

Abstract
Cellular reprogramming and generation of induced pluripotent stem cells (iPSCs) from adult cell types has enabled the creation of patient-specific stem cells for use in disease modeling. To date, many iPSC lines have been generated from a variety of disorders, which have then been differentiated into disease-relevant cell types. When a disease-specific phenotype is detectable in such differentiated cells, the reprogramming technology provides a new opportunity to identify aberrant disease-associated pathways and drugs that can block them. Here, we highlight recent progress as well as limitations in the use of iPSCs to recapitulate disease phenotypes and to screen for therapeutics in vitro.
Introduction
Understanding the molecular basis of many diseases have been hampered by the lack of appropriate in vitro cell culture models that accurately reflect the disease phenotypes. The availability of such models is also crucial for the discovery and development of therapeutics. However, primary human cells and especially disease-bearing ones are difficult to obtain and propagate in culture for extended periods of time. Since their discovery by Takahashi and Yamanaka and application to the human context, induced pluripotent stem cells (iPSCs) have emerged as a way to generate disease-specific cell types [1–5]. With this technology, somatic cells isolated from patients (in most cases fibroblasts from skin biopsies), can be reprogrammed into a pluripotent state by overexpressing four transcription factors (Oct4, Sox2, Klf4 and c-Myc)[1]. The resulting iPSCs can be expanded in culture virtually indefinitely, and then be differentiated into cell types of interest.
The first disease-specific iPSCs were derived from patients with familial amyotrophic lateral sclerosis (ALS) and a number of genetic diseases with either Mendelian or complex inheritance [6,7]. Since then an ever-growing number of disease-specific iPSCs are being generated from primary cell samples from patients afflicted with a variety of genetically inherited and sporadic diseases affecting virtually every organ system (Table 1). In general, patient-specific iPSCs are then differentiated to the key cell types that are affected in the disease in question. The next step is the identification of disease-associated phenotypes that are readily detectable by molecular and/or cellular assays. Once a robust assay is identified, larger scale screens can be undertaken to discover key disease pathways and therapeutics (Figure 1).
Table 1. Summary of published human disease-specific iPSCs.
| Disease | Molecular defect | Disease phenotype observed / References |
|---|---|---|
| ADA-severe combined immunodeficiency | Mutations in ADA | Not determined [7]. |
| Adrenoleukodystrophy, X-linked | Mutation in ABCD1 | Very long chain fatty acid accumulation in oligodendrocytes [50]. |
| Alzheimer's disease, familial | Mutation in PS1 or PS2, Duplication of APP | Increased amyloid β42 secretion[51]. Higher levels of the pathological markers amyloid-β(1–40), phospho-tau and active glycogen synthase kinase-3β [52]. |
| Alzheimer's disease, sporadic | Unknown | Higher levels of the pathological markers in one out of two patient-derived lines [52]. |
| Amyotrophic lateral sclerosis (ALS), familial | Mutation in SOD1 or VAPB | Not determined [6]. VAPB protein levels are reduced in ALS8-derived motor neurons [53]. |
| Angelman syndrome | Loss of maternal UBE3A allele | Reduced UBE3A expression upon neuronal differentiation [28]. |
| Atypical Werner syndrome | Mutation in LMNA | Alterations in the nuclear membrane, slow proliferation, senescence [54]. |
| α1-antitrypsin deficiency | Mutation in α1-antitrypsin | Loss of α1-antitrypsin expression [10][55]. |
| Becker muscular dystrophy | Mutation in DMD | Not determined [7]. |
| β-thalassaemia | Deletion in β-globin gene | Not determined [56] [57]. |
| Chronic granulomatous, X-linked | CYBB deficiency | Neutrophils differentiated from X-CGD iPSCs lack ROS production [44]. |
| Crigler–Najjar syndrome | Mutation in UGT1A1 | Not determined [58]. |
| Cystic fibrosis | Mutations in CFTR | Not determined [55]. |
| Diabetes, Type1 | Multifactorial; unknown | Not determined [7][59]. |
| Down syndrome | Trisomy 21 | Not determined [7]. |
| Dyskeratosis congenita | Mutations in DKC1, TERT or TCAB1 | Increased expression of telomerase components [30]. Progressive telomere shortening, loss of self-renewal [31]. |
| Duchenne muscular dystrophy | Mutation in DMD | Not determined [7]. Loss of dystrophin expression in iPSC-derived muscle tissue [13]. |
| Dystrophic epidermolysis bullosa | Mutations in COL7A1 | Lack of expression of type VII collagen [60][61]. |
| Emanuel Syndrome | Partial trisomy 11;22 | Not determined [62] |
| Fanconi Anemia | FANCA or FANCC deficiency | Corrected loss of FANCA function [20][21]. |
| Familial dysautonomia | Mutation in IKBKAP | Defects in neural crest differentiation migration [17]. |
| Familial hypercholesterolaemia | Mutation in gene encoding LDL receptor | Reduced ability to incorporate LDL in iPSC-derived hepatoctyes [10]. |
| Fragile X syndrome | Trinucleotide (CGG) expansion, silencing of FMR1 | Not determined [27][29]. |
| Friedreich's ataxia (FRDA) | Trinucleotide GAA repeat expansion in FXN | Reduced FXN mRNA expression [63]. |
| Gaucher's disease | Mutation in β-glucocerebrosidase | Impaired lysosomal protein degradation, accumulation of α-synuclein, and neurotoxicity [64]. |
| Glycogen storage disease type 1A | Deficiency in glucose-6-phosphate | Hyperaccumulation of glycogen [10] [58]. |
| Gyrate atrophy | Mutation in OAT | Not determined [65]. |
| Hereditary tyrosinaemia type 1 | Mutation in FAH | Not determined [58]. |
| Huntington's disease | Trinucleotide expansion in huntingtin gene | Not determined [7][66]. |
| Hutchinson–Gilford progeria syndrome | Mutation in LMNA | Increased senescence, progerin accumulation, DNA damage, nuclear abnormalities [67][68][54]. |
| Inherited dilated cardiomyopathy | Mutation in LMNA | Increased senescence and apoptosis in iPSC-derived fibroblasts, nuclear abnormalities [54]. |
| LEOPARD Syndrome | Mutation in PTPN11 | Increased cardiomyocyte size, decreased MAPK signaling [69]. |
| Lesch–Nyhan syndrome (carrier) | Heterozygous for HPRT1 | Not determined [7]. |
| Long QT syndromes 1 and 2 | Mutations in KCNQ1 or KCNH2 | Increased cardiomyocyte depolarization [11][18] |
| MPS type I (Hurler syndrome) | IDUA deficiency | Not determined [70]. |
| MPS type IIIB | α-N-acetylglucosaminidase deficiency | Defects in storage vesicles and Golgi apparatus [71]. |
| Osteogenesis imperfect | Mutations in COL1A1 or COL1A2 | Not determined [72] |
| Parkinson's disease, sporadic | Unknown | Not determined [7][15][16][73]. |
| Parkinson's disease, familial | Mutations in LRRK2 or PINK1 or triplication of SNCA | Sensitivity to oxidative stress in LRRK2-mutant neurons[74]. Impaired mitochondrial function in PINK1-mutant dopaminergic neurons[75]. Increased α-synuclein protein in neurons[76]. |
| Patau syndrome | Trisomy 13 | Not determined [62]. |
| Polycythaemia vera | Mutation in JAK2 | Enhanced erythropoiesis [77] |
| Pompe disease | Mutations in GAA | High levels of glycogen and defective cellular respiration in iPSC-derived cardiomycyte-like cells [78]. |
| Prader-Willi syndrome | Paternal deletion of 15q11-q13 | Imprint disorder [79][28]. |
| Progressive familial hereditary cholestasis | Unknown | Not determined [58]. |
| Retinitis pigmentosa | Mutations in RP1, RP9, PRPH2 or RHO | Decreased numbers of differentiated rod cells [80]. |
| Rett syndrome | Mutation in MECP2 | Decreased synapse number, reduced spine density [9][81][82]. |
| Schizophrenia | Unknown | Decreased neuronal connectivity, neurite number, and PSD95 and glutamate receptor expression [19]. |
| Scleroderma | Unknown | Not determined [55]. |
| Shwachman–Bodian– Diamond syndrome | Mutations in SBDS | Not determined [7]. |
| Sickle cell anaemia | Mutation in β-globin gene | Not determined [55][46]. |
| Spinal muscular atrophy (SMA) | Mutations in SMN1 | Loss of SMN gene expression, reduced size and number of motor neurons [8]. |
| Spinocerebellar ataxia type 3 | Trinucleotide expansion in MJD1 gene | Neuron-specific formation of SDS-insoluble aggregates [14]. |
| Timothy Syndrome | Mutations in CACNA1C | Increased cardiomyocyte depolarization [12]. |
| Turner Syndrome | Monosomy X | Not determined [62]. |
| Wilson's disease | Mutations in ATP7B | Defective copper transport in iPSC-derived hepatocyte-like cells [83]. |
| Warkany Syndrome 2 | Trisomy 8 | Not determined [62]. |
Figure 1. Disease modeling and drug discovery using patient-derived iPSCs.
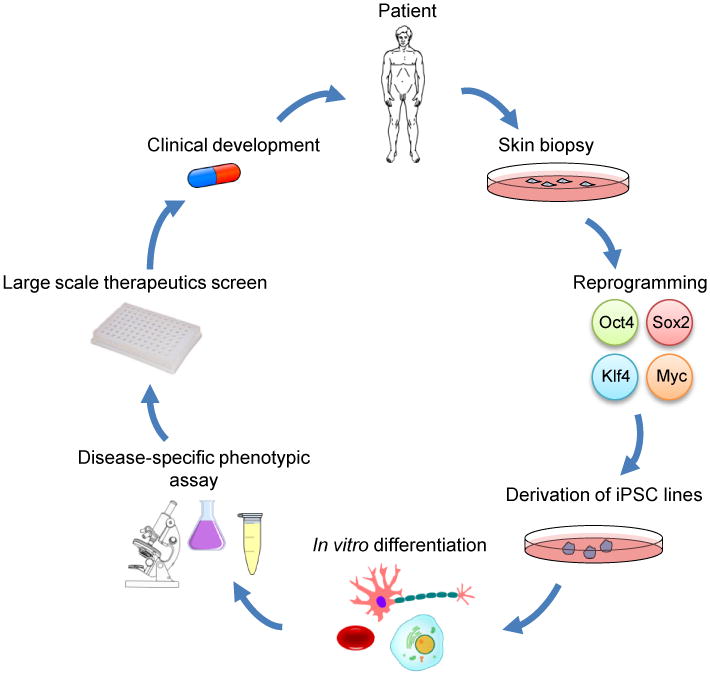
Generation of an iPSC-based disease model starts with cells isolated from patients, usually by a skin punch biopsy. Upon reprogramming, several iPSC clones are selected, expanded and characterized. High-quality iPSCs are then differentiated into mature cell types exhibiting a disease-specific phenotype that is readily detected by cellular and/or molecular assays. High-throughput screens based on such assays can be carried out to discover therapeutics that reverse the disease phenotypes. Hits from these screens are candidates for lead optimization by medicinal chemistry, and then further preclinical studies.
In this review we will highlight some of the salient themes that emerge from studies reported to date and point out some of the promises and limitations of using iPSCs in disease modeling.
Identification of disease relevant cellular phenotypes
For iPSC-based disease modeling to be meaningful, a relevant cellular or molecular phenotype must be observable in either the derived iPSCs or their differentiated progeny. Monogenic diseases affecting a specific cell type that can efficiently be derived from pluripotent stem cells are the best candidates for this approach. Some examples include neurological diseases such as spinal muscular atrophy (SMA) and Rett syndrome, metabolic diseases such as α1-antitrypsin deficiency, familial hypercholesterolemia and glycogen storage disease type 1A, and cardiovascular diseases such as Timothy syndrome and Type 1 and 2 Long QT syndrome. Upon differentiation into disease-relevant cell types, most of these iPSCs manifest an observable phenotype. SMA-specific iPSCs displayed progressive loss of motor neurons upon differentiation, which may be similar to the developmental loss of this cell type during disease progression[8]. Neurons derived from Rett syndrome-specific iPSCs had reduced spine density, smaller soma size and altered calcium signaling when compared to controls[9]. For the metabolic diseases mentioned above, differentiation of iPSCs into hepatocyte-like cells resulted in several phenotypes including aggregation of misfolded α1-antitrypsin in the endoplasmic reticulum, deficient LDL receptor-mediated cholesterol uptake, and elevated lipid and glycogen accumulation[10]. In the modeling of cardiovascular diseases, disease-specific phenotypes manifest themselves in the differentiated cardiomyocytes such as the prolonged action potentials of Long QT and Timothy syndrome-specific cells [11,12]. In certain cases, the disease phenotype could also be observed upon spontaneous differentiation of iPSCs in teratoma assays, as recently shown for the absence of dystrophin expression in muscle-like tissues derived from Duchenne muscular dystrophy-specific iPSCs [13].
A recent study by Brustle and colleagues highlights some of the advantages of iPSCs-based disease modeling. Spinocerebellar ataxia (also called Machado-Joseph disease) is a late-onset neurodegenerative disease caused by the expansion of polyglutamine (CAG) repeats in the MJD1 gene. Upon proteolytic cleavage, the MJD1 gene product ataxin3 (ATXN3) is thought to cause SDS-insoluble protein aggregates which then have a critical role in neurodegeneration. To explore the role of neuron specific proteolysis in initiating the disease process, Koch et al. first derived iPSCs from MJD patients and showed that the expanded MJD allele was expressed in both the pluripotent cells and their differentiated neuron-like progeny[14]. They then showed that glutamate-induced excitation of these differentiated cells initiates Ca2+ - dependent proteolysis of ATXN3, which is followed by aggregate formation. Interestingly, this observed phenotype could be abrogated by the inhibition of Ca2+ -dependent calpain proteases. This cell model manifest a neuron-specific phenotype; aggregate formation was not observed in the iPSCs, fibroblasts or glia, thus providing an explanation for the neuron-specific manifestation of the disease phenotype[14]. Although the molecular mechanisms linking microaggregate formation to late-stage neurodegeneration remains elusive, this study shows how aspects of a late-onset disease can also be modeled in a cell-type specific manner.
Unfortunately, there have been numerous studies in which disease-specific iPSCs were generated but no phenotype has been assessed or observed (Table 1). For some diseases, this is most likely due to lack of suitable differentiation protocols to the affected cell type and/or specific assays to detect the molecular defect. For example, modeling hematological disorders such as sickle-cell anemia or β-Thalassemia, and ADA-SCID has been hampered by a lack of pure in vitro derivation protocols for the specific blood lineages most relevant to these conditions (beta-globin expressing adult red blood cells, and T cells, respectively). In another example, iPSCs generated from sporadic Parkinson's disease patients could in fact be efficiently differentiated into dopaminergic neurons, but the resulting cells did not show significant differences in various in vitro assays, when compared to control iPSC-derived cells [15,16]. It remains to be seen whether other late-onset diseases that have complex environmental and genetic causes will yield detectable in vitro phenotypes.
Disease-specific iPSC-derived cells as discovery and screening platforms
When a robust disease phenotype is identified in patient-specific iPSCs or their differentiated progeny, such cells can then be used to discover new therapeutic compounds or test existing candidates. One of the first demonstrations that disease-specific iPSCs can be used to test therapeutic compounds came from iPSCs generated from Familial dysautonomia (FD), a rare genetic disorder affecting sensory and autonomic neurons due to mutations in the I-κ-B kinase complex-associated protein (IKBKAP) gene. These mutations result in a tissue-specific splicing defect that lowers the levels of IKAP protein. Lee et al. derived iPSCs from a FD patient which they then differentiated into neural precursors cells that exhibited three FD-associated phenotypes: defective IKBKAP splicing, decreased rate of neurogenesis, and reduced migration[17]. Such iPSC-derived cells were then used to test a number of candidate compounds for their ability to affect any of these phenotypes. One compound, kinetin, could partially reverse the aberrant splicing and the neurogenic differentiation and migration defects[17].
A growing number of disease-specific IPSC-derived cells are being used to test candidate small molecules. For example, motor neuron survival defects observed in SMA-specific cells were partially abrogated by the application of two compounds, valproic acid and tobramycin, which increased the level of SMN protein in the patient-derived iPSCs [8]. In Rett-iPSC derived neurons, IGF1 treatment increased synapse formation[9]. In Long QT syndromes, cardiomyocyte-specific defects could be attenuated by beta-blockers and several ion channel blockers [11,18]. The antipsychotic loxapine has been shown to ameliorate the diminished neuronal connectivity and decreased neurite number phenotype of Schizophrenia-iPSC-derived neurons[19]. All in all, these examples show that the stage is set for the use of disease-specific iPSC-derived cells to be used in larger screens aimed to discover new therapeutic agents that will block the disease-associated phenotypes. Given the disease-relevance of human iPSC-based drug screens, there is considerable hope for therapeutic development.
Limitations of iPSCs
As enumerated above, iPSC-based disease modeling offers significant potential in drug discovery. However, as it currently stands, there are also limitations to this technology, which may preclude its use for some types of diseases. Certain genetic lesions inhibit or may even preclude the derivation of iPSCs from patients by interfering with the reprogramming process itself. A case in point comes from the attempt to model Fanconi anaemia (FA), the most common genetic bone marrow failure syndrome that is caused by recessive autosomal or X-linked mutation in one of 13 genes in the FA pathway. This pathway plays an important role in DNA repair, and it was recently shown that reprogramming leads to activation of the FA pathway, increased DNA double strand breaks and senescence [20]. Initial attempts to reprogram FA fibroblasts were unsuccessful unless the genetic defect was first corrected with a viral vector [21]. Therefore it was concluded that restoration of the FA pathway is a prerequisite for iPSC generation from FA patients [21]. Subsequent work has shown that it is possible to reprogram FA fibroblasts, albeit at a very low efficiency [20]. Nevertheless, these studies suggest that in cases where the diseases caused by mutations that affect DNA repair and senescence pathways, it may be difficult to generate iPSCs from patient cells. It should be noted that a number of small molecules have been shown to improve reprogramming efficiency of human fibroblasts and could therefore prove useful in such cases [22–26].
Another example of the limitation of iPSC-based disease modeling comes from studies of Fragile X syndrome, the most common form of inherited mental retardation. Fragile X is caused by silencing of the FMR1 gene due to CGG triplet expansion in its 5′UTR, which then results in aberrant DNA methylation and accumulation of repressive histone marks. Urbach et al. successfully derived iPSC lines from Fragile X patients, and observed that the FMR1 gene remained inactive and was thus refractory to factor-based epigenetic reprogramming [27]. This contrasted to the active FMR1 gene expression and down-regulation following differentiation in embryonic stem cell (ESC) lines derived from embryos affected by Fragile X [27]. Thus, in this context, Fragile X-iPSCs could not model the silencing of the FMR1 gene that occurs during development, but ESC lines could. There is also evidence indicating that other epigenetic disorders such as imprinting defects are not reset by somatic cell reprogramming [28]. Sheridan et al. recently derived additional Fragile X iPSCs and observed clonal variation among the iPSC lines with respect to CGG-repeat length, CpG methylation and expression levels of FMR1 suggesting that iPSCs may not faithfully reproduce these phenotypes present in the original fibroblasts [29]. Furthermore, in one case, the patient derived fibroblast line was a heterogeneous mixture of normal and expanded-repeat cells, and this line yielded individual iPSC clones bearing either the normal or expanded-repeat lengths, suggesting that reprogramming can capture single target cells with variable properties [29]. This point highlights the clone-to-clone variability that can confound and complicate disease models while also being potentially useful as a platform for line-to-line comparison from specific individuals.
Whether clonal variation among different iPSC lines from patients influences the observed disease phenotype remains an important concern. Agarwal et al. and Batista et al. both reprogrammed fibroblasts from patients afflicted with dyskeratosis congenita (DC), a premature aging syndrome caused by mutations in telomerase components [30,31]. While both groups observed increases in expression of multiple members of the core telomerase machinery in the reprogrammed iPS cells, there was clonal variation with respect to maintenance of telomere length. Agarwal et al. observed that some iPSCs recovered enough telomerase activity to regrow telomeres, whereas in the study by Batista et al. DC-specific iPSCs continued to show telomere decay [32]. Given these different outcomes, one must remain wary of the clonal variation that results from the technical inefficiency and infidelity of reprogramming when iPSC-based disease models are generated. In general, the number and location of viral integrations, heterogeneity in the starting somatic cell populations, passage numbers and culture conditions as well as genetic alterations introduced during reprogramming are all likely to contribute to the clonal variability among iPSC lines[33][34]. In addition, there is growing evidence that epigenetic remodeling can be incomplete, thus influencing the consistency of disease phenotypes [35-39]. Therefore, multiple clones from multiple patients will most likely need to assessed before a truly representative and faithfully reprogrammed iPSC-based disease model is established.
One potential way to overcome the variability among disease-specific iPSC lines, especially with respect to monogenic diseases, is to generate genetically defined sibling cell lines. Engineered zinc finger nucleases (ZFNs) and transcription activator-like effector nucleases (TALENs) have emerged as important tools to achieve “genome editing” in pluripotent stem cells [40-42]. This technology can be used to correct the disease-specific mutations by gene targeting or generate de novo disease-specific mutations in wild-type lines. This approach has recently been to used to model susceptibility variants for familial Parkinson's disease by genetically modifying single base pairs in the alpha-synuclein gene[43]. Genome editing has also been employed for X-linked chronic granulomatous disease where a wild-type gene has been inserted into a ZFN-induced break induced at a potential “safe harbor” locus in iPSCs and for sickle-cell anemia in which case correction of mutations in the beta-globin gene has been achieved by either ZFNs or by homologous recombination [44-46]. Such panels of isogenic disease and control cell lines that differ only at precise disease-associated loci are going to be useful in studying the effects of the mutations in question.
Modeling complex diseases using iPSCs will be especially challenging. First of all, robust differentiation protocols for disease-relevant cell types may be lacking. Modeling of diabetes would be greatly facilitated by the discovery of reliable differentiation methods for generating pancreatic β-cells in vitro from iPSCs. Although progress has been made on this front, routine production of β-cells from diabetic patients to be used in genetic and drug screens are still some time away [47]. In addition, certain disease-associated phenotypes might arise from non-cell autonomous interactions between two different cell types. In such cases, it will be essential to differentiate the iPSCs into multiple different cell lineages and to generate in vitro co-culture assays. For example, Di Giorgio et al. have shown that in a minority of ALS cases resulting from SOD1 mutations, neurodegeneration may be due to the non-cell autonomous effect of the glial cells on the motor neurons [48]. Finally, for late-onset diseases that have large environmental components, it will be important to identify ways by which disease latency can be shortened or environmental conditions recreated by the applications of exogenous stresses [49].
Conclusions
The growing number of studies reporting the generation of disease-specific iPSCs is a testament to the potential of this technology in drug and pathway discovery. However, it is also becoming increasingly apparent that iPSC-based disease modeling will have to incorporate more reliable protocols for directed differentiation into mature cell types in order to realize its full potential. Furthermore, more robust disease phenotypes, especially for complex and common diseases, will have to be detected and assayed. Despite the progress described above, it should be noted that all the published studies to date have tested the effects of previously reported candidate compounds on disease-specific iPSCs. As such, these studies constitute only proof-of-principle demonstrations for larger higher throughput screens to discover novel agents that can potentially reverse the disease phenotypes observed in iPSCs-derived cells. The full benefit of the iPSC platform for disease modeling and drug development, although promising, remains to be proven.
Acknowledgments
We thank Tugba Bagci-Onder for critical reading of the manuscript and graphical design. Research was funded by grants from the US National Institutes of Health (NIH) to G.Q.D., and the CHB Stem Cell Program.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
* of special interest
** of outstanding interest
- **1.Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. [Internet] Cell. 2006;126:663–76. doi: 10.1016/j.cell.2006.07.024. This is the first study to demonstrate that reprogramming of differentiated cells to a pluripotent state can be achieved by the expression of four transcription factors, Oct4, Sox2, c-Myc, and Klf4. [DOI] [PubMed] [Google Scholar]
- **2.Takahashi K, Tanabe K, Ohnuki M, Narita M, Ichisaka T, Tomoda K, Yamanaka S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. [Internet] Cell. 2007;131:861–72. doi: 10.1016/j.cell.2007.11.019. This study, along with references 3-5, proved the feasibility of reprogramming human somatic cells to a pluripotent state. [DOI] [PubMed] [Google Scholar]
- 3.Yu J, Vodyanik MA, Smuga-Otto K, Antosiewicz-Bourget J, Frane JL, Tian S, Nie J, Jonsdottir GA, Ruotti V, Stewart R, et al. Science. Vol. 318. New York, N.Y.: 2007. Induced pluripotent stem cell lines derived from human somatic cells; pp. 1917–20. Internet. [DOI] [PubMed] [Google Scholar]
- 4.Park IH, Zhao R, West Ja, Yabuuchi A, Huo H, Ince Ta, Lerou PH, Lensch MW, Daley GQ. Reprogramming of human somatic cells to pluripotency with defined factors. Nature. 2008;451:141–6. doi: 10.1038/nature06534. Internet. [DOI] [PubMed] [Google Scholar]
- 5.Lowry WE, Richter L, Yachechko R, Pyle AD, Tchieu J, Sridharan R, Clark AT, Plath K. Generation of human induced pluripotent stem cells from dermal fibroblasts. Proceedings of the National Academy of Sciences of the United States of America; 2008; pp. 2883–8. Internet. [DOI] [PMC free article] [PubMed] [Google Scholar]
- **6.Dimos JT, Rodolfa KT, Niakan KK, Weisenthal LM, Mitsumoto H, Chung W, Croft GF, Saphier G, Leibel R, Goland R, et al. Science. Vol. 321. New York, N.Y.: 2008. Induced pluripotent stem cells generated from patients with ALS can be differentiated into motor neurons. [Internet] pp. 1218–21. This study demonstrated that patient-specific iPSCs can be used to generate disease-relevant cells types, in the particular case of ALS, motor neurons. [DOI] [PubMed] [Google Scholar]
- **7.Park IH, Arora N, Huo H, Maherali N, Ahfeldt T, Shimamura A, Lensch MW, Cowan C, Hochedlinger K, Daley GQ. Disease-specific induced pluripotent stem cells. [Internet] Cell. 2008;134:877–86. doi: 10.1016/j.cell.2008.07.041. This study was the first study to describe the generation of iPSCs from patients with a variety of genetic diseases with either Mendelian or complex inheritance. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ebert AD, Yu J, Rose FF, Mattis VB, Lorson CL, Thomson JA, Svendsen CN. Induced pluripotent stem cells from a spinal muscular atrophy patient. Nature. 2009;457:277–80. doi: 10.1038/nature07677. Internet. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Marchetto MCN, Carromeu C, Acab A, Yu D, Yeo GW, Mu Y, Chen G, Gage FH, Muotri AR. A model for neural development and treatment of Rett syndrome using human induced pluripotent stem cells. Cell. 2010;143:527–39. doi: 10.1016/j.cell.2010.10.016. Internet. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rashid ST, Corbineau S, Hannan N, Marciniak SJ, Miranda E, Alexander G, Huang-Doran I, Griffin J, Ahrlund-Richter L, Skepper J, et al. Modeling inherited metabolic disorders of the liver using human induced pluripotent stem cells. The Journal of clinical investigation. 2010;120:3127–36. doi: 10.1172/JCI43122. Internet. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Moretti A, Bellin M, Welling A, Jung CB, Lam JT, Bott-Flügel L, Dorn T, Goedel A, Höhnke C, Hofmann F, et al. Patient-specific induced pluripotent stem-cell models for long-QT syndrome. The New England journal of medicine. 2010;363:1397–409. doi: 10.1056/NEJMoa0908679. Internet. [DOI] [PubMed] [Google Scholar]
- 12.Paşca SP, Portmann T, Voineagu I, Yazawa M, Shcheglovitov A, Paşca AM, Cord B, Palmer TD, Chikahisa S, Nishino S, et al. Using iPSC-derived neurons to uncover cellular phenotypes associated with Timothy syndrome. Nature medicine. 2011;17:1657–62. doi: 10.1038/nm.2576. Internet. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kazuki Y, Hiratsuka M, Takiguchi M, Osaki M, Kajitani N, Hoshiya H, Hiramatsu K, Yoshino T, Kazuki K, Ishihara C, et al. Complete genetic correction of ips cells from Duchenne muscular dystrophy. Molecular therapy: the journal of the American Society of Gene Therapy. 2010;18:386–93. doi: 10.1038/mt.2009.274. Internet. [DOI] [PMC free article] [PubMed] [Google Scholar]
- **14.Koch P, Breuer P, Peitz M, Jungverdorben J, Kesavan J, Poppe D, Doerr J, Ladewig J, Mertens J, Tüting T, et al. Excitation-induced ataxin-3 aggregation in neurons from patients with Machado-Joseph disease. [Internet] Nature. 2011;480:543–6. doi: 10.1038/nature10671. This study showed that iPSCs enable the study of aberrant protein processing associated with late-onset neurodegenerative disorders in patient-specific neurons in a cell-type specific manner. [DOI] [PubMed] [Google Scholar]
- *15.Soldner F, Hockemeyer D, Beard C, Gao Q, Bell GW, Cook EG, Hargus G, Blak A, Cooper O, Mitalipova M, et al. Parkinson's disease patient-derived induced pluripotent stem cells free of viral reprogramming factors. [Internet] Cell. 2009;136:964–77. doi: 10.1016/j.cell.2009.02.013. The authors were able to derive Transgene-free iPSCs from idiopathic Parkinson's disease patients using Cre-recombinase excisable viruses and subsequently differentiated them into dopaminergic neurons. Interestingly no disease-specific phenotype was discernible with differentiation assays. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hargus G, Cooper O, Deleidi M, Levy A, Lee K, Marlow E, Yow A, Soldner F, Hockemeyer D, Hallett PJ, et al. Differentiated Parkinson patient-derived induced pluripotent stem cells grow in the adult rodent brain and reduce motor asymmetry in Parkinsonian rats. Proceedings of the National Academy of Sciences of the United States of America; 2010; pp. 15921–6. Internet. [DOI] [PMC free article] [PubMed] [Google Scholar]
- **17.Lee G, Papapetrou EP, Kim H, Chambers SM, Tomishima MJ, Fasano CA, Ganat YM, Menon J, Shimizu F, Viale A, et al. Modelling pathogenesis and treatment of familial dysautonomia using patient-specific iPSCs. [Internet] Nature. 2009;461:402–6. doi: 10.1038/nature08320. This is the first study to use iPSCs for validating the potency of candidate drugs in a specific disease model. The authors demonstrate that the plant hormone kinetin reverses the aberrant splicing of IKBKAP gene and ameliorates the neuronal differentiation and migration of familial dysautonomia-specific neural crest precursors. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Itzhaki I, Maizels L, Huber I, Zwi-Dantsis L, Caspi O, Winterstern A, Feldman O, Gepstein A, Arbel G, Hammerman H, et al. Modelling the long QT syndrome with induced pluripotent stem cells. Nature. 2011;471:225–9. doi: 10.1038/nature09747. Internet. [DOI] [PubMed] [Google Scholar]
- *19.Brennand KJ, Simone A, Jou J, Gelboin-Burkhart C, Tran N, Sangar S, Li Y, Mu Y, Chen G, Yu D, et al. Modelling schizophrenia using human induced pluripotent stem cells. [Internet] Nature. 2011;473:221–5. doi: 10.1038/nature09915. Starting from schizophrenia patients' fibroblasts this study demonstrates that a complex genetic psychiatric disorder can be modeled using iPSCs and that disease-specific neuronal phenotypes and gene expression changes can be observed in the resulting cells. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Müller LUW, Milsom MD, Harris CE, Vyas R, Brumme KM, Parmar K, Moreau LA, Schambach A, Park IH, London WB, et al. Overcoming Reprogramming Resistance of Fanconi Anemia Cells. Blood. 2012 doi: 10.1182/blood-2012-02-408674. Internet. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Raya A, Rodríguez-Pizà I, Guenechea G, Vassena R, Navarro S, Barrero MJ, Consiglio A, Castellà M, Río P, Sleep E, et al. Disease-corrected haematopoietic progenitors from Fanconi anaemia induced pluripotent stem cells. Nature. 2009;460:53–9. doi: 10.1038/nature08129. Internet. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Huangfu D, Maehr R, Guo W, Eijkelenboom A, Snitow M, Chen AE, Melton Da. Induction of pluripotent stem cells by defined factors is greatly improved by small-molecule compounds. Nature biotechnology. 2008;26:795–7. doi: 10.1038/nbt1418. Internet. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mali P, Chou BK, Yen J, Ye Z, Zou J, Dowey S, Brodsky RA, Ohm JE, Yu W, Baylin SB, et al. Stem cells. Vol. 28. Dayton, Ohio: 2010. Butyrate greatly enhances derivation of human induced pluripotent stem cells by promoting epigenetic remodeling and the expression of pluripotency-associated genes; pp. 713–20. Internet. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li W, Zhou H, Abujarour R, Zhu S, Young Joo J, Lin T, Hao E, Schöler HR, Hayek A, Ding S. Stem cells. Vol. 27. Dayton, Ohio: 2009. Generation of human-induced pluripotent stem cells in the absence of exogenous Sox2; pp. 2992–3000. Internet. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Esteban MA, Wang T, Qin B, Yang J, Qin D, Cai J, Li W, Weng Z, Chen J, Ni S, et al. Vitamin C enhances the generation of mouse and human induced pluripotent stem cells. Cell stem cell. 2010;6:71–9. doi: 10.1016/j.stem.2009.12.001. Internet. [DOI] [PubMed] [Google Scholar]
- 26.Onder TT, Kara N, Cherry A, Sinha AU, Zhu N, Bernt KM, Cahan P, Mancarci OB, Unternaehrer J, Gupta PB, et al. Chromatin-modifying enzymes as modulators of reprogramming. Nature. 2012;483:598–602. doi: 10.1038/nature10953. Internet. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *27.Urbach A, Bar-Nur O, Daley GQ, Benvenisty N. Differential modeling of fragile X syndrome by human embryonic stem cells and induced pluripotent stem cells. [Internet] Cell stem cell. 2010;6:407–11. doi: 10.1016/j.stem.2010.04.005. This study reprots a significant difference between ESC and iPSC models of fragile X syndrome with regard to their expression of the FMR1 gene suggesting that factor based reprogramming may not reset certain epigenetic aberrations. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chamberlain SJ, Chen PF, Ng KY, Bourgois-Rocha F, Lemtiri-Chlieh F, Levine ES, Lalande M. Induced pluripotent stem cell models of the genomic imprinting disorders Angelman and Prader-Willi syndromes. Proceedings of the National Academy of Sciences of the United States of America; 2010; pp. 17668–73. Internet. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sheridan SD, Theriault KM, Reis SA, Zhou F, Madison JM, Daheron L, Loring JF, Haggarty SJ. Epigenetic characterization of the FMR1 gene and aberrant neurodevelopment in human induced pluripotent stem cell models of fragile X syndrome. PloS one. 2011;6:e26203. doi: 10.1371/journal.pone.0026203. Internet. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *30.Agarwal S, Loh YH, McLoughlin EM, Huang J, Park IH, Miller JD, Huo H, Okuka M, Dos Reis RM, Loewer S, et al. Telomere elongation in induced pluripotent stem cells from dyskeratosis congenita patients. Nature. 2010;464:292–6. doi: 10.1038/nature08792. Internet. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *31.Batista LFZ, Pech MF, Zhong FL, Nguyen HN, Xie KT, Zaug AJ, Crary SM, Choi J, Sebastiano V, Cherry A, et al. Telomere shortening and loss of self-renewal in dyskeratosis congenita induced pluripotent stem cells. Nature. 2011;474:399–402. doi: 10.1038/nature10084. Internet. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Agarwal S, Daley GQ. Telomere dynamics in dyskeratosis congenita: the long and the short of iPS. Cell research. 2011;21:1157–60. doi: 10.1038/cr.2011.120. Internet. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *33.Gore A, Li Z, Fung HL, Young JE, Agarwal S, Antosiewicz-Bourget J, Canto I, Giorgetti A, Israel MA, Kiskinis E, et al. Somatic coding mutations in human induced pluripotent stem cells. [Internet] Nature. 2011;471:63–7. doi: 10.1038/nature09805. Exome seqeuncing of multiple iPSC lines resulted in the identification of reprogramming-associated mutations highlighting the importance of extensive genetic screening of particular lines before clinical use. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *34.Hussein SM, Batada NN, Vuoristo S, Ching RW, Autio R, Närvä E, Ng S, Sourour M, Hämäläinen R, Olsson C, et al. Copy number variation and selection during reprogramming to pluripotency. [Internet] Nature. 2011;471:58–62. doi: 10.1038/nature09871. This study shows that copy number of variations are abundant in early passage iPSC lines but that extended passaging in culture selects rapidly against mutated cells rendering the lines more similar to human ES cells. [DOI] [PubMed] [Google Scholar]
- *35.Kim K, Doi A, Wen B, Ng K, Zhao R, Cahan P, Kim J, Aryee MJ, Ji H, Ehrlich LIR, et al. Epigenetic memory in induced pluripotent stem cells. [Internet] Nature. 2010;467:285–90. doi: 10.1038/nature09342. Along with reference 36 and 37, this paper shows that iPSCs harbor residual DNA methylation signatures characteristic of their somatic tissue of origin which may favor their differentiation along lineages related to the donor cell, while restricting alternative cell fates. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *36.Polo JM, Liu S, Figueroa ME, Kulalert W, Eminli S, Tan KY, Apostolou E, Stadtfeld M, Li Y, Shioda T, et al. Cell type of origin influences the molecular and functional properties of mouse induced pluripotent stem cells. Nature biotechnology. 2010;28:848–55. doi: 10.1038/nbt.1667. Internet. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *37.Lister R, Pelizzola M, Kida YS, Hawkins RD, Nery JR, Hon G, Antosiewicz-Bourget J, O'Malley R, Castanon R, Klugman S, et al. Hotspots of aberrant epigenomic reprogramming in human induced pluripotent stem cells. Nature. 2011;471:68–73. doi: 10.1038/nature09798. Internet. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ohi Y, Qin H, Hong C, Blouin L, Polo JM, Guo T, Qi Z, Downey SL, Manos PD, Rossi DJ, et al. Incomplete DNA methylation underlies a transcriptional memory of somatic cells in human iPS cells. Nature cell biology. 2011;13:541–9. doi: 10.1038/ncb2239. Internet. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kim K, Zhao R, Doi A, Ng K, Unternaehrer J, Cahan P, Huo H, Loh YH, Aryee MJ, Lensch MW, et al. Donor cell type can influence the epigenome and differentiation potential of human induced pluripotent stem cells. Nature biotechnology. 2011;29:1117–9. doi: 10.1038/nbt.2052. Internet. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Urnov FD, Rebar EJ, Holmes MC, Zhang HS, Gregory PD. Genome editing with engineered zinc finger nucleases. Nature reviews Genetics. 2010;11:636–46. doi: 10.1038/nrg2842. Internet. [DOI] [PubMed] [Google Scholar]
- 41.Hockemeyer D, Soldner F, Beard C, Gao Q, Mitalipova M, DeKelver RC, Katibah GE, Amora R, Boydston EA, Zeitler B, et al. Efficient targeting of expressed and silent genes in human ESCs and iPSCs using zinc-finger nucleases. Nature biotechnology. 2009;27:851–7. doi: 10.1038/nbt.1562. Internet. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hockemeyer D, Wang H, Kiani S, Lai CS, Gao Q, Cassady JP, Cost GJ, Zhang L, Santiago Y, Miller JC, et al. Genetic engineering of human pluripotent cells using TALE nucleases. Nature biotechnology. 2011;29:731–4. doi: 10.1038/nbt.1927. Internet. [DOI] [PMC free article] [PubMed] [Google Scholar]
- **43.Soldner F, Laganière J, Cheng AW, Hockemeyer D, Gao Q, Alagappan R, Khurana V, Golbe LI, Myers RH, Lindquist S, et al. Generation of isogenic pluripotent stem cells differing exclusively at two early onset Parkinson point mutations. [Internet] Cell. 2011;146:318–31. doi: 10.1016/j.cell.2011.06.019. By combining zinc finger nuclease-mediated genome editing and reprogramming, the authors generated sets of isogenic disease and control human iPSCs and genetically correct the disease-causing point mutations in patient-derived iPSCs. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zou J, Sweeney CL, Chou BK, Choi U, Pan J, Wang H, Dowey SN, Cheng L, Malech HL. Oxidase-deficient neutrophils from X-linked chronic granulomatous disease iPS cells: functional correction by zinc finger nuclease-mediated safe harbor targeting. Blood. 2011;117:5561–72. doi: 10.1182/blood-2010-12-328161. Internet. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sebastiano V, Maeder ML, Angstman JF, Haddad B, Khayter C, Yeo DT, Goodwin MJ, Hawkins JS, Ramirez CL, Batista LFZ, et al. Stem cells. Vol. 29. Dayton, Ohio: 2011. In situ genetic correction of the sickle cell anemia mutation in human induced pluripotent stem cells using engineered zinc finger nucleases; pp. 1717–26. Internet. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zou J, Mali P, Huang X, Dowey SN, Cheng L. Site-specific gene correction of a point mutation in human iPS cells derived from an adult patient with sickle cell disease. Blood. 2011;118:4599–608. doi: 10.1182/blood-2011-02-335554. Internet. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cohen DE, Melton D. Turning straw into gold: directing cell fate for regenerative medicine. Nature reviews Genetics. 2011;12:243–52. doi: 10.1038/nrg2938. Internet. [DOI] [PubMed] [Google Scholar]
- *48.Di Giorgio FP, Carrasco MA, Siao MC, Maniatis T, Eggan K. Non-cell autonomous effect of glia on motor neurons in an embryonic stem cell-based ALS model. [Internet] Nature neuroscience. 2007;10:608–14. doi: 10.1038/nn1885. By deriving multiple cell types from disease-specific iPSCs and using co-cultures, the authors show a non-cell autonomous effect of glial cells on the neurodegenerative phenotypes of ALS-specific motor neurons. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cherry ABC, Daley GQ. Reprogramming Cellular Identity for Regenerative Medicine. Cell. 2012;148:1110–1122. doi: 10.1016/j.cell.2012.02.031. Internet. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jang J, Kang HC, Kim HS, Kim JY, Huh YJ, Kim DS, Yoo JE, Lee JA, Lim B, Lee J, et al. Induced pluripotent stem cell models from X-linked adrenoleukodystrophy patients. Annals of neurology. 2011;70:402–9. doi: 10.1002/ana.22486. Internet. [DOI] [PubMed] [Google Scholar]
- 51.Yagi T, Ito D, Okada Y, Akamatsu W, Nihei Y, Yoshizaki T, Yamanaka S, Okano H, Suzuki N. Modeling familial Alzheimer's disease with induced pluripotent stem cells. Human molecular genetics. 2011;20:4530–9. doi: 10.1093/hmg/ddr394. Internet. [DOI] [PubMed] [Google Scholar]
- *52.Israel MA, Yuan SH, Bardy C, Reyna SM, Mu Y, Herrera C, Hefferan MP, Van Gorp S, Nazor KL, Boscolo FS, et al. Probing sporadic and familial Alzheimer's disease using induced pluripotent stem cells. [Internet] Nature. 2012;482:216–20. doi: 10.1038/nature10821. This study demonstrates that in certain cases (one sporadic Alzheimer case) patient specific-iPSCs can be used to observe disease-relevant phenotypes for a disease that can take decades to manifest in patients. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mitne-Neto M, Machado-Costa M, Marchetto MCN, Bengtson MH, Joazeiro CA, Tsuda H, Bellen HJ, Silva HCA, Oliveira ASB, Lazar M, et al. Downregulation of VAPB expression in motor neurons derived from induced pluripotent stem cells of ALS8 patients. Human molecular genetics. 2011;20:3642–52. doi: 10.1093/hmg/ddr284. Internet. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ho JCY, Zhou T, Lai WH, Huang Y, Chan YC, Li X, Wong NLY, Li Y, Au KW, Guo D, et al. Generation of induced pluripotent stem cell lines from 3 distinct laminopathies bearing heterogeneous mutations in lamin A/C. Aging. 2011;3:380–90. doi: 10.18632/aging.100277. Internet. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Somers A, Jean JC, Sommer CA, Omari A, Ford CC, Mills JA, Ying L, Sommer AG, Jean JM, Smith BW, et al. Stem cells. Vol. 28. Dayton, Ohio: 2010. Generation of transgene-free lung disease-specific human induced pluripotent stem cells using a single excisable lentiviral stem cell cassette; pp. 1728–40. Internet. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ye L, Chang JC, Lin C, Sun X, Yu J, Kan YW. Induced pluripotent stem cells offer new approach to therapy in thalassemia and sickle cell anemia and option in prenatal diagnosis in genetic diseases. Proceedings of the National Academy of Sciences of the United States of America; 2009; pp. 9826–30. Internet. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Papapetrou EP, Lee G, Malani N, Setty M, Riviere I, Tirunagari LMS, Kadota K, Roth SL, Giardina P, Viale A, et al. Genomic safe harbors permit high β-globin transgene expression in thalassemia induced pluripotent stem cells. Nature biotechnology. 2011;29:73–8. doi: 10.1038/nbt.1717. Internet. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ghodsizadeh A, Taei A, Totonchi M, Seifinejad A, Gourabi H, Pournasr B, Aghdami N, Malekzadeh R, Almadani N, Salekdeh GH, et al. Generation of liver disease-specific induced pluripotent stem cells along with efficient differentiation to functional hepatocyte-like cells. Stem cell reviews. 2010;6:622–32. doi: 10.1007/s12015-010-9189-3. Internet. [DOI] [PubMed] [Google Scholar]
- 59.Maehr R, Chen S, Snitow M, Ludwig T, Yagasaki L, Goland R, Leibel RL, Melton DA. Generation of pluripotent stem cells from patients with type 1 diabetes. Proceedings of the National Academy of Sciences of the United States of America; 2009; pp. 15768–73. Internet. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Tolar J, Xia L, Riddle MJ, Lees CJ, Eide CR, McElmurry RT, Titeux M, Osborn MJ, Lund TC, Hovnanian A, et al. Induced pluripotent stem cells from individuals with recessive dystrophic epidermolysis bullosa. The Journal of investigative dermatology. 2011;131:848–56. doi: 10.1038/jid.2010.346. Internet. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Itoh M, Kiuru M, Cairo MS, Christiano AM. Generation of keratinocytes from normal and recessive dystrophic epidermolysis bullosa-induced pluripotent stem cells. Proceedings of the National Academy of Sciences of the United States of America; 2011; pp. 8797–802. Internet. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Li W, Wang X, Fan W, Zhao P, Chan YC, Chen S, Zhang S, Guo X, Zhang Y, Li Y, et al. Modeling abnormal early development with induced pluripotent stem cells from aneuploid syndromes. Human molecular genetics. 2012;21:32–45. doi: 10.1093/hmg/ddr435. Internet. [DOI] [PubMed] [Google Scholar]
- 63.Liu J, Verma PJ, Evans-Galea MV, Delatycki MB, Michalska A, Leung J, Crombie D, Sarsero JP, Williamson R, Dottori M, et al. Generation of induced pluripotent stem cell lines from Friedreich ataxia patients. Stem cell reviews. 2011;7:703–13. doi: 10.1007/s12015-010-9210-x. Internet. [DOI] [PubMed] [Google Scholar]
- 64.Mazzulli JR, Xu YH, Sun Y, Knight AL, McLean PJ, Caldwell GA, Sidransky E, Grabowski GA, Krainc D. Gaucher disease glucocerebrosidase and α-synuclein form a bidirectional pathogenic loop in synucleinopathies. Cell. 2011;146:37–52. doi: 10.1016/j.cell.2011.06.001. Internet. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Howden SE, Gore A, Li Z, Fung HL, Nisler BS, Nie J, Chen G, McIntosh BE, Gulbranson DR, Diol NR, et al. Genetic correction and analysis of induced pluripotent stem cells from a patient with gyrate atrophy. Proceedings of the National Academy of Sciences of the United States of America; 2011; pp. 6537–42. Internet. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zhang N, An MC, Montoro D, Ellerby LM. Characterization of Human Huntington's Disease Cell Model from Induced Pluripotent Stem Cells. PLoS currents. 2010;2:RRN1193. doi: 10.1371/currents.RRN1193. Internet. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zhang J, Lian Q, Zhu G, Zhou F, Sui L, Tan C, Mutalif RA, Navasankari R, Zhang Y, Tse HF, et al. A human iPSC model of Hutchinson Gilford Progeria reveals vascular smooth muscle and mesenchymal stem cell defects. Cell stem cell. 2011;8:31–45. doi: 10.1016/j.stem.2010.12.002. Internet. [DOI] [PubMed] [Google Scholar]
- 68.Liu GH, Barkho BZ, Ruiz S, Diep D, Qu J, Yang SL, Panopoulos AD, Suzuki K, Kurian L, Walsh C, et al. Recapitulation of premature ageing with iPSCs from Hutchinson-Gilford progeria syndrome. Nature. 2011;472:221–5. doi: 10.1038/nature09879. Internet. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Carvajal-Vergara X, Sevilla A, D'Souza SL, Ang YS, Schaniel C, Lee DF, Yang L, Kaplan AD, Adler ED, Rozov R, et al. Patient-specific induced pluripotent stem-cell-derived models of LEOPARD syndrome. Nature. 2010;465:808–12. doi: 10.1038/nature09005. Internet. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Tolar J, Park IH, Xia L, Lees CJ, Peacock B, Webber B, McElmurry RT, Eide CR, Orchard PJ, Kyba M, et al. Hematopoietic differentiation of induced pluripotent stem cells from patients with mucopolysaccharidosis type I (Hurler syndrome) Blood. 2011;117:839–47. doi: 10.1182/blood-2010-05-287607. Internet. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lemonnier T, Blanchard S, Toli D, Roy E, Bigou S, Froissart R, Rouvet I, Vitry S, Heard JM, Bohl D. Modeling neuronal defects associated with a lysosomal disorder using patient-derived induced pluripotent stem cells. Human molecular genetics. 2011;20:3653–66. doi: 10.1093/hmg/ddr285. Internet. [DOI] [PubMed] [Google Scholar]
- 72.Deyle DR, Khan IF, Ren G, Wang PR, Kho J, Schwarze U, Russell DW. Normal collagen and bone production by gene-targeted human osteogenesis imperfecta iPSCs. Molecular therapy: the journal of the Amecan Society of Gene Therapy. 2012;20:204–13. doi: 10.1038/mt.2011.209. Internet. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Swistowski A, Peng J, Liu Q, Mali P, Rao MS, Cheng L, Zeng X. Stem cells. Vol. 28. Dayton, Ohio: 2010. Efficient generation of functional dopaminergic neurons from human induced pluripotent stem cells under defined conditions; pp. 1893–904. Internet. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Nguyen HN, Byers B, Cord B, Shcheglovitov A, Byrne J, Gujar P, Kee K, Schüle B, Dolmetsch RE, Langston W, et al. LRRK2 mutant iPSC-derived DA neurons demonstrate increased susceptibility to oxidative stress. Cell stem cell. 2011;8:267–80. doi: 10.1016/j.stem.2011.01.013. Internet. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Seibler P, Graziotto J, Jeong H, Simunovic F, Klein C, Krainc D. Mitochondrial Parkin recruitment is impaired in neurons derived from mutant PINK1 induced pluripotent stem cells. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2011;31:5970–6. doi: 10.1523/JNEUROSCI.4441-10.2011. Internet. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Devine MJ, Ryten M, Vodicka P, Thomson AJ, Burdon T, Houlden H, Cavaleri F, Nagano M, Drummond NJ, Taanman JW, et al. Parkinson's disease induced pluripotent stem cells with triplication of the α-synuclein locus. Nature communications. 2011;2:440. doi: 10.1038/ncomms1453. Internet. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Ye Z, Zhan H, Mali P, Dowey S, Williams DM, Jang YY, Dang CV, Spivak JL, Moliterno AR, Cheng L. Human-induced pluripotent stem cells from blood cells of healthy donors and patients with acquired blood disorders. Blood. 2009;114:5473–80. doi: 10.1182/blood-2009-04-217406. Internet. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Huang HP, Chen PH, Hwu WL, Chuang CY, Chien YH, Stone L, Chien CL, Li LT, Chiang SC, Chen HF, et al. Human Pompe disease-induced pluripotent stem cells for pathogenesis modeling, drug testing and disease marker identification. Human molecular genetics. 2011;20:4851–64. doi: 10.1093/hmg/ddr424. Internet. [DOI] [PubMed] [Google Scholar]
- 79.Yang J, Cai J, Zhang Y, Wang X, Li W, Xu J, Li F, Guo X, Deng K, Zhong M, et al. Induced pluripotent stem cells can be used to model the genomic imprinting disorder Prader-Willi syndrome. The Journal of biological chemistry. 2010;285:40303–11. doi: 10.1074/jbc.M110.183392. Internet. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Jin ZB, Okamoto S, Osakada F, Homma K, Assawachananont J, Hirami Y, Iwata T, Takahashi M. Modeling retinal degeneration using patient-specific induced pluripotent stem cells. PloS one. 2011;6:e17084. doi: 10.1371/journal.pone.0017084. Internet. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Muotri AR, Marchetto MCN, Coufal NG, Oefner R, Yeo G, Nakashima K, Gage FH. L1 retrotransposition in neurons is modulated by MeCP2. Nature. 2010;468:443–6. doi: 10.1038/nature09544. Internet. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Hotta A, Cheung AYL, Farra N, Vijayaragavan K, Séguin CA, Draper JS, Pasceri P, Maksakova IA, Mager DL, Rossant J, et al. Isolation of human iPS cells using EOS lentiviral vectors to select for pluripotency. Nature methods. 2009;6:370–6. doi: 10.1038/nmeth.1325. Internet. [DOI] [PubMed] [Google Scholar]
- 83.Zhang S, Chen S, Li W, Guo X, Zhao P, Xu J, Chen Y, Pan Q, Liu X, Zychlinski D, et al. Rescue of ATP7B function in hepatocyte-like cells from Wilson's disease induced pluripotent stem cells using gene therapy or the chaperone drug curcumin. Human molecular genetics. 2011;20:3176–87. doi: 10.1093/hmg/ddr223. Internet. [DOI] [PubMed] [Google Scholar]