

Abstract
The coordination chemistry of hydrated and solvated vanadium(III), oxovanadium(IV), and dioxovanadium(V) ions in the oxygen donor solvents water, dimethylsulfoxide (dmso) and N,N′-dimethylpropyleneurea (dmpu) has been studied in solution by EXAFS and large angle X-ray scattering (LAXS) and in solid state by single crystal X-ray diffraction and EXAFS. The hydrated vanadium(III) ion has a regular octahedral configuration with a mean V-O bond distance of 1.99 Å. In the hydrated and dimethylsulfoxide solvated oxovanadium(IV) ions vanadium binds strongly to an oxo group at ca. 1.6 Å. The solvent molecule trans to the oxo group is very weakly bound, at ca. 2.2 Å, while the remaining four solvent molecules, with a mean V-O bond distance of 2.0 Å, form a plane slightly below the vanadium atom; the mean O=V-Operp bond angle is ca. 98°. In the dmpu solvated oxovanadium(IV) ion, the space demanding properties of the dmpu molecule leaving no solvent molecule in the trans position to the oxo group which reduces the coordination number to 5. The O=V-O bond angle is consequently much larger, 106°, and the mean V=O and V-O bond distances decrease to 1.58 and 1.97 Å, respectively. The hydrated and dimethylsulfoxide solvated dioxovanadium(V) ions display a very distorted octahedral configuration with the oxo groups in cis position with mean V=O bond distances of 1.6 Å and a O=V=O bond angle of ca. 105°. The solvent molecules trans to the oxo groups are weakly bound, at ca. 2.2 Å, while the remaining two have bond distances of 2.02 Å. The experimental studies of the coordination chemistry of hydrated and solvated vanadium(III,IV,V) ions are complemented by summarizing previously reported crystal structures to yield a comprehensive description of the coordination chemistry of vanadium with oxygen donor ligands.
Introduction
Vanadium plays a number of roles in biological systems, especially in marine environments. It is e.g. present in haloperoxidases as vanadium(V),1–5 as vanadium(III) or vanadium(IV) complexes in tunicate blood cells.6,7 and also as amavadine, a vanadium(IV) complex produced the mushroom genus Amanita.8–10 Some species in the family Ascidiidae accumulate vanadium at concentrations in excess of 350 mmol·dm−3, corresponding to about 107 times that found in seawater,11 and the transport function of vanadium into ascidians has recently been described.12 The role of vanadium in biological systems is both structural, seen by maintenance of various biological structures, and functional, thereby bringing key reactivity to active centers of proteins, enzymes and co-enzymes.7 Vanadium(IV) salts have been utilized in the treatment of diabetes for some time, and the insulin-mimetic behavior of vanadium(IV,V) coordination compounds has been studied been extensively.13–19 Vanadium toxicity is often associated to the production of reactive oxygen species, causing a lipid peroxidation and an alteration in the enzymatic antioxidant defence.20 The coordination chemistry and role of vanadium in biological systems have been reviewed in many publications and books.1,5–10,21–23 Vanadium compounds have received increased attention as catalysts in a number of applications as recently reviewed.24
Vanadium(III) has well-defined coordination chemistry retaining octahedral complexes in most systems, while the coordination chemistry of vanadium(IV) and vanadium(V) is much more flexible with several configurations and coordination numbers reported.23
Vanadium(V) has different speciation and coordination depending on the level of pH in the aqueous solution.25 At low pH vanadium(V) is present as the cationic vanadyl(V) ion, VO2+, while in neutral and alkaline solution it is present as tetrahedral vanadate(V) ions, HnVO4(3−n)−. The dominating vanadium(V) species in weak acidic solution, 2 ≤ pH ≤ 6, is the decamer [V10O28]6−. The acidic constants of vanadic acid, H3VO4, pKa1 = 3.5, pKa2 = 7.8, and pKa3 = 12.5, respectively,6 resemble closely those of phosphoric acid, H3PO4, pKa1 = 2.1, pKa2 = 7.2, and pKa3 = 12.7.26 This makes the chemical properties of orthophosphoric and orthovanadic acid systems very similar in neutral and alkaline aqueous solution, though the vanadate(V) ion is slightly larger than the phosphate ion. The mean V-O and P-O bond distances are 1.72 and 1.54 Å, respectively, and no evidence for the presence of orthovanadic acid has been put forward.27,28 A more detailed overview of the hydrolysis and polymerization of vanadium(V) is given elsewhere.25
No structures of the hydrated dioxovanadium(V) ion have been reported so far in the solid state, but one five-coordinate neutral solvate29 and a few additional five- and six-coordinate anionic hydrated and solvated complexes have been reported,30–32 Tables S4a–e. In six-coordinate dioxovanadium(V) complexes the bonds trans to the oxo groups are significantly longer than those in cis position.31,32 In the five-coordinate trigonal bipyramidal complexes, where no true trans position exists, all three V-O bond distances are similar.29,30
The characteristic of the coordination chemistry of oxovanadium(IV) complexes is the strong V=O double bond.33 The uniquely stable oxovanadium(IV) (vanadyl(IV)) ion, VO2+, possibly the most stable diatomic ion known,34 can withstand many types of redox reactions and form complexes with a large variety of ligands. Vanadium(IV) starts to hydrolyze and dimerize at pH ≈ 4; the pKa value of the [VO(H2O)5]2+ ion has been reported to be in the range 5.3–6.0.35 At pH = 5 insoluble {VO(OH)2}n starts to form,36 which converts to water soluble [(VO)2(OH)5]− and [VO(OH)3]2− complexes in strong alkaline aqueous solution.37,38
Nine pentaaquaoxovanadium(IV) structures have been reported in the solid state with a strong V=O bond, mean bond distance dV=O = 1.586 Å. They all feature four near-perpendicularly bound waters, mean dV-O = 2.024 Å, and a fifth water molecule in trans position to the V=O bond more weakly bound, mean dV-O = 2.189 Å, forming a distorted octahedral structure,39 Table S3a. Additionally, four dmso and urea solvates of oxovanadium(IV) display a very similar coordination pattern as the hydrates with mean bond distances 1.583, 2.022, and 2.183 Å,40 respectively, Table S3b.
The hydrated vanadium(II) and vanadium(III) ions, with d3 and d2 electron configuration, respectively, have regular octahedral configuration based on the structures in solid state and from theoretical simulations,39–43 though it has not yet been confirmed by experimental structural studies in aqueous solution. A limited number of solid state structures containing hexaaquavanadium(II) and hexaaquavanadium(III) ions have been reported with mean V-O bond distances of 2.130 and 1.995 Å, respectively, Tables S1a and S2a. Similarly, the reported structures containing solvated vanadium(II) and vanadium(III) ions are also octahedral with similar mean V-O bond distances as in the hydrates, 2.132 and 1.982 Å respectively, Tables S1b and S2b. The hydrolysis properties of vanadium(III) were recently studied, showing that vanadium(III) starts to hydrolyze already at about pH = 1, with maximal dominance of [VOH]2+(aq) and [V(OH)2]+(aq) at pH = 3 and 4, respectively.44 This shows that vanadium(III) starts to hydrolyze at somewhat lower pH than previously reported.25
The objective of the present work is to investigate the structures of the hydrated and solvated cationic vanadium(III), oxovanadium(IV) and dioxovanadium(V) ions in the oxygen donor solvents water, dimethylsulfoxide (dmso), and N,N′-dimethylpropyleneurea (dmpu) in both solution and the solid state. The structures of solid [VO(OH2)5](CF3SO3)2, [VO(dmso)5](ClO4)2, [VO(dmpu)4](CF3SO3)2, and [VO2(dmso)4]CF3SO3, have been determined by single crystal X-ray diffraction and EXAFS at room temperature, with some additional information on solid dioxovanadium(V) perchlorate, VO2ClO4, obtained through powder diffraction. The structures of the hydrated and hydrolyzed vanadium(III) ions in aqueous solution, and the hydrates and solvates of the oxovanadium(IV) and dioxovanadium(V) ions in aqueous, dimethylsulfoxide and N,N′-dimethylpropyleneurea solution have been determined by means of the large angle X-ray scattering (LAXS) and EXAFS. The studies performed here are complemented with the information obtained from the two crystallographic databases Cambridge Structure Database (CSD)41 and Inorganic Crystal Structure Database (ICSD),42 yielding a comprehensive description of the coordination chemistry of cationic vanadium ions with oxygen donor ligands. Additionally, the UV-Vis spectra of respective solution were recorded providing spectrophotometric characterization of the studied solutions.
Experimental
Solvents
Deionized water has been used throughout this study. Dimethylsulfoxide (dmso), (CH3)2SO, (Merck) and N,N′-dimethylpropyleneurea (dmpu), C6H12N2O, (BASF) were distilled over calcium hydride, CaH2 (Fluka), and stored in dark bottles over 3 Å molecular sieves.
Chemicals
Vanadium(III) chloride, VCl3, (Merck), vanadium(IV) sulfate pentahydrate, VOSO4·5H2O, S1,(Merck), vanadium(V) oxide, V2O5, (Merck), barium perchlorate, Ba(ClO4)2, (Kebo), barium trifluoromethanesulfonate, Ba(CF3SO3)2, (Aldrich), trifluoromethanesulfonic acid, CF3SO3H (Riedel-de Haën) and perchloric acid, (BDH, 70%) were used as purchased.
Solids
Pentaaquaoxovanadium(IV) trifluoromethanesulfonate, [VO(OH2)5](CF3SO3)2, 1. A dilute trifluoromethanesulfonic acidic aqueous oxovanadium(IV) trifluoromethanesulfonate solution was prepared by mixing aqueous solutions of oxovanadium(IV) sulfate pentahydrate, and barium trifluoromethanesulfonate in a molar ratio 1:1 as described elsewhere.45 The formed barium sulfate was filtered off after cooling the mixture in a refrigerator for one week. The volume of the filtered solution was reduced to a final concentration of ca. 4 mol·dm−3. This concentrated solution was placed in refrigerator and crystals of 1 of sufficient quality for single crystal X-ray diffraction studies were obtained.
Pentakis(dmso)oxovanadium(IV) perchlorate, [VO(OS(CH3)2)5](ClO4)2, 2a, and pentakis(dmso)oxovanadium(IV) trifluoromethanesulfonate, [VO(OS(CH3)2)5](CF3SO3)2, 2b. Dimethylsulfoxide was added to a concentrated aqueous solution of oxovanadium(IV) perchlorate or trifluoromethanesulfonate. Water and most of the excess of dimethylsulfoxide were evaporated off under reduced pressure. The salts crystallized after cooling in refrigerator, and they were recrystallized twice from freshly distilled dimethylsulfoxide. Tetrakis(dmpu)oxovanadium(IV) trifluoromethanesulfonate, [VO(OCN2(CH3)2(CH2)3)4]-(CF3SO3)2, 3, was prepared in an analogous way as described for 2b above, using dmpu instead of dmso.
Dioxovanadium(V) perchlorate and trifluoromethanesulfonate hydrates, 4. Vanadium(V) oxide, V2O5, was dissolved in dilute sulfuric acid, equimolar amounts of barium perchlorate or triflouromethanesulfonate, dissolved in water, was added, and the formed barium sulfate was filtered off. The obtained solutions were evaporated under reduced pressure until a fine-grained deep red precipitate was formed, and the solutions were put in refrigerator for continued crystallization. The precipitates were filtered off and studied by X-ray powder diffraction. The powders were found to be only partially crystalline, as indicated by the powder diffraction data of the perchlorate salt shown in Figure S6. Tetrakis(dmso)dioxovanadium(V) trifluoromethanesulfonate, [VO2(OS(CH3)2)4]CF3SO3, 5. A dmso solution of dioxovanadium(V) trifluoromethanesulfonate was prepared by adding trifluoromethanesulfonate acid (Riedel-de Haën) to a vanadium(V) oxide/dmso slurry. The mixture was stirred and heated to 363 K for about 48 h until most of the vanadium(V) oxide had reacted. The excess of dimethylsulfoxide was removed by slow evaporation under reduced pressure of a filtered solution. 5 crystallized after cooling in refrigerator, and it was recrystallized once from pure dmso. Caution! Note that perchlorate salts cannot be prepared by this route due to the explosive reaction between dmso and aqueous perchloric acid.
Solutions
An aqueous solution of vanadium(III) chloride, L0, was prepared by dissolving anhydrous vanadium(III) chloride in 0.1 mol·dm−3 perchloric acid degassed with nitrogen for 15 minutes. Concentrated perchloric acid was added drop-wise until a clear blue solution of vanadium(III) was obtained; pH was measured to 0.60. A hydrolyzed vanadium(III) solution, L0hyd, was prepared by adjusting pH with dilute sodium hydroxide solution until a significant color change to bluish-green was observed; pH was measured to 1.22 in this solution. Vanadium(III) seems to be perfectly stable for weeks under these conditions. The UV-Vis spectra of the aqueous vanadium(III) solutions studied, Fig. S1, are in full agreement with those reported by Kanamori.46
Aqueous solutions of oxovanadium(IV) perchlorate, VO(ClO4)2, L1a-c, were prepared by mixing aqueous solutions of vanadium(IV) oxide sulfate (Merck) and barium perchlorate (Merck), and the barium sulfate precipitate was filtered off. The final solutions were prepared by removing water by evaporation to the desired concentration. The dmso solutions of oxovanadium(IV) perchlorate, L2a-b, were prepared by dissolving 2a in freshly distilled dmso.
A dmpu solution of oxovanadium(IV) trifluoromethanesulfonate, L3, was prepared by adding the stoichiometric amount of the trifluoromethanesulfonic acid (Riedel-de Haën) to a vanadium(IV) oxide/dmpu slurry. The mixture was stirred and heated to 373 K for about 60 h until the most of the vanadium(IV) oxide had reacted, and remaining starting material was filtered off.
Acidic aqueous solutions of dioxovanadium(V) sulfate, L4a-b, were prepared by adding the considerable excess of highly concentrated sulfuric acid (98% by weight, Merck) to a vanadium(V) oxide/water slurry heated to 350 K. The mixture was stirred and heated to 360 K until all vanadium(V) oxide had reacted. The obtained solution was cooled and diluted to the desired concentration.
Acidic dmso solutions of dioxovanadium(V) sulfate, L5a-b, were prepared by adding dmso to a dioxovanadium(V) sulfate solution in sulfuric acid prepared as described above.
A dmpu solution of dioxovanadium(V) trifluoromethanesulfonate, L6, was prepared by adding the stoichiometric amount of the trifluoromethanesulfonic acid (Riedel-de Haën) to a vanadium(V) oxide/dmpu slurry as described above.
The concentrations of all ions and solvents, wherever appropriate, for all solutions studied by EXAFS and LAXS are summarized in Table 1. UV-Vis spectra of the studied solutions after appropriate dilution are shown in Fig. S1.
Table 1.
Concentrations of all ions and, wherever appropriate, solvent medium in mol·dm−3 of the vanadium solutions studied by LAXS (L) and EXAFS (E), including their densities ρ, linear absorption coefficient μ, and label. xs denotes excess solvent
| Sample | [Vtot] | [Cl−] | [ClO4−] | [CF3SO3−] | [SO42−] | [solvent] | ρ/g·cm−3 | μ/cm−1 | pH | Note |
|---|---|---|---|---|---|---|---|---|---|---|
| V3+ in water, pH=0.6 | 0.50 | 1.50 | 0.10 | xs | 0.60 | E, L0 | ||||
| V3+ in water, pH=1.2 | 0.50 | 1.50 | 0.10 | xs | 1.22 | E, L0hyd | ||||
| VO2+ in water | 1.228 | 2.556 | 53.228 | 1.273 | 4.123 | 0.53 | L, L1a | |||
| VO2+ in water | 4.433 | 8.866 | 11.512 | 1.386 | 10.880 | 0.4 | L, L1b | |||
| VO2+ in water | 0.70 | 1.40 | xs | 0.5 | E, L1c | |||||
| VO2+ in dmso | 0.369 | 0.738 | 13.282 | 1.136 | 5.460 | L, L2a | ||||
| VO2+ in dmso | 0.18 | 0.36 | xs | E, L2b | ||||||
| VO2+ in dmpu | 0.18 | 0.36 | xs | E, L3 | ||||||
| VO2+ in water | 1.833 | 7.237 | 32.953 | 1.449 | 6.181 | <0 | L, L4a | |||
| VO2+ in water | 0.87 | 3.435 | xs | <0 | E, L4b | |||||
| VO2+ in dmso | 1.374 | 3.822 | 11.693 | 1.401 | 7.503 | L, L5a | ||||
| VO2+ in dmso | 0.50 | 0.50 | xs | E, L5b | ||||||
| VO2+ in dmpu | 0.20 | 0.20 | xs | E, L6 |
Single-Crystal X-ray Diffraction
The crystals were taken out of the mother liquid and mounted in glass capillaries, which were sealed by burning immediately after mounting. Data collections were performed on a Bruker SMART platform equipped with a CCD area detector47 and a graphite monochromator using Mo-Kα (λ = 0.7107 Å) radiation at ambient room temperature. A hemisphere dataset was collected for each structure using the ω scan method, with 1271 frames and a crystal-to-detector distance of 5.0 cm for all compounds studied, 1, 2a, 3 and 5. Appropriate absorption correction was applied using overlapping multi-scans. The structures were solved by direct methods in SHELXL,48 and refined using full-matrix least squares on F2. Hydrogens were added to the water molecules in 1 allowing the distance to be refined, whereas hydrogen atoms were calculated in ideal positions riding on their respective carbon atom in 2a, 3 and 5. Selected bond distances and angles are given in the Supplementary Material section, and the Crystallographic Information Files (CIFs) have been deposited at the Cambridge Crystallographic Data Centre; CCDC 864570–864573.
EXAFS - Data Collection
Vanadium K edge X-ray absorption spectra were recorded at the wiggler beam line 4-1 (old station) at the Stanford Synchrotron Radiation Lightsource (SSRL). The EXAFS station was equipped with a Si[220] double crystal monochromator. SSRL operated at 3.0 GeV and a maximum current of 100 mA. The data collection was performed in transmission mode and at ambient temperature. Higher order harmonics were reduced by detuning the second monochromator to 40% of maximum intensity at the end of the scans. The solutions were placed in cells with Mylar foil windows and 1–5 mm Teflon spacers. The energy scale of the X-ray absorption spectra was calibrated by assigning the first inflection point of the K edge of a vanadium foil to 5465.2 eV.49 For each sample 3–6 scans were averaged, giving satisfactory data (k3-weighted) in the k range 2–13 Å−1. The EXAFSPAK program package was used for the primary data treatment.50
EXAFS - Data Analysis
The EXAFSPAK program package was used for the data treatment,50 including refinement of structure parameters using FEFF calculated EXAFS parameters.51 The standard deviations reported for the refined parameters in Table 2 are obtained from k3 weighted least squares refinements of the EXAFS function χ(k), and do not include systematic errors of the measurements. These statistical error values allow reasonable comparisons of e.g. the significance when comparing relative shifts in the distances. However, the variations in the refined parameters, including the shift in the Eo value (for which k = 0), using different models and data ranges, indicate that the absolute accuracy of the distances given for the separate complexes is within ±0.005 to 0.02 Å for well-defined interactions. The “estimated standard deviations” given in the text have been increased accordingly to include estimated additional effects of systematic errors.
Table 2.
Crystallographic data and structure refinement details for vanadium compounds 1, 2a, 3 and 5.
| 1 | 2a | 3 | 5 | |
|---|---|---|---|---|
| Structure formula | [VO(H2O)5](CF3SO3)2 | [VO(dmso)5](ClO4)2 | [VO(dmpu)4](CF3SO3)2 | [VO2(dmso)4]CF3SO3 |
| Sum formula | C2H10F6O12S2V | C10H30Cl2O14S5V | C26H48F6N8O11S2V | C9H24F3O9S5V |
| MW | 455.16 | 656.48 | 877.80 | 544.55 |
| Diffractometer system | Bruker Smart CCD | Bruker Smart CCD | Bruker Smart CCD | Bruker Smart CCD |
| Radiation, λ/Å | 0.71073 | 0.71073 | 0.71073 | 0.71073 |
| Crystal system | Monoclinic | Monoclinic | Monoclinic | Monoclinic |
| Space group | P21/m (No. 11) | P21/c (No. 14) | P21/c (No. 14) | P21/c (No. 14) |
| a/Å | 9.7598(11) | 11.0482(15) | 16.590(4) | 13.756(3) |
| b/Å | 7.8650(9) | 10.8796(15) | 27.137(6) | 6.4360(13) |
| c/Å | 10.2154(11) | 24.928(3) | 18.215(4) | 24.660(5) |
| α/° | 90 | 90 | 90 | 90 |
| β/° | 102.555(2) | 109.297(5) | 98.667(5) | 92.291(4) |
| γ/° | 90 | 90 | 90 | 90 |
| V/Å3 | 765.39(15) | 2828.0(7) | 8107(3) | 2181.5(7) |
| Z | 2 | 4 | 8 | 4 |
| T/K | 295(2) | 295(2) | 295(2) | 295(2) |
| Dc/g cm−1 | 1.975 | 1.542 | 1.438 | 1.658 |
| F2 | 454 | 1356 | 3656 | 1120 |
| μ/mm−1 | 1.043 | 0.962 | 0.436 | 0.993 |
| crystal size/mm3 | 0.50·0.40·0.30 | 0.60·0.30·0.14 | 0.55·0.45·0.40 | 0.40·0.22·0.20 |
| θ range/° | 2.0–25.7 | 1.7–25.0 | 1.4–25.7 | 1.5–25.7 |
| index ranges | −8 ≤ h ≤ 11 | −13 ≤ h ≤ 12 | −20 ≤ h ≤ 19 | −16 ≤ h ≤ 10 |
| −9 ≤ k ≤ 9 | −12 ≤ k ≤ 12 | −33 ≤ k ≤ 24 | −7 ≤ k ≤ 7 | |
| −12 ≤ l ≤ 12 | −29 ≤ l ≤ 26 | −22 ≤ l ≤ 21 | −29 ≤ l ≤ 29 | |
| Measured reflections | 4087 | 13771 | 42469 | 11340 |
| Unique reflections | 1567 | 4950 | 15320 | 4116 |
| Obs. refl., I > 2σ(Rint) | 1275 (0.028) | 2477 (0.054) | 6205 (0.077) | 1717 (0.161) |
| Data/Restraints/Params | 1561/11/143 | 4950/78/337 | 15320/49/1012 | 4116/0/285 |
| Goodness of fit | 0.99 | 0.93 | 0.93 | 0.98 |
| Refinement method | Full-matrix least-squares on F2 Full-matrix least-squares on F2 Full-matrix least-squares on F2 | |||
| Final R1, wR2 [I > 2σ(I)]a | 0.0392, 0.1062 | 0.0735, 0.2255 | 0.0904, 0.2948 | 0.0795, 0.1402 |
| Largest diff. peak/hole eÅ−3 | −0.44, 0.49 | −0.45, 0.68 | −0.54, 1.72 | −0.35, 0.33 |
R values are defined as: R1 = Σ||Fo|−|Fc||/Σ|Fo|, wR2 = [Σ[w(Fo2 − Fc2)2]/Σ[w(Fo2)2]]0.5
Large-Angle X-ray Scattering
A large-angle θ-θ diffractometer was used to measure the scattering of Mo Kα radiation (λ = 0.7107 Å) on the free surface of aqueous, dmso, and dmpu solutions of vanadyl(IV) and vanadyl(V) perchlorate. The solutions were contained in a Teflon cuvette inside radiation shield with beryllium windows. After monochromatization of scattered radiation, by means of a focusing LiF crystal, the intensity was measured at 450 discrete points in the range 1 < θ < 65° (the scattering angle is 2θ). A total of 100,000 counts were accumulated at each angle and the whole angular range was scanned twice, which corresponds to a statistical uncertainty of about 0.3%. The divergence of the primary X-ray beam was limited by 1 or ¼° slits for different θ regions with overlapping some part of data for scaling purposes.
All data treatment was carried out by using the KURVLR program52 which was previously described in more detail.53 The experimental intensities were normalized to a stoichiometric unit of volume containing one vanadium atom, using the scattering factors f for neutral atoms, including corrections for anomalous dispersion, Δf′ and Δ f″,54 and values for Compton scattering.55,56 To receive a better alignment of the intensity function, a Fourier back-transformation was applied to eliminate spurious peaks below 1.2 Å not related to any interatomic distances in the radial distribution function.57 Least-squares refinements of the model parameters were performed by means of the STEPLR program58 to minimizing the error square sum U = Σw(s)·[iexp(s)−ical(s)]2.
Results and Discussion
The hydrated and hydrolyzed vanadium(III) in aqueous solution, L0 and L0hyd
The EXAFS data of the hydrated and hydrolyzed vanadium(III) ion in aqueous solution display simple back-scattering patterns typical for regular octahedral complexes with only one coordination shell of oxygen atoms as back-scatterer, Fig. 1. The hydrolyzed vanadium(III) complex, most likely present as [V(OH)(H2O)5]2+,44 has a significantly shorter phase than the hydrated vanadium(III) ion, Fig. 1. Refinement of the structure parameters gave mean V-O bond distances of 1.992(3) and 2.014(5) Å for hydrated and hydrolyzed vanadium(III), respectively, and with matching linear multiple scattering path lengths within the octahedral VO6 entities, Table 3. The longer mean V-O bond distance of the hydrolyzed vanadium(III) complex is due to the larger radius of the oxygen in the hydroxide ion, 1.37 Å, than in the coordinated water, 1.34 Å.59,60 The observed difference in V-O bond distance between hydrated and hydrolyzed complexes is therefore expected.
Figure 1.

Experimental EXAFS data (black lines) fitted with a model (Table 3) formed by ab initio-calculated scattering paths from the FEFF program (red lines), a/ [V(H2O)6]3+ in water, b/ [V(OH)3(H2O)3] in water, c/ [VO(H2O)5]2+ in water, d/ solid [VO(H2O)5]SO4, e/ [VO(OSMe2)5]2+ in dmso, f/ solid [VO(OSMe2)5](CF3SO3)2, g/ [VO(dmpu)4]2+ in dmpu, h/ [VO2(H2O)4]+ in water, i/ [VO2(OSMe2)4]+ in dmso, j/ solid [VO2(OSMe2)4]CF3SO3, and k/ [VO2(OSMe2)3]+ in dmpu.
Table 3.
Mean bond distances, d/Å, number of distances, N, and temperature coefficients, b/Å2 (LAXS) or Debye-Waller coefficients, σ/Å2 (EXAFS), from the LAXS (L) and EXAFS (E) studies of the hydrated and dimethylsulfoxide and N,N′-dimethylpropylene-urea solvated vanadium(III), oxovanadium(IV) and dioxovanadium(V) ions in solution and solid state at room temperature.
| Species | Interaction | N | d | b/σ2 | Method |
|---|---|---|---|---|---|
| V3+ in perchloric acidic water, 0.50 mol·dm−3, L0 | |||||
| [V(H2O)6]3+ | V-O | 6 | 1.992(1) | 0.0038(1) | E |
| MS (VO6) | 3·6 | 3.998(11) | 0.012(2) | ||
| Hydrolyzed vanadium(III) in water, 0.50 mol·dm−3, L0hyd | |||||
| [V(OH)3(H2O)3] | V-O | 6 | 2.014(2) | 0.0039(1) | E |
| MS (VO6) | 3·6 | 4.026(12) | 0.011(2) | ||
|
| |||||
| VO2+ in water, 1.228 mol·dm−3, L1a | |||||
| [VO(H2O)5]2+ | V=O | 1 | 1.615(5) | 0.0024(5) | L |
| V-Oeq | 4 | 2.024(7) | 0.0061(5) | ||
| V-Oax | 1 | 2.20(5) | 0.020(3) | ||
| V···OII,eq | 8 | 4.06(2) | 0.040(3) | ||
| ClO4−/aq | Cl-O | 4 | 1.453(5) | 0.0022(4) | |
| Water bulk | O···O | 2 | 2.890(6) | 0.0200(8) | |
| VO2+ in water, 4.433 mol·dm−3, L1b | |||||
| [VO(H2O)5]2+ | V=O | 1 | 1.607(5) | 0.0020(5) | L |
| V-Oeq | 4 | 1.993(7) | 0.0061(5) | ||
| V-Oax | 1 | 2.18(5) | 0.021(3) | ||
| V···OII,eq | 8 | 4.06(2) | 0.040(3) | ||
| ClO4−/aq | Cl-O | 4 | 1.436(4) | 0.0022(4) | |
| Water bulk | O···O | 2 | 2.860(5) | 0.0098(8) | |
| VO2+ in water, 0.700 mol·dm−3, L1c | |||||
| [VO(H2O)5]2+/aq | V=O | 1 | 1.588(1) | 0.0006(2) | E |
| V-Oeq | 4 | 2.027(2) | 0.0041(1) | ||
| V-Oax | 1 | 2.21(5) | 0.015(5) | ||
| Solid [VO(H2O)5]SO4 | |||||
| [VO(H2O)5]2+/aq | V=O | 1 | 1.585(2) | 0.0004(2) | E |
| V-Oeq | 4 | 2.009(2) | 0.0028(2) | ||
| V-Oax | 1 | 2.21(5) | 0.015(5) | ||
| MS (VO4,eq) | 3·4 | 4.00(4) | 0.042(10) | ||
| V···S | 1 | 2.883(7) | 0.0083(8) | ||
| V-O-S | 2 | 3.120(4) | 0.0053(4) | ||
| VO2+ in dimethylsulfoxide, 0.369 mol·dm−3, L2a | |||||
| [VO(OS(CH3)2)5]2+ | V=O | 1 | 1.605(8) | 0.0064(4) | L |
| V-Oeq | 4 | 2.010(9) | 0.0056(10) | ||
| V-Oax | 1 | 2.24(4) | 0.015(5) | ||
| V···Seq | 4 | 3.236(8) | 0.0127(11) | ||
| V···Sax | 1 | 3.36(3) | 0.011(4) | ||
| ClO4− | Cl-O | 4 | 1.425(4) | 0.003(1) | |
| VO2+ in dimethylsulfoxide, 0.18 mol·dm−3, L2b | |||||
| [VO(OS(CH3)2)5]2+ | V=O | 1 | 1.583(2) | 0.0051(3) | E |
| V-Oeq | 4 | 2.021(2) | 0.0043(2) | ||
| V-Oax | 1 | 2.20(4) | 0.013(2) | ||
| V···Seq | 4 | 3.189(6) | 0.0146(7) | ||
| V-Oeq-Seq | 4 | 3.338(13) | 0.014(2) | ||
| MS(VO4) | 3·4 | 4.06(2) | 0.026(4) | ||
| Solid [VO(OS(CH3)2)5](ClO4)2, 2a | |||||
| [VO(OS(CH3)2)5]2+ | V=O | 1 | 1.615(3) | 0.0075(5) | E |
| V-Oeq | 4 | 2.004(2) | 0.0068(3) | ||
| V-Oax | 1 | 2.220(12) | 0.013(2) | ||
| V···Seq | 4 | 3.166(3) | 0.0102(3) | ||
| V-Oeq-Seq | 8 | 3.325(6) | 0.0070(8) | ||
| MS (VO4,eq) | 3·4 | 4.08(2) | 0.030(5) | ||
| VO2+ in N,N-dimethylpropyleneurea, 0.20 mol·dm−3, L3 | |||||
| [VO(dmpu)4]2+ | V=O | 1 | 1.579(2) | 0.0052(3) | E |
| V-Oeq | 4 | 1.971(1) | 0.0033(2) | ||
| V···Ceq | 4 | 2.965(10) | 0.0121(14) | ||
| V-Oeq-Ceq | 8 | 3.072(11) | 0.020(3) | ||
|
| |||||
| VO2+ in sulfuric acid, 1.833 mol·dm−3, L4a | |||||
| [VO2(H2O)4]+/aq | V=O | 2 | 1.621(5) | 0.0024(5) | L |
| V-Ocis | 2 | 1.987(13) | 0.0054(7) | ||
| V-Otrans | 2 | 2.215(17) | 0.019(3) | ||
| V···OII,cis | 4 | 4.16(2) | 0.019(3) | ||
| V···OII,trans | 4 | 4.27(2) | 0.029(4) | ||
| V···Ssulfate | 2 | 3.556(3) | 0.0197(5) | ||
| SO42−/aq | S-O | 4 | 1.495(6) | 0.0027(4) | |
| VO2+ in dilute sulfuric acid, 0.87 mol·dm−3, L4b | |||||
| [VO2(H2O)4]+/aq | V=O | 2 | 1.628(2) | 0.0012(5) | E |
| V-Ocis | 2 | 2.004(2) | 0.0021(3) | ||
| V-Otrans | 2 | 2.224(17) | 0.0063(4) | ||
| VO2+ in dimethylsulfoxide, 1.833 mol·dm−3, L5a | |||||
| [VO2(dmso)4]+ | V=O | 2 | 1.628(5) | 0.0024(5) | L |
| V-Ocis | 2 | 2.014(13) | 0.0125(7) | ||
| V-Otrans | 2 | 2.25(17) | 0.0175(13) | ||
| V···Scis | 4 | 3.205(2) | 0.020(3) | ||
| V···Strans | 4 | 3.393(2) | 0.025(4) | ||
| V···Ssulfate | 2 | 3.556(3) | 0.0197(5) | ||
| Solid [VO2(OS(CH3)2)5](CF3SO4)2, 5 | |||||
| [VO2(dmso)4]+ | V=O | 2 | 1.646(3) | 0.0010(5) | E |
| V-Ocis | 2 | 2.046(7) | 0.0017(10) | ||
| V-Otrans | 2 | 2.22(1) | 0.006(2) | ||
| V···S | 2 | 3.175(9) | 0.0068(10) | ||
| V-O-S | 4 | 3.26(2) | 0.0016(3) | ||
| VO2+ in N,N′-dimethylpropyleneurea, 0.20 mol·dm−3,L6 | |||||
| [VO2(dmpu)3]+ | V=O | 2 | 1.652(3) | E | |
| V-O | 3 | 2.049(2) | |||
| V--C | 3 | 3.016(6) | |||
| V-O-C | 6 | 3.148(9) | |||
Confirmation of crystal structure of VO(H2O)5(CF3SO3)2 (1)
The crystal structure of pentaaquaoxovanadium(IV) triflouromethanesulfonate, 1, has been reported previously.39i The reported structure, crystallizing in space group P21/m (No. 11), is confirmed in this study with near-identical structure parameters, Table 2 and Figure S2. Selected bond distances and bond angles are listed in Tables S5a and S5b, respectively.
Structure of the hydrated oxovanadium(IV) ion in aqueous solution
The EXAFS and LAXS data show that the structure of the hydrated oxovanadium(IV) ion in aqueous solution is in principle the same as observed in the solid state. The refinement of the EXAFS data gave mean V=O, V-Operp and V-Otrans bond distances of 1.588(3), 2.026(3) and 2.24(6) Å, respectively, Table 3 and Figures 1 and 2. The RDF from the LAXS experiments on the aqueous oxovanadium(IV) perchlorate solutions reveal a total of four peaks or shoulders at 1.5, 2.1, 2.9 and 4.1 Å, Figure 3. The peak at 1.5 Å corresponds to the Cl-O bond distance in the perchlorate ion and the V=O bond in the oxovanadium(IV) ion. The structure of the hydrated perchlorate ion is identical to those reported in previous studies of perchlorate salts in aqueous solution, with a Cl-O bond distance of 1.453(5) and 1.436(6) Å, in the dilute and concentrated solution, respectively.61 The peak/shoulder at 2.05 Å corresponds to the perpendicularly bound water molecules, while the peak at 4.1 Å corresponds with the second hydration shell of water molecules hydrogen bound to perpendicularly bound waters. The peak at 2.9 Å corresponds to O···O distances in the aqueous bulk. The refined structure parameters from the LAXS measurements are summarized in Table 3. The V=O bond distance of the strongly bound oxo group in hydrated oxovanadyl(IV) ion is 1.61(1) Å, the water molecules bound perpendicular to the V=O bond display a V-O bond distance of 2.03(2) Å, while the water molecule trans to the oxo group is significantly more weakly bound at ca. 2.20 Å, Table 3.
Figure 2.

Experimental (black lines) and modeled (red lines) Fourier transforms of the EXAFS data, a/ [V(H2O)6]3+ in water, b/ [V(OH)3(H2O)3] in water, c/ [VO(H2O)5]2+ in water, d/ solid [VO(H2O)5]SO4, e/ [VO(OSMe2)5]2+ in dmso, f/ solid [VO(OSMe2)5](CF3SO3)2, g/ [VO(dmpu)4]2+ in dmpu, h/ [VO2(H2O)4]+ in water, i/ [VO2(OSMe2)4]+ in dmso, j/ solid [VO2(OSMe2)4]CF3SO3, and k/ [VO2(OSMe2)3]+ in dmpu.
Figure 3.

(Top) LAXS radial distribution curves for a 1.228 mol·dm−3 aqueous solution of oxovanadium(IV) perchlorate, L1a. Upper part: Separate model contributions (offset: 18) of the hydrated oxovanadium(IV) ion (green line), the perchlorate ion (purple line) and aqueous bulk structure (orange line). (Middle) Experimental RDF: D(r)−4πr2 ρo (red line); sum of model contributions (black line); difference (blue line).
(Bottom) Reduced LAXS intensity functions s·i(s) (black line); model s·icalc(s) (red line).
The dimethylsulfoxide solvated oxovanadyl(IV) ion, VO(dmso)52+
The structure of solid pentakis(dmso)oxovanadium perchlorate, 2a, was solved and refined in the monoclinic space group P21/c (no. 14) with improved structural parameters compared to a previously reported determination of the same compound using the non-standard setting P21/n (no. 14).40b The sole crystallographically independent pentakis(dmso)oxovanadium(IV) structural unit has a short V=O bond, dV=O = 1.575(5) Å, four longer ones perpendicular to the V=O bond in the range of 1.999–2.021 Å, mean 2.007 Å, while the fifth dmso molecule, trans to the V=O bond is much more weakly bound, dV-O = 2.168(5) Å, Figure 4. The deviation from the horizontal plane is within 7.5±1.0° for the four dmso ligands, and the trans O=V-O bond angle is 178°. In one of the dmso ligands the sulfur atom has an alternate position with a refined 50:50 distribution. One of the trifluoromethanesulfonate ions was refined with a structural disorder with two superimposed ions sharing a common S-C bond. Crystallographic data and selected bond distances and angles are listed in Tables 2, S6a and S6b, respectively.
Figure 4.
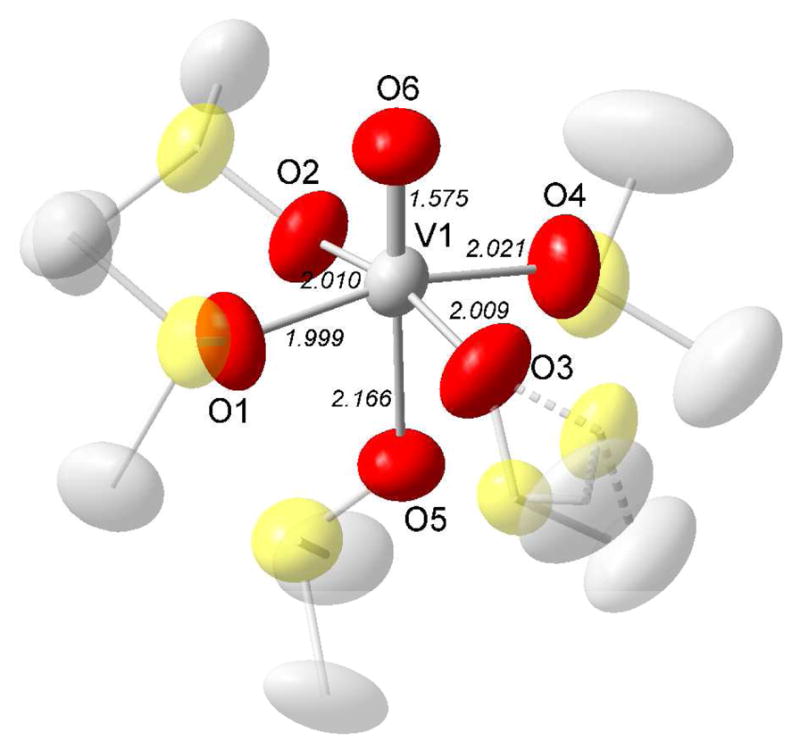
Representation of the oxovanadium(IV) cation complex in 2a. The thermal ellipsoids are set at 50% probability. The carbon and sulfur atoms have been faded, and the hydrogens and counter ions removed for clarity. The alternate position of one of the sulfur atoms is indicated by a dashed bond (faded).
The EXAFS and LAXS data show that the dmso solvated oxovanadium(IV) ion has the same structure in solution as in the solid pentakis(dmso)oxovanadium(IV) perrhenate,40d perchlorate,40b and iodide40c salts reported previously, Table S3b, and in this study. The refined structure parameters of the dmso solvated oxovanadium(IV) are given in Table 3, and the fits of the experimental data are given in Figures 1, 2 and 5.
Figure 5.

(Top) LAXS radial distribution curves for a 0.369 mol·dm−3 dimethylsulf-oxide solution of oxovanadium(IV) perchlorate, L2a. Upper part: Separate model contributions (offset: 40) of the hydrated oxovanadium(IV) ion (green line), the perchlorate ion (purple line) and dimethylsulfoxide (yellow line). (Middle) Experimental RDF: D(r)−4πr2ρo (red line); sum of model contributions (black line); difference (blue line).
(Bottom) Reduced LAXS intensity functions s·i(s) (black line); model s·icalc(s) (red line).
The N,N′-dimethylpropyleneurea solvated oxovanadium(IV) ion, VO(dmpu)42+
The structure of the tetrakis(dmpu)oxovanadium trifluoromethanesulfonate, 3, was solved and refined in the monoclinic space group P21/c (no. 14). The structure consists of discrete tetrakis(dmpu)oxovanadium(IV) and trifluoromethanesulfonate ions. Due to the space-demanding properties of the dmpu ligand, the oxovanadium(IV) ion binds only four dmpu molecules at a mean V-O bond distance of 1.971 Å, and the V=O distance is 1.576 Å, Figure 6. The trans position is unoccupied due to the space required by the four dmpu molecules which also cause or allow the equatorial V-O bonds to be slightly shorter than in the pentasolvated oxovanadium(IV) ions. Furthermore, the O=V-O bond angle deviation from a theoretical horizontal plane of a regular square pyramidal structure is much larger with a mean value of about 17°. In the structural refinement, one point of unusually high residual electron density (1.7 e) was located trans to the V1=O10 bond, but cannot be logically attributed to any real atom. Crystallographic data and selected bond distances and angles are listed in Tables 2, S7a and S7b, respectively. The structure of the dmpu solvated oxovanadium(IV) ion is maintained in dmpu solution with mean V=O and V-O bond distances of 1.580(5) and 1.971(3) Å, respectively. The refined structure parameters of the dmpu solvated oxovanadium(IV) are given in Table 3, and the fits of the experimental data are given in Figures 1 and 2.
Figure 6.

Representation of the two crystallographically independent oxovanadium(IV) cation complex in 3. The thermal ellipsoids are set at 30% probability. The carbon and nitrogen atoms have been faded, and the hydrogens and counter ions removed for clarity.
The hydrated dioxovanadium(V) ion
The hydrated dioxovanadium(V) ion, VO2+(aq), has been studied in acidic aqueous solution. Several attempts to crystallize the hydrated dioxovanadium(V) ion with perchlorate and trifluoromethanesulfonate as counter ions failed, see Experimental section. The hydrated dioxovanadium(V) ion seems to be stabilized in concentrated sulfuric acid; the high sulfate concentration allows complexation with sulfate or hydrogen sulfate ions. The Fourier transform of hydrated dioxovanadium(V) ion shows three different V-O distances, 1.6, 2.0 and 2.1 Å, corresponding to the oxo groups, which from other oxovanadium(V) complexes are shown to be in cis configuration, and to the water molecules in cis and trans position to the oxo groups, respectively. The same structure modeled the LAXS data very well. The refined structure parameters from the EXAFS and LAXS studies are summarized in Table 3, and the fit of the experimental data are given in Figures 1, 2 and 7.
Figure 7.

(Top) LAXS radial distribution curves for a 1.833 mol·dm−3 aqueous solution of dioxovanadium(IV) sulfate, L4a. Upper part: Separate model contributions (offset: 18) of the hydrated dioxovanadium(V) ion (green line), the sulfate ion (purple line), aqueous bulk structure (yellow line) and V···S distances (magenta line). (Middle) Experimental RDF: D(r)−4πr2ρo (red line); sum of model contributions (black line); difference (blue line).
(Bottom) Reduced LAXS intensity functions s·i(s) (black line); model s·icalc(s) (red line).
The dmso solvated dioxovanadyl(V) ion, VO2(dmso)4+
The crystal structure of tetrakis(dmso)dioxovanadium(V) trifluoromethanesulfonate, 5, was solved and refined in the space group P21/c (no. 14). The oxovanadium(V) cation is solvated by four dimethylsulfoxide molecules through their oxygen atoms. The coordination around vanadium is a highly distorted octahedron with the strongly bound oxo groups in cis configuration with V=O bond distances of 1.591(5) and 1.594(5) Å, and an O=V=O angle of 104.0(3)°, Figure 8. These V=O distances are slightly shorter than analogous distances in dioxovanadium(V) compounds reported before, which usually have values longer than 1.6 Å in complexes with inorganic as well organic oxygen donor ligands, Table S4.23–26 However, the current compound is, to the best our knowledge, the first such example of a discrete dioxovanadium(V) cation being six-coordinate with non-charged ligands. The V-O distances trans to the oxo groups are relatively long, 2.201 and 2.206 Å, while the dmso molecules perp to both oxo groups have V-O bond distances of 1.985 and 2.048 Å. These two perp dmso molecules are disordered and split into two alternative positions, (O13 and O13b) and (O16 and O16b) which were refined with occupancy factors of 55 and 45 per cent for each pair, respectively. All O-V-O angles deviate significantly from 90 and 180°. In addition, the vanadium atom is displaced from the center of the octahedron towards the two terminal oxo ligands. The O=V=O angle in 5, 104.0(3)°, is in close agreement those observed in [VO2(SO4)2(OH2)2]3− at 103.80°,31c and VO2IO3(H2O)2 at 104.82°.30b In comparison, in the dioxovanadium(V) complexes with organic molecules the cis O=V=O angle is generally 3–4° larger23–26 than reported here.
Figure 8.

Representation of the oxovanadium(IV) cation complex in 5. The thermal ellipsoids are set at 50% probability. The carbon and sulfur atoms have been faded, and the hydrogens and counter ions removed for clarity. The alternate position of one of the sulfur atoms is indicated by a dashed bond (faded).
The intramolecular bond distances within the dmso molecules in 5 are in good agreement with distances found for other compounds with oxygen-coordinated dmso ligands.41 The mean V-O-S angle in 5, 120.2°, is significantly smaller than in 2a, 128.4°, which may indicate a higher degree of covalency in the VV-O bonds than the corresponding VIV-O ones. The atomic positions of the trifluoromethanesulfonate ions are fairly well-defined and give bond distances in good agreement with other determinations.41
The radial distribution function (RDF) of the dmso solution of dioxovanadium(V) sulfate studies by LAXS is shown in Fig. 9. The S-O interactions from the sulfate anion and the S-O and C-S interactions from the dimethylsulfoxide molecule are all contained within the broad unresolved peak around 1.6 Å. The broad peak at about 2.2 Å corresponds to the V-O bonds interactions but also to the non-bonding interactions within the sulfate and dmso species. The small peak at 3.15 Å is due to the V···S distance from coordinated dimethylsulfoxide molecules and coordinated sulfate ions. Dimethylsulfoxide is known to reveal a strong proton accepting ability.62 In case of presence of considerable excess amounts of sulfuric acid in this solution, the bis-dimethylsulfoxide cations, (dmso)2H+, are formed, which results in an extremely short O···O distance at about 2.5 Å.63
Figure 9.

(Top) LAXS radial distribution curves for a 0.50 mol·dm−3 dimethylsulfoxide solution of oxovanadium(IV) perchlorate, L5a. Upper part: Separate model contributions (offset: 18) of the hydrated oxovanadium(IV) ion (green line), the perchlorate ion (purple line) and dimethylsulfoxide (yellow line). (Middle) Experimental RDF: D(r)−4πr2 ρo (red line); sum of model contributions (black line); difference (blue line).
(Bottom) Reduced LAXS intensity functions s·i(s) (black line); model s·icalc(s) (red line).
The N,N′-dimethylpropyleneurea solvated dioxovanadium(V) ion
The structure of the N,N′-dimethylpropyleneurea solvated dioxovanadium(V) ion, VO2(dmpu)3+, has been studied in dmpu solution by EXAFS, L6. The mean V=O and V-O bond distances are 1.652(2) and 2.048(3) Å, respectively, which clearly indicates that vanadium is strongly solvated by three dmpu molecules in a similar way previously shown for triphenylphosphine oxide, another ligand known to be space-demanding upon coordination.28 The V···C and V-O-C scattering paths are 3.02(1) and 3.15(2) Å, giving a V-O-C bond angle of 130°. Several attempts to crystallize the dmpu solvated dioxovanadium(V) ion with both perchlorate and trifluoromethanesulfonate as counter ions unfortunately failed.
Conclusions
The hydrated vanadium(III) ion has regular octahedral geometry with a mean V-O bond distance of 1.99 Å, yielding ionic radius of 0.65 Å, which is in excellent agreement with the value reported by Shannon.59 Hydrated oxovanadium(IV) ions have distorted octahedral configuration with a mean V=O bond distance to the oxo group of 1.586 Å, a mean V-O bond distance to the four water molecules almost perpendicular to the V=O bond of 2.02 Å, while the V-O bond trans to the oxo group is only weakly bound at ca. 2.2 Å. The solvated oxovanadium(IV) ions with small solvent molecules display structures very similar to the hydrate, while solvates with solvent molecules space demanding at coordination, as N,N′-dimethylpropyleneurea, are five-coordinate with a distorted square-pyramidal geometry. The V=O and V-O bond distances of the dmpu solvated oxovanadium(IV) ion are 1.58 and 1.97 Å, respectively. The hydrated and solvated dioxovanadium(V) ions have very distorted octahedral configurations with O=V=O angles in the range 103–109°. The mean V=O bond distance is 1.63 Å, and the V-O bond distances in the cis and trans positions to the oxo groups are 1.99 and 2.20 Å, respectively. A solvate of oxovanadium(V) with a space demanding solvent molecule, as dmpu and triphenyl phosphate, show a bicapped trigonal configuration with mean V=O and V-O bond distances of 1.617 and 2.020 Å, respectively.
Supplementary Material
Acknowledgments
We acknowledge the assistance from Ann-Sofi Ullström in obtaining most of the crystallographic data and Lars Eriksson at Stockholm University for obtaining the powder diffraction data. Portions of this research were carried out at the Stanford Synchrotron Radiation Lightsource (SSRL), and the MAX-Lab synchrotron radiation source at Lund University, Sweden, which are acknowledged for the allocation of beam time and laboratory facilities. Portions of this research were carried out at the Stanford Synchrotron Radiation Lightsource, a Directorate of SLAC National Accelerator Laboratory and an Office of Science User Facility operated for the U.S. Department of Energy Office of Science by Stanford University. The SSRL Structural Molecular Biology Program is supported by the DOE Office of Biological and Environmental Research, and by the National Institutes of Health, National Center for Research Resources, Biomedical Technology Program (P41RR001209). Portions of this research were carried out at beam-line I811, MAX-lab synchrotron radiation source, Lund University, Sweden. Funding for the beam-line I811 project was kindly provided by the Swedish Research Council and “Knut och Alice Wallenbergs Stiftelse.
Footnotes
Supporting Information Available: Summary of reported V-O and V=O bond distances of hydrate and solvate complexes of vanadium in the oxidation states +II, +III, +IV, and +V are given in Tables S1–S4, respectively. Selected bond distances and angles of 1, 2a, 3 and 5 are given in Tables S5–S8, and the packing of the unit cells of 1, 2a, 3 and 5 are shown in Figures S2–S5, respectively. The powder diffractogram of 4 is given in Figure S5. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Schneider CJ, Pecoraro VL. ACS Symposium Series 2. 2007;Ch 12:974. [Google Scholar]
- 2.Van Pee K-H, Keller S, Wage T, Wynands I, Schnerr H, Zehner S. Biol Chem. 2000;381:1–5. doi: 10.1515/BC.2000.001. [DOI] [PubMed] [Google Scholar]
- 3.Kirk O, Conrad LS. Angew Chem, Int Ed. 1999;38:977–979. doi: 10.1002/(SICI)1521-3773(19990401)38:7<977::AID-ANIE977>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- 4.Vilter H. Met Ions Biol Syst. 1995;31:325–362. [PubMed] [Google Scholar]
- 5.Winter JM, Bradley SM. J Biol Chem. 2009;284:18577–18581. doi: 10.1074/jbc.R109.001602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chasteen ND, editor. Vanadium in Biological Systems. Kluwer Academic Publishers; Boston: 1990. [Google Scholar]
- 7.Crans DC, Smee JJ, Gaidamauskas E, Yang Luqin. Chem Rev. 2004;104:849–902. doi: 10.1021/cr020607t. and references therein. [DOI] [PubMed] [Google Scholar]
- 8.Garner CD, Armstrong EM, Berry RE, Beddoes RL, Collison D, Cooney JJA, Ertok NS, Helliwell M. J Inorg Biochem. 2000;80:17–20. doi: 10.1016/s0162-0134(00)00034-9. [DOI] [PubMed] [Google Scholar]
- 9.Slebodnick C, Hamstra BJ, Pecoraro VL. Struct Bonding. 1997;89:51–108. [Google Scholar]
- 10.Bayer E. Met Ions Biol Syst. 1995;31:407–422. [Google Scholar]
- 11.Michibata H, Yamaguchi N, Uyama T, Ueki T. Coord Chem Rev. 2003;237:41–51. [Google Scholar]
- 12.Ueki T, Furuno N, Michibata H. Biochim Biophys Acta. 2011;1810:457–464. doi: 10.1016/j.bbagen.2010.12.006. [DOI] [PubMed] [Google Scholar]
- 13.Thompson KH, Orvig C. J Inorg Biochem. 2006;100:1925–1935. doi: 10.1016/j.jinorgbio.2006.08.016. [DOI] [PubMed] [Google Scholar]
- 14.Sakurai H, Fujisawa Y, Fujimoto S. J Trace Elements in Exp Med. 1999;12:393–401. [Google Scholar]
- 15.Poucheret P, Verma S, Grynpas MD, McNeill JH. Mol Cellular Biochem. 1998;188:73–80. [PubMed] [Google Scholar]
- 16.Wei Y, Zhang C, Zhao P, Yang X, Wang K. J Inorg Biochem. 2011;105:1081–1085. doi: 10.1016/j.jinorgbio.2011.05.008. [DOI] [PubMed] [Google Scholar]
- 17.Rehder D, Costa Pessoa J, Geraldes CFGC, Kabanos T, Kiss T, Meier B, Micera G, Pettersson L, Rangel M, Salifoglou A, Turel I, Wang D. J Biol Inorg Chem. 2002;7:384–396. doi: 10.1007/s00775-001-0311-5. [DOI] [PubMed] [Google Scholar]
- 18.Gätjens J, Meier B, Adachi Y, Sakurai H, Rehder D. Eur J Inorg Chem. 2006:3575–3585. [Google Scholar]
- 19.Maurya MR, Agarwal S, Abid M, Azam A, Bader C, Ebel M, Rehder D. Dalton Trans. 2006:937–947. doi: 10.1039/b512326g. [DOI] [PubMed] [Google Scholar]
- 20.Marouane W, Soussi A, Murat J-C, Bezzine S, El Feki A. Lipids Health Disease. 2011;10:65. doi: 10.1186/1476-511X-10-65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wever R, Renirie R, Hasan Z. Encyclopedia of Inorganic Chemistry. John Wiley & Sons; 2006. Vanadium in Biology. [Google Scholar]
- 22.Nechay BR. Ann Rev Pharmacol, Toxicol. 1984;24:501–524. doi: 10.1146/annurev.pa.24.040184.002441. [DOI] [PubMed] [Google Scholar]
- 23.Rehder D. Coord Chem Rev. 1999;182:297–322. [Google Scholar]
- 24.Ma L, Yang S. Adv Mater Res. 2012;396–398:267–273. [Google Scholar]
- 25.Baes CF, Jr, Mesmer RE. The Hydrolysis of Cations. Chap 10.2.5–10.2.8. Krieger Publishing Company; Malabar, Fl. USA: 1976. [Google Scholar]
- 26.Dissociation Constants of Acids and Bases CRC Handbook of Chemistry and Physics. (92) 2011–2012;5 [Google Scholar]
- 27.Pettersson L, Andersson I, Ingri N. Chem Scr. 1983;22:254–264. [Google Scholar]
- 28.Pettersson L, Andersson I, Hedman B. Chem Scr. 1985;25:309–317. [Google Scholar]
- 29.Davis MF, Jura M, Leung A, Levason W, Littlefield B, Reid G, Webster M. Dalton Trans. 2008:6265–6273. doi: 10.1039/b811422f. [DOI] [PubMed] [Google Scholar]
- 30.a) Leclaire A, Borel MM, Raveau B. J Solid State Chem. 2002;163:534–539. [Google Scholar]; b) Bingping Yang, Chunli Hu, Xiang Xu, Fu Sun Chuan, Han Zhang Jian, Gao Mao Jiang. Chem Mat. 2010;22:1545–1550. [Google Scholar]; c) Bircsak Z, Harrison WTA. J Solid State Chem. 1998;140:272–277. [Google Scholar]
- 31.a) Zubkov VG, Tyutyunnik AP, Berger IF, Krasil’nikov VN, Perelyaeva LA, Baklanova IV. Russ J Inorg Chem. 2007;52:1415–1423. [Google Scholar]; b) Meschede W, Mattes R. Z Anorg Allg Chem. 1976;419:25–30. [Google Scholar]; c) Hashimoto M, Kubata M, Yagasaki A. Acta Crystallogr, Sect C. 2000;56:1411–1412. doi: 10.1107/s0108270100012063. [DOI] [PubMed] [Google Scholar]; d) Leclaire A, Borel MM, Chardon J, Raveau B. Solid State Sci. 2000;2:293–297. [Google Scholar]; e) Hyejin Kim, Yoonsuk Cho, Hoseop Yun, Junghwan Do. Z Anorg Allg Chem. 2007;633:473–477. [Google Scholar]; f) Drew RE, Einstein FWB, Gransden SE. Can J Chem. 1974;52:2184–2189. [Google Scholar]
- 32.a) Scheidt WR, Tsai C, Hoard JL. J Am Chem Soc. 1971;93:3867–3872. [Google Scholar]; b) Atovmyan LO, Sokolova YA. Russ J Struct Chem. 1971;12:863–864. [Google Scholar]; c) Stomberg R. Acta Chem Scand, Ser A. 1986;40:168–176. [Google Scholar]; e) Jakusch T, Dean A, Oncsik T, Csaba Benyei A, Di Marco V, Kiss T. Dalton Trans. 2009;39:212–220. doi: 10.1039/b914849c. [DOI] [PubMed] [Google Scholar]
- 33.Selbin J. Chem Rev. 1965;65:153–175. [Google Scholar]
- 34.Selbin J. Coord Chem Rev. 1966;1:293–314. [Google Scholar]
- 35.a) Vilas Boas LV, Costa Pessoa J. In: Comprehensive Coordination Chemistry. Wilkinson G, Gillard RD, McCleverty JA, editors. Vol. 3 Pergamon Press; New York: 1987. [Google Scholar]; b) Franchavilla J, Chasteen ND. Inorg Chem. 1975;14:2860–2862. [Google Scholar]; c) Henry RP, Mitchell PC, Prue JE. J Chem Soc, Dalton Trans. 1973:1156–1159. [Google Scholar]; d) Rossotti FJC, Rossotti HS. Acta Chem Scand. 1955;9:1177–1192. [Google Scholar]; d) Mateo S, Brito F. An Quim. 1966;62B:123, 193. [Google Scholar]
- 36.Francavilla J, Chasteen ND. Inorg Chem. 1975;14:2860–2862. [Google Scholar]
- 37.Komura A, Hayashi M, Imanaga H. Bull Chem Soc Jpn. 1977;50:2927–2931. [Google Scholar]
- 38.Iannuzzi MM, Rieger PH. Inorg Chem. 1975;14:2895–2899. [Google Scholar]
- 39.a) Tézé A, Marchal-Roch C, So H, Fournier MT, Hervé G. Solid State Sci. 2001;3:329–338. [Google Scholar]; b) Hawthorne FC, Schindler M, Grice JD, Haynes P. Can Min. 2001;39:1325–1331. [Google Scholar]; c) Tachez M, Theobald FR, Watson KJ, Mercier R. Acta Crystallogr, Sect B. 1979;35:1545–1550. [Google Scholar]; d) Tachez M, Theobald FR. Acta Crystallogr, Sect B. 1980;36:249–254. [Google Scholar]; e) Tachez M, Theobald FR. Acta Crystallogr, Sect B. 1980;36:1757–1761. [Google Scholar]; f) Hjorth M, Norrestam R, Johansen H. Acta Crystallogr, Sect B. 1990;46:1–7. [Google Scholar]; g) Li Lianzhi, Xu Tao, Wang Daqi, Niu Meiju. Acta Crystallogr, Sect E. 2004;60:m1374–m13775. [Google Scholar]; h) De Munno G, Bazzicalupi C, Faus J, Lloret FL, Julve M. J Chem Soc, Dalton Trans. 1994:1879–1884. [Google Scholar]; i) Magnussen M, Brock-Nannestad T, Bendix J. Acta Crystallogr, Sect E. 2007;63:m51–m52. doi: 10.1107/S010827010605219X. [DOI] [PubMed] [Google Scholar]
- 40.Sabirov VH, Khodzhaev OF, Litvinov IA, Musaev ZM, Usmankhodzhaeva Y. Koord Khim. 1991;17:325–329. [Google Scholar]; b) Khodashova TS, Porai-Koshits MA, Sergienko VS, Butman LA, Uslaev YA, Kovalev VV, Kuznetsova AA. Koord Khim. 1978;4:1909–1911. [Google Scholar]; c) Seifert HJ, Uebach J. Z Anorg Allg Chem. 1981;479:32–40. [Google Scholar]; d) Khodashova TS, Porai-Koshits MA, Buslaev YA, Kuznetsova AA, Kobalev VV. Koord Khim. 1977;3:38–40. [Google Scholar]
- 41.Allen FH. Acta Crystallogr, Sect B. 2002;58:380–388. doi: 10.1107/s0108768102003890. Cambridge Structure Database (Conquest 1.13) [DOI] [PubMed] [Google Scholar]
- 42.Inorganic Crystal Structure Database 1.4.6, Release 2011–2, FIZ Karlsruhe, 2011.
- 43.Loeffler H, Iglesias Yagüe J, Rode BM. Chem Phys Lett. 2002;363:367–371. [Google Scholar]
- 44.Buglyó P, Crans DC, Nagy EM, Lindo RL, Yang Luqin, Smee JJ, Jin Wenzheng, Chi Lai-Har, Godzala ME, Willsky GR. Inorg Chem. 2005;44:5416–5427. doi: 10.1021/ic048331q. [DOI] [PubMed] [Google Scholar]
- 45.Kanamori K. Coord Chem Rev. 2003;237:147–161. [Google Scholar]
- 46.Hedwig GR, Parker AJ. J Am Chem Soc. 1974;96:6589–6593. [Google Scholar]
- 47.Bruker, Bruker SMART and SAINT, Area Detector Control and Integration Software. Bruker Analytical X-ray Systems; Madison WI, USA: 1995. [Google Scholar]
- 48.Sheldrick GM. SHELXL-97. University of Göttingen; Germany: 1997. [Google Scholar]
- 49.Thompson A, Attwood D, Gullikson E, Howells M, Kim K-J, Kirz J, Kortright J, Lindau I, Pianatta P, Robinson A, Scofield J, Underwood J, Vaughan D, Williams G, Winick H. LBNL/PUB-490 Rev 2. Lawrence Berkeley National Laboratory; Berkeley, California 94720: 2001. X-ray Data Booklet. [Google Scholar]
- 50.George GN, Pickering IJ. EXAFSPAK – A Suite of Computer Programs for Analysis of X-ray Absorption Spectra. SSRL; Stanford, CA: 1993. [Google Scholar]
- 51.a) Zabinsky SI, Rehr JJ, Ankudinov A, Albers RC, Eller MJ. Phys Rev B. 1995;52:2995–3009. doi: 10.1103/physrevb.52.2995. [DOI] [PubMed] [Google Scholar]; b) Ankudinov AL, Rehr JJ. Phys Rev B. 1997;56:R1712–R1715. the FEFF program is available at http://feff.phys.washington.edu/feff/ [Google Scholar]
- 52.Johansson G, Sandström M. Chem Scr. 1973;4:195–197. [Google Scholar]
- 53.Stålhandske CMV, Persson I, Sandström M, Kamienska-Piotrowicz E. Inorg Chem. 1997;36:3174–3182. doi: 10.1021/ic961339i. [DOI] [PubMed] [Google Scholar]
- 54.Wilson AJC. International Tables for X-ray Crystallography. C4. Kluwer Academic Publishers; Dordrecht, The Netherlands: Kynoch Press; Birmingham: 1995. 1974. [Google Scholar]
- 55.Cromer DT. J Chem Phys. 1969;50:4857–4859. [Google Scholar]
- 56.Cromer DT, Mann JB. J Chem Phys. 1967;47:1892–1894. [Google Scholar]
- 57.Levy HA, Danford MD, Narten AH. Report ORNL-3960. Oak Ridge National Laboratory; Oak Ridge: 1966. Data Collection and Evaluation with an X-Ray Diffractometer Designed for the Study of Liquid Structure. [Google Scholar]
- 58.Molund M, Persson I. Chem Scr. 1985;25:197–197. [Google Scholar]
- 59.Shannon RD. Acta Crystallogr, Sect A. 1976;32:751–767. [Google Scholar]
- 60.Beattie JK, Best SP, Skelton BW, White AH. J Chem Soc, Dalton Trans. 1981:2105–2111. [Google Scholar]
- 61.Persson I, Eriksson L, Lindqvist-Reis P, Persson P, Sandström M. Chem Eur J. 2008;14:6687–6696. doi: 10.1002/chem.200800225. [DOI] [PubMed] [Google Scholar]
- 62.Caesar B, Johnsson M, Persson I. Inorg Chim Acta. 1987;128:L23–L24. [Google Scholar]
- 63.a) Cherkasova TG, Goryunova IP. Zh Neorg Khim (Russ J Inorg Chem) 2004;49:26. [Google Scholar]; b) Lobadyuk VI, Spevak VN, Skvortsov NK, Stash AI, Bel’sky VK. Zh Obshch Khim (Russ J Gen Chem) 1996;66:705. [Google Scholar]; c) Kotel’nikova AS, Osmanov NS, Koz’min PA, Surazhskaya MD, Larina TB, Abbasova TA, Lebedev VG. Zh Neorg Khim (Russ J Inorg Chem) 1988;33:2681. [Google Scholar]; d) Rudnitskaya OV, Buslaeva TM, Koteneva NA, Stash AI. Izv Akad Nauk SSSR, Ser Khim(Russ)(Russ Chem Bull) 1994:1333. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.