoma. J Cutan Pathol. 2004;31(1):26–34. doi: 10.1046/j.0303-6987.2004.0132.x. [DOI] [PubMed] [Google Scholar]
- Pantanowitz L, Schwartz EJ, Dezube BJ, Kohler S, Dorfman RF, Tahan SR. C-Kit (CD117) expression in AIDS-related, classic, and African endemic Kaposi sarcoma. Appl Immunohistochem Mol Morphol. 2005;13(2):162–6. doi: 10.1097/00129039-200506000-00009. [DOI] [PubMed] [Google Scholar]
- Phelps WC, Yee CL, Munger K, Howley PM. The human papillomavirus type 16 E7 gene encodes transactivation and transformation functions similar to those of adenovirus E1A. Cell. 1988;53(4):539–47. doi: 10.1016/0092-8674(88)90570-3. [DOI] [PubMed] [Google Scholar]
- Riou G, Barrois M, Sheng ZM, Duvillard P, Lhomme C. Somatic deletions and mutations of c-Ha-ras gene in human cervical cancers. Oncogene. 1988;3(3):329–33. [PubMed] [Google Scholar]
- Scheffner M, Werness BA, Huibregtse JM, Levine AJ, Howley PM. The E6 oncoprotein encoded by human papillomavirus types 16 and 18 promotes the degradation of p53. Cell. 1990;63(6):1129–36. doi: 10.1016/0092-8674(90)90409-8. [DOI] [PubMed] [Google Scholar]
- Sharma-Walia N, Krishnan HH, Naranatt PP, Zeng L, Smith MS, Chandran B. ERK1/2 and MEK1/2 induced by Kaposi’s sarcoma-associated herpesvirus (human herpesvirus 8) early during infection of target cells are essential for expression of viral genes and for establishment of infection. J Virol. 2005;79(16):10308–29. doi: 10.1128/JVI.79.16.10308-10329.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uccini S, Ruco LP, Monardo F, Stoppacciaro A, Dejana E, La Parola IL, Cerimele D, Baroni CD. Co-expression of endothelial cell and macrophage antigens in Kaposi’s sarcoma cells. J Pathol. 1994;173(1):23–31. doi: 10.1002/path.1711730105. [DOI] [PubMed] [Google Scholar]
- Uccini S, Sirianni MC, Vincenzi L, Topino S, Stoppacciaro A, Lesnoni La Parola I, Capuano M, Masini C, Cerimele D, Cella M, Lanzavecchia A, Allavena P, Mantovani A, Baroni CD, Ruco LP. Kaposi’s sarcoma cells express the macrophage-associated antigen mannose receptor and develop in peripheral blood cultures of Kaposi’s sarcoma patients. Am J Pathol. 1997;150(3):929–38. [PMC free article] [PubMed] [Google Scholar]
- Vieira J, O’Hearn PM. Use of the red fluorescent protein as a marker of Kaposi’s sarcoma-associated herpesvirus lytic gene expression. Virology. 2004;325(2):225–40. doi: 10.1016/j.virol.2004.03.049. [DOI] [PubMed] [Google Scholar]
- Wandzioch E, Edling CE, Palmer RH, Carlsson L, Hallberg B. Activation of the MAP kinase pathway by c-Kit is PI-3 kinase dependent in hematopoietic progenitor/stem cell lines. Blood. 2004;104(1):51–7. doi: 10.1182/blood-2003-07-2554. [DOI] [PubMed] [Google Scholar]
- Wang HW, Trotter MW, Lagos D, Bourboulia D, Henderson S, Makinen T, Elliman S, Flanagan AM, Alitalo K, Boshoff C. Kaposi sarcoma herpesvirus-induced cellular reprogramming contributes to the lymphatic endothelial gene expression in Kaposi sarcoma. Nat Genet. 2004;36(7):687–93. doi: 10.1038/ng1384. [DOI] [PubMed] [Google Scholar]
- Weninger W, Partanen TA, Breiteneder-Geleff S, Mayer C, Kowalski H, Mildner M, Pammer J, Sturzl M, Kerjaschki D, Alitalo K, Tschachler E. Expression of vascular endothelial growth factor receptor-3 and podoplanin suggests a lymphatic endothelial cell origin of Kaposi’s sarcoma tumor cells. Lab Invest. 1999;79(2):243–51. [PubMed] [Google Scholar]
- Xu H, Edwards JR, Espinosa O, Banerji S, Jackson DG, Athanasou NA. Expression of a lymphatic endothelial cell marker in benign and malignant vascular tumors. Hum Pathol. 2004;35(7):857–61. doi: 10.1016/j.humpath.2004.02.009. [DOI] [PubMed] [Google Scholar]