

Abstract
The immune surveillance hypothesis proposed over 50 years ago that many precancerous lesions are eliminated without a histological trace due to immunological pressure. Since then, it has become apparent that both the tumor and the anti-cancer immune response evolve over a long period to allow the eventual escape of nascent precancerous lesions into full-blown tumors. Although primarily focusing on loss of antigenicity, the immunoediting hypothesis has gradually evolved to appreciate the role of active immunosuppression in tumor progression, where myeloid leukocytes are increasingly recognized as the major driving force. This review highlights recent studies implicating how myeloid cells with antigen-presenting capabilities are co-opted by tumors to promote malignant progression. Because at least some advanced tumors remain significantly immunogenic, these new studies add a tweak to the immunoediting hypothesis as well as a rationale to block immunosuppressive mechanisms as a first-line intervention in cancer patients.
Keywords: Tumor microenvironment, immunosuppression, immunoediting, immunotherapy, tumor immunology, dendritic cell, immune surveillance
1. INTRODUCTION
Burnet [1] and Thomas [2] originally posited that nascent tumor lesions were eliminated by the immune system without a pathological trace. Since then, overwhelming experimental evidence demonstrates that both the innate and adaptive immune systems play a non-redundant role in the prevention or promotion of tumorigenesis. Immune recognition of tumor antigens lead to the formulation of the cancer immunoediting hypothesis, which supports that immune pressure - primarily mediated by T cells - results in progressive loss of antigens (editing) by tumor cells, eventually allowing them to escape from accumulating immune pressure [3]. Loss of natural, spontaneous (relevant) antigens has been conclusively demonstrated in carcinogen-induced tumor models [4]. However, T cell infiltration is clearly associated with superior outcomes in patients with many different tumors [5–8], while clinically relevant responses have been achieved against many tumors using T cell based immunotherapies [9–11]. Most importantly, emerging clinical evidence indicates that blockade of immunosuppressive signals such as CTLA4 and, especially, PD-1/PD-L1, allows the immune system to regain control of the progression of a variety of tumors [12, 13]. These clinical data and recent experimental evidence produced by our group [14] support that advanced tumors remain sufficiently immunogenic for effective control by the immune system, adding weight to the role of immunosuppression as a major driver of malignant progression. Pathological expansion of a heterogeneous population of immature myeloid cells with immunosuppressive activity is a hallmark of virtually all solid tumor-bearing hosts, and these cells are emerging as key players of immune regulation in the tumor microenvironment (TME) [11]. Paradoxically, myeloid leukocytes with antigen-presenting capabilities are required for the orchestration of tumor-specific T cell responses. Correspondingly, we recently identified a progressive phenotypic and numerical switch in dendritic cell (DC) populations in tumor-draining lymph nodes, parallel to both malignant progression and the abrogation of T cell-mediated protection [14]. The pivotal interplay between lymphoid and myeloid cells in the TME for preventing tumorigenesis vs. dampening the anti-tumor immune responses, and how to modulate it in vivo to control established tumors, will be the focus of this review.
2. Innate and adaptive immunity during tumor initiation and malignant progression
Studies using mice deficient in immune effector molecules have emphasized the critical role of innate and adaptive immunity in tumor initiation and malignant progression. Challenge of these immune-deficient mice with chemical carcinogens such as methylcholanthrene (MCA) or 7,12-dimenthylbenz[a]-anthracen (DMBA)/12-O-tetradecanoyl phorbol-13 acetate (TPA), resulted in accelerated generation of sarcomas or skin tumors compared to control WT mice with fully functional immune effector molecules (reviewed in [15]). Innate cells such as NK, NKT cells, γδ T cells, eosinophils [15, 16] and neutrophils [17–19] mediate immune protective or tumor promoting functions in experimental models for cancers, similar to what was observed in cancer patients. NK cells in particular appear to be critical for the rejection of nascent tumors [20]. However, so far only T cells in the TME have been associated with clinically relevant immune pressure against the progression of the established tumors that are detectable in the clinic. Thus, although it is theoretically possible that NK cells “hit and run”, still being important although absent from tumor locations, current clinical evidence supports that the adaptive immune system, and in particular T cells, are the crucial effector immune cells that remain able to exert some significant (although obviously suboptimal) anti-tumor activity in advanced malignancies.
The specific T cell subsets empowered with anti-tumor activity are the subject of intense debate, because both γδ T cells and CD4 αβ lymphocytes include cells with enough spontaneous regulatory activity to dampen protective immunity [21, 22]. Consequently, some studies have restricted the protective role of tumor-infiltrating lymphocytes to CD8 T cells [23], although both CD4 and γδ T cells are known to contribute to the orchestration and maintenance of adaptive effective immune responses through a variety of cytotoxic [24–26] and non-cytotoxic mechanisms [27–29]. Nevertheless, the prognostic value of T cell responses implies that antigen-presenting cells are able to effectively prime T lymphocytes at some point during tumor progression. Consequently, it has been shown that CD8α+ DCs are important in cross-presenting the tumor antigens to CD8+ T cells, so that in Batf3-deficient CD8α+ DCs, T cell mediated tumor rejection is impaired [30]. Furthermore, we have shown that the elimination of DCs in nascent tumor-bearing hosts dramatically accelerates malignant progression in an ovarian cancer model, which is restrained by CD8 T cells [14]. Because T cell infiltration is clinically relevant, and also because antibodies against tumor antigens are detected in a variety of cancer patients (which requires the activation of at least CD4 T cells), effective cooperation between T cells and DCs presenting tumor antigens appears to be taken place at initial stages of tumor progression. However, despite the fact that immune system can mount strong anti-tumor responses, tumors still evade the immune pressure [31]. Although the “self” nature of non-viral tumor antigens partially explains suboptimal T cell responses, the activity of tumor-specific T cells is further paralyzed in the TME through multiple complementary mechanisms.
3. Tumor immunoediting
Seminal studies by Schreiber and colleagues using chemical carcinogens such as MCA in immunocompromised mice conclusively demonstrated that tumors developed in the absence of adaptive immune system are more immunogenic in subsequent transplantation into WT mice [32]. IFNγ was found to be the principle molecule involved in tumor cell editing and both CD4 and CD8 T cells are the mediators of this strong anti-tumor response [32]. These studies are the cornerstone of the immunoediting hypothesis, which is the current framework accepted by most tumor immunologists [3, 33, 34]. The immunoediting hypothesis proposes that adaptive immune response not only regulates the quantity but also the quality of anti-tumor immunity, and has three important windows in which anti-tumor immune responses occurs. It starts with an elimination phase, during which cells of the innate and adaptive immune system eliminate cells undergoing transformation. If this elimination is complete, tumors disappear at this stage. Though experimental data supports this elimination phase, it cannot be characterized in humans because these events take place before tumors become detectable, if they ever become established. If tumor cells escape immune rejection, tumor progression goes through an equilibrium phase whereby tumors are kept under the control of effective immune responses, primarily mediated by components of the adaptive immune system. This phase culminates with three possible outcomes: First, the immune system can override the tumor cells and eliminate them. Second, this phase is continual and individuals remain free of clinically relevant tumors for their life-time. The third possibility is that adaptive immunity edits the tumors in such a way that new tumor cell variants develop, for which no T cell clones exist in the immune system. In that the case, edited tumor cells were proposed to evade the immune pressure, leading to accelerated expansion and, subsequently, development of clinical symptoms [3, 33–35].
While progressive loss of antigenicity has been experimentally supported and has provided a valuable framework for years, most data supporting the editing hypothesis derive from chemically (MCA or DMBA/TPA) induced tumors, or cell lines derived from them (reviewed in [15]). The value of artificial antigens that do not reflect the mild responses induced by tumor antigens (such as ova [36]) should be interpreted with more caution. The issue associated with chemically induced tumor models is that high degree of variability in mutated antigens between each mouse in the study groups as chemicals induce random mutations. Therefore, immune responses as well as editing will be variable in these models. Furthermore, experiments in which secondary transfer of transplanted tumor cells result in tumor escape may be due to deregulation and enhancement of the proliferative capacities of tumor cells [37], and not necessarily alterations in the antigenic repertoire. A seminal step to define these mechanisms was provided by a recent cancer exome analysis of MCA induced sarcomas, which identified spectrin-b2 as a potential tumor rejection antigen in MCA and T cells selectively exclude the cells expressing this mutations during the course of tumor evasion [4]. However, the fact that multiple established tumors become at least partially immunologically controlled simply by blocking T cell checkpoints [12], indicates that tumors cells can be still recognized by the immune system and therefore retain the expression of relevant antigens, which is further supported by our recent experimental observations [14]. The cancer immunoediting hypothesis has correspondingly evolved to integrate immunosuppression (in addition to loss of immunogenicity) as a relevant mechanism behind the escape phase of tumor progression [38]. The question is which is the predominant mechanism in the oncogene-driven tumors that take place in humans?
4. T cell unresponsiveness in the tumor microenvironment
Recently the hallmarks of cancer have been modified to incorporate additional characteristics of cancer in the context of how cancer subverts the immune system. These include tumor-promoting inflammation, reprogramming energy metabolism, and evasion of the immune system [39]. Thus, solid tumors maintain an immunosuppressive, hypoxic and hostile environment that directly affects the effector function of T cells. Sustained exposure to suboptimal antigen levels and multiple suppressive factors can result in unresponsiveness through T cell exhaustion, anergy or senescence, 3 mechanisms that use different molecular pathways [40]. Studies in chronic viral infections have unveiled that T cell exhaustion is characterized by a progressive weakening of effector activity, expression of inhibitory receptors (e.g., PD-1 TIM3, LAG-3 and CTLA-4 (reviewed in [41]) and a transcriptional state that includes the overexpression of Blimp-1 and T-bet, along with up-regulation of NFAT2 in the presence of suboptimal levels of AP1 [40]. An identical phenotype is identified in the microenvironment of many tumors, particularly in CD8 T cells, where the expression of inhibitory receptors is required for induction and maintenance of T cells in exhausted state. Ligands for these receptors are generally expressed by regulatory DCs and myeloid derived suppressor cells (MDSCs), in addition to tumor cells.
In contrast to the progressive nature of T cell exhaustion, anergy is rapidly initiated at the time of priming, and is characterized by the up-regulation of Rnf128, Egr2 and Egr3, and diminished Ras activation, along with excessive NFAT [42]. Maintenance of anergy is antigen independent while maintenance of exhaustion dependents on persistent antigen availability/TCR signaling [43]. Importantly, both exhaustion and anergy can be reversed through, respectively, the blockade of inhibitory pathways and cytokines [42][44, 45]. As commented above, emerging clinical evidence supports the promise of blocking some of these inhibitory receptors [12].
In addition to anergic and exhausted T cells, senescent lymphocytes with shortened telomeres that have reached their terminal replicative potential are also found in the TME, particularly in elderly patients. These cells are characterized by the expression of CD57 and the absence of CD28 [46], and unresponsiveness is considered to be permanent. Irreversible cell cycle arrest can also be caused by a signal transduction program induced by cellular stress [47], although these molecular pathways remain largely uninvestigated in T cells.
Besides intrinsic transcriptional programs leading to T cell unresponsiveness, many factors in the TME abrogate the activity of effector T cells. Interestingly, some of these mediators are not only produced by tumor cells, but also by DCs that, rather than promoting anti-tumor immunity, are transformed into immunosuppressive players. Those factors include Indoleamine 2,3-dioxygenase (IDO) and L-arginase, enzymes secreted by tumor cells, CD8α+ DCs with tolerogenic phenotypes and MDSCs [48, 49]. These enzymes deplete Amino Acids that are required for T cell functions from the TME [49, 50]. IDO catalyzes the tryptophan degradation in the kynurenine pathway [51]. Both the reduction in tryptophan concentration as well as accumulation of tryptophan metabolites is immunosuppressive. In addition, tumor-infiltrating DCs and MDSCs actively contribute to the suppression of anti-tumor CD8 T cells through the production of L-arginase [49, 52, 53]. Other potent immunosuppressive factors are secreted by both myeloid leukocytes and tumor cells, including TGFβb [41]. Therefore, tumor immune evasion is the outcome of complex immunosuppressive mechanisms paradoxically driven by myeloid leukocytes, which eventually paralyze protective T cell responses.
5. Myeloid leukocytes and tumor-induced immunosuppression
The presence of exhausted tumor-specific T cells and (CD4-dependent) tumor antigen- specific antibodies in most cancer patients indicates that at least a fraction of tumor-reactive lymphocytes are effectively primed at early stages of tumor progression. So, how are myeloid leukocytes responsible for the orchestration of adaptive immune responses turned into immunosuppressive cells in tumor-bearing hosts? The answer is that a hallmark of virtually all advanced solid tumors is excessive mobilization of bone marrow precursors of myeloid leukocytes (including macrophages, dendritic cells and granulocytes), in response to multiple inflammatory cytokines [54–57]. This heterogeneous population, globally termed MDSCs, home to tumor locations in response to multiple chemokines, but they also exert immunosuppressive activity beyond the TME (reviewed in [58]). Among the multiple tolerogenic mechanisms that they promote, nitration of tyrosines in TCR-CD8 complex appears to be particularly relevant [56, 59]. Once inside the TME, maturation of these myeloid cells into immunocompetent antigen-presenting cells is derailed, resulting in diminished adaptive immunity and eventual tumor escape. Thus, under hypoxic conditions, Ly6C+ MDSCs differentiate into immunosuppressive macrophages and DCs in solid tumors [60]. Correspondingly, the categorization of the highly heterogeneous myeloid populations that massively accumulate in solid tumors is complicated by a high degree of phenotypic overlap, different stages of differentiation, predominant inflammatory signals produced by every specific tumor, and the location and histological type of the tumor itself, among other factors. Applying the markers and functional attributes of leukocytes categorically defined under steady-state conditions to immune cells in the TME is therefore very challenging. Nevertheless, immunosuppressive, pro-angiogenic CD11b+CD68+MHC-II+ macrophages are represented in virtually all solid tumors. In addition, we have repeatedly demonstrated that the predominant leukocyte subset found in solid ovarian tumors (but not in human tumor ascites) co-expresses determinants of bona fide DCs, including CD11c, DEC205, CD86 and MHC-II, and in at least a third of clinical specimens lacks the macrophage markers CD11b and CD14 [61–67]. From their perivascular location, these myeloid cells abrogate the activity of anti-tumor T cells extravassating from blood vessels into the tumor microenvironment [66]. The expression of PD-L1 by ovarian cancer-associated DCs appears to be particularly relevant immunosuppressive mechanism, based on multiple converging lines of evidence [23, 63, 68]. Additionally, pDCs isolated from tumors in the prostate expressed high amounts of IDO and TGFβ to promote immune suppression and VEGF-A, and IL-6 to promote angiogenesis and metastasis [69]. Therefore, although the role of other immunosuppressive leukocyte subsets such as Treg is also relevant for tumor progression [21], the abundance and per cell immunosuppressive activity of myeloid cells in the TME indicates that this heterogeneous population is the major driving force for the abrogation of anti-tumor immunity in the TME. In addition, how and to what extent myeloid leukocytes control the conversion of inducible Treg remains largely uninvestigated.
6. Tumor mediated escape: Dendritic cell conversion
DCs and macrophages are sentinels in immunity, and are required to respond rapidly to infection or to be able to quickly modulate robust inflammatory responses. Because of this plasticity in function and phenotype, myeloid-derived cells are vulnerable to the polarizing signals elicited by the tumor and tumor microenvironment. For instance, mobilization of monocytes from the periphery due to recognition of bacterial ligands [70] or during inflammation in the intestine [71], can give rise to inflammatory dendritic cells and possibly conventional CD103+ DCs capable of inducing potent T cell responses. Conversely, tumor associated fibroblasts, through depletion of GMCSF in the tumor microenvironment, are capable of converting CD11c+ dendritic cells into macrophages with potent immunosuppressive capabilities [72]. To investigate the dynamics of plastic antigen-presenting cells from tumor initiation to terminal malignant progression, we recently generated an inducible model of ovarian carcinoma driven by mutations in oncogenes and suppressor genes, as it happens in humans [14]. As expected, we found that measurable tumor-specific T cell responses are orchestrated shortly after tumor initiation by immunocompetent DCs. These responses were enough to keep tumors as microscopic lesions for relatively long periods. Correspondingly, depletion of DCs 7 days after tumor challenge resulted in a dramatic acceleration of tumor growth.
Paradoxically, the initiation of malignant macroscopic expansion was dependent upon the accumulation of CD11c+DEC205+MHC-II+ DCs within the TME. However, these cells were not only unable to effectively present tumor antigens, but also abrogated the robust priming of T cells elicited by different immunocompetent DCs. Consistently, depletion of DCs at advanced stages of tumor progression significantly delayed tumor growth, allowing the immune system to regain control of tumors, again in the absence of any direct intervention on tumor cells [73]. These results demonstrate that myeloid leukocytes, and in particular DCs in ovarian tumors, govern malignant progression, as tumor growth can be modulated in opposite directions simply by eliminating this microenvironmental cell type at different stages. Our data also support that advanced tumors remain immunogenic, because cells from advanced tumors were able to induce significant T cell responses, particularly in lymphocytes derived from early tumors. Most importantly, our results provide a framework to understand the progression of aggressive epithelial tumors, whereby transition from microscopic lesions to exponentially growing masses could be occurring without a pre-malignant or dormant detectable lesion.
So, what factors induce the conversion of immunosuppressive DCs? Our data indicate that tumor cell derived PGE2 and TGFβ are sufficient to initiate the switch in DC phenotype from an immunostimulatory to immunosuppressive phenotype [73]. Ongoing studies should clarify which one is the predominant mechanism of tolerization in vivo. Other pathways potentially involved in the immunosuppressive activity of advanced tumor DCs include hypoxia within the TME, which induces DCs that are capable of presenting peptides but have impaired antigen processing capabilities and express significantly higher levels of VEGF-A, CXCL1, and CXCL8; chemokines all implicated in promoting angiogenesis in multiple forms of cancers [74]. Although we do not find high levels of IL-10 in human or mouse ovarian cancers, secretion of IL10 can also lead to DC-inhibition of maturation in different tumors [75], in addition to inducing the expression of immunosuppressive OX40 ligand via production of thymic stromal lymphopoietin (TSLP) [76]. Finally, increased lipid accumulation in tumor associated DCs in both humans and mice resulted in a diminished ability to process and load antigen onto MHC, resulting in ineffective antigen presentation to T cells within the TME [77].
Other tumor-derived factors that could be significant contributors to DC conversion include IL6, which induce Socs3 upregulation in tumor-associated DCs leading to inhibition of pyruvate kinase M2 (M2-PK) [78], an enzyme involved in aerobic glycolysis [79]. S100A8/A9 [80], which induce the massive recruitment of immune cells and prevent their differentiation within the TME [81], could also participate in this process.
7. Back to normal: In vivo re-programming of tumor DCs
Due to their massive accumulation and suppressive power, macrophages and DCs in the TME emerge as major therapeutic targets. Importantly, we have demonstrated that when these leukocytes receive certain activating signals, at least in mouse models, they can process full-length OVA in vitro[61] and in vivo [63, 67], and effectively present processed SIINFEKL to T cells. Therefore, interventions that achieve effective re-programing of immunosuppressive myeloid leukocytes in vivo into immunocompetent antigen-presenting cells, could be much more effective than their mere depletion, by simultaneously eliminating a major immunosuppressive driving force and boosting anti-tumor immunity in situ at tumor locations.
Ovarian cancer represents an ideal disease for these interventions because the TME is both compartmentalized and accessible. Supporting the feasibility of this approach in preclinical models, we have demonstrated that agonistic (and clinically available) CD40 and TLR agonists synergize to transform ovarian cancer-associated myeloid cells from an immunosuppressive to an immunostimulatory cell type [67]. Building on the insight of these studies, and by taking advantage of the enhanced endocytic pathways of tumor-associated DCs [63], we have more recently combined the synergy between the intrinsic TLR agonistic activity of double-stranded RNA and CD40 activation with the immunostimulatory activity of miR-155. Thus, we demonstrated that Dicer substrates mimicking the sequence and structure of endogenous miR-155 are selectively taken-up by tumor-associated CD11c+MHC-II+ DCs in mice growing aggressive orthotopic ovarian tumors when combined with biocompatible polymers, which synergizes with CD40 agonists [82]. Two important observations can be drawn from these studies; one, that DCs are major orchestrators of the immunosuppressive microenvironment, and two, in situ delivery of a potent antigenic stimulus is sufficient to reverse the tolerogenic phenotype, which provides a rationale for subsequent clinical testing.
8. Conclusions and future perspectives
Accelerated malignant growth coincides with the massive accumulation of immature myeloid leukocytes into the TME, which eventually breaks the dynamic equilibrium between protective T cell responses and proliferating tumor cells. Although tumors also lose recognizable antigens during their progression, they appear to remain significantly immunogenic to be controlled by existing anti-tumor T cells when inhibitory checkpoints are neutralized, as supported by experimental and clinical evidence. Because of their plasticity, myeloid leukocytes are highly susceptible to endogenous and exogenous signals within the tumor milieu. The presence of these cells within the tumor microenvironment is sufficient to tip the balance in favor of exponential tumor progression and escape from the immune pressure. However, myeloid cells (DCs) initially orchestrate measurable adaptive immune responses that can keep tumors in check for relatively long periods. Consequently, emerging evidence indicates that myeloid leukocytes govern cancer progression. Most importantly, partial reversal of the immunosuppressive genetic program of tumor DCs can be achieved in vivo and in situ by combining immunostimulatory agonists and delivering immune-activating miRNA mimetics. Understanding the genetic pathways and secretory factors that influence the mobilization of myeloid precursors and how to transform them from an immature to immunosuppressive phenotype should open new avenues for effective control of established tumors, besides iterations of chemotherapeutic drugs directly targeting tumor cells.
Figure 1. Dendritic cell plasticity influences tumor progression.
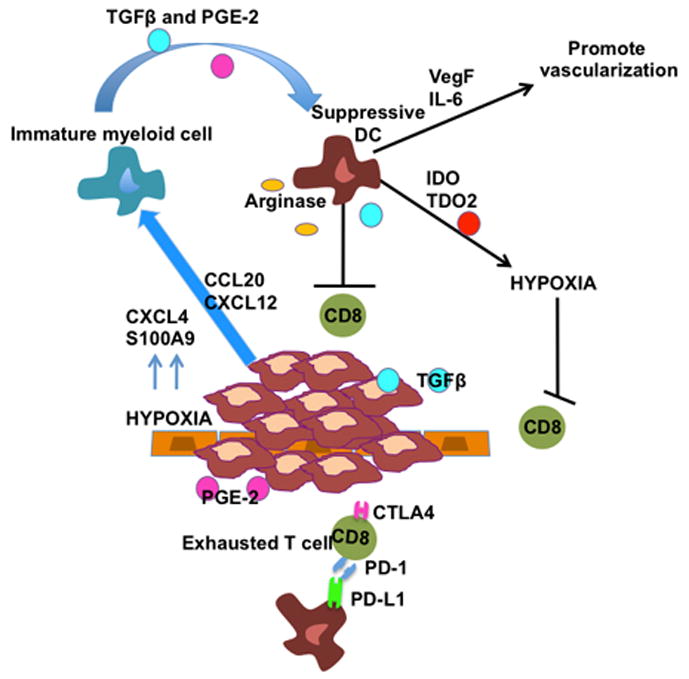
During aggressive malignant expansion of tumor cells, immature myeloid cells are recruited into the tumor microenvironment (TME) by CCL20 and CXCL12 produced by tumor cells or CXCL4 and S100A9 (upregulated in hypoxic environments). In the TME immature myeloid cells are converted into suppressive regulatory DCs by TGFβ and PGE-2 produced by the tumor cells. Suppressive DCs cooperate with the developing tumor mass to promote escape by secreting VegF and Il-6 (supporting angiogenesis), producing IDO and TDO2 (establishing a more hypoxic and immunosuppressive microenvironment) and secretion of immunosuppressive factors such as TGFβ and arginase (directly impeding T cell function). T cells become exhausted characterized by upregulation of inhibitory receptors such as PD-1 and CTLA4. This immunosuppressive tumor microenvironment impairs CD8 T cell anti-tumor responses, resulting in tumor escape.
Figure 2. In situ reversal of the immature phenotype of suppressive DCs with potent antigenic stimulus.
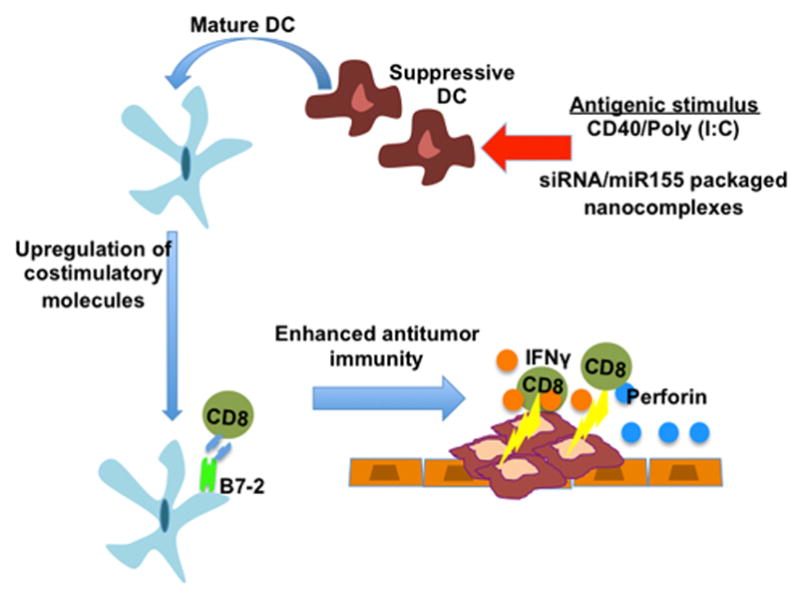
Reversal of DC phenotype can be achieved by delivery of potent antigenic stimulation using agonistic CD40 and poly(I:C) or by delivery of immunostimulatory nanoparticles complexed with a mimetic for miR155, resulting in the conversion of immunosuppressive DCs into immunostimulatory DCs. Following stimulation, costimulatory molecules such as B7-2 are upregulated and resulting in enhanced effector T cell function and inhibition of tumor progression.
HIGHLIGHTS.
Measurable T cell responses control tumor progression from very early stages
Accelerated malignant expansion is driven by immunosuppressive myeloid leukocytes
Tumor cells remain immunogenic during the escape phase of tumor progression
Neutralizing inhibitory checkpoints in T cells restores immune control of tumors
Immunosuppressive leukocytes can be re-programmed in vivo and in situ
Acknowledgments
This work was supported by NCI Grants CA157664 and CA124515, and by DoD grant OC100059.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Melanie R Rutkowski, Email: mrutkowski@wistar.org.
Tom-Li Stephen, Email: tstephen@wistar.org.
References
- 1.Burnet M. Cancer; a biological approach. I. The processes of control. Br Med J. 1957;1:779–786. doi: 10.1136/bmj.1.5022.779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Thomas L. Cellular and Humoral Aspects of the Hypersensitive States. Hoeber-Harper; New York: 1959. [Google Scholar]
- 3.Dunn GP, Bruce AT, Ikeda H, Old LJ, Schreiber RD. Cancer immunoediting: from immunosurveillance to tumor escape. Nat Immunol. 2002;3:991–998. doi: 10.1038/ni1102-991. [DOI] [PubMed] [Google Scholar]
- 4.Matsushita H, Vesely MD, Koboldt DC, Rickert CG, Uppaluri R, Magrini VJ, Arthur CD, White JM, Chen YS, Shea LK, Hundal J, Wendl MC, Demeter R, Wylie T, Allison JP, Smyth MJ, Old LJ, Mardis ER, Schreiber RD. Cancer exome analysis reveals a T-cell-dependent mechanism of cancer immunoediting. Nature. 2012;482:400–404. doi: 10.1038/nature10755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yoshimoto M, Sakamoto G, Ohashi Y. Time dependency of the influence of prognostic factors on relapse in breast cancer. Cancer. 1993;72:2993–3001. doi: 10.1002/1097-0142(19931115)72:10<2993::aid-cncr2820721022>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- 6.Galon J, Costes A, Sanchez-Cabo F, Kirilovsky A, Mlecnik B, Lagorce-Pages C, Tosolini M, Camus M, Berger A, Wind P, Zinzindohoue F, Bruneval P, Cugnenc PH, Trajanoski Z, Fridman WH, Pages F. Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science. 2006;313:1960–1964. doi: 10.1126/science.1129139. [DOI] [PubMed] [Google Scholar]
- 7.Clemente CG, Mihm MC, Jr, Bufalino R, Zurrida S, Collini P, Cascinelli N. Prognostic value of tumor infiltrating lymphocytes in the vertical growth phase of primary cutaneous melanoma. Cancer. 1996;77:1303–1310. doi: 10.1002/(SICI)1097-0142(19960401)77:7<1303::AID-CNCR12>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 8.Zhang L, Conejo-Garcia JR, Katsaros D, Gimotty PA, Massobrio M, Regnani G, Makrigiannakis A, Gray H, Schlienger K, Liebman MN, Rubin SC, Coukos G. Intratumoral T cells, recurrence, and survival in epithelial ovarian cancer. N Engl J Med. 2003;348:203–213. doi: 10.1056/NEJMoa020177. [DOI] [PubMed] [Google Scholar]
- 9.Kalos M, Levine BL, Porter DL, Katz S, Grupp SA, Bagg A, June CH. T cells with chimeric antigen receptors have potent antitumor effects and can establish memory in patients with advanced leukemia. Sci Transl Med. 2011;3:95ra73. doi: 10.1126/scitranslmed.3002842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Porter DL, Levine BL, Kalos M, Bagg A, June CH. Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. N Engl J Med. 2011;365:725–733. doi: 10.1056/NEJMoa1103849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dudley ME, Wunderlich JR, Robbins PF, Yang JC, Hwu P, Schwartzentruber DJ, Topalian SL, Sherry R, Restifo NP, Hubicki AM, Robinson MR, Raffeld M, Duray P, Seipp CA, Rogers-Freezer L, Morton KE, Mavroukakis SA, White DE, Rosenberg SA. Cancer regression and autoimmunity in patients after clonal repopulation with antitumor lymphocytes. Science. 2002;298:850–854. doi: 10.1126/science.1076514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pardoll D, Drake C. Immunotherapy earns its spot in the ranks of cancer therapy. J Exp Med. 2012;209:201–209. doi: 10.1084/jem.20112275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, Powderly JD, Carvajal RD, Sosman JA, Atkins MB, Leming PD, Spigel DR, Antonia SJ, Horn L, Drake CG, Pardoll DM, Chen L, Sharfman WH, Anders RA, Taube JM, McMiller TL, Xu H, Korman AJ, Jure-Kunkel M, Agrawal S, McDonald D, Kollia GD, Gupta A, Wigginton JM, Sznol M. Safety, Activity, and Immune Correlates of Anti-PD-1 Antibody in Cancer. N Engl J Med. 2012 doi: 10.1056/NEJMoa1200690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Scarlett UK, Rutkowski MR, Rauwerdink AM, Fields J, Escovar-Fadul X, Baird J, Cubillos-Ruiz JR, Jacobs AC, Gonzalez JL, Weaver J, Fiering S, Conejo-Garcia JR. Ovarian cancer progression is controlled by phenotypic changes in dendritic cells. J Exp Med. 2012;209:495–506. doi: 10.1084/jem.20111413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vesely MD, Kershaw MH, Schreiber RD, Smyth MJ. Natural innate and adaptive immunity to cancer. Annu Rev Immunol. 2011;29:235–271. doi: 10.1146/annurev-immunol-031210-101324. [DOI] [PubMed] [Google Scholar]
- 16.Prestwich RJ, Errington F, Hatfield P, Merrick AE, Ilett EJ, Selby PJ, Melcher AA. The immune system--is it relevant to cancer development, progression and treatment? Clin Oncol (R Coll Radiol) 2008;20:101–112. doi: 10.1016/j.clon.2007.10.011. [DOI] [PubMed] [Google Scholar]
- 17.Fridlender ZG, Albelda SM. Tumor-associated neutrophils: friend or foe? Carcinogenesis. 2012;33:949–955. doi: 10.1093/carcin/bgs123. [DOI] [PubMed] [Google Scholar]
- 18.Granot Z, Henke E, Comen EA, King TA, Norton L, Benezra R. Tumor entrained neutrophils inhibit seeding in the premetastatic lung. Cancer Cell. 2011;20:300–314. doi: 10.1016/j.ccr.2011.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fridlender ZG, Sun J, Kim S, Kapoor V, Cheng G, Ling L, Worthen GS, Albelda SM. Polarization of tumor-associated neutrophil phenotype by TGF-beta: “N1” versus “N2” TAN. Cancer Cell. 2009;16:183–194. doi: 10.1016/j.ccr.2009.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Koebel CM, Vermi W, Swann JB, Zerafa N, Rodig SJ, Old LJ, Smyth MJ, Schreiber RD. Adaptive immunity maintains occult cancer in an equilibrium state. Nature. 2007;450:903–907. doi: 10.1038/nature06309. [DOI] [PubMed] [Google Scholar]
- 21.Curiel TJ, Coukos G, Zou L, Alvarez X, Cheng P, Mottram P, Evdemon-Hogan M, Conejo-Garcia JR, Zhang L, Burow M, Zhu Y, Wei S, Kryczek I, Daniel B, Gordon A, Myers L, Lackner A, Disis ML, Knutson KL, Chen L, Zou W. Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat Med. 2004;10:942–949. doi: 10.1038/nm1093. [DOI] [PubMed] [Google Scholar]
- 22.Peng G, Wang HY, Peng W, Kiniwa Y, Seo KH, Wang RF. Tumor-infiltrating gammadelta T cells suppress T and dendritic cell function via mechanisms controlled by a unique toll-like receptor signaling pathway. Immunity. 2007;27:334–348. doi: 10.1016/j.immuni.2007.05.020. [DOI] [PubMed] [Google Scholar]
- 23.Sato E, Olson SH, Ahn J, Bundy B, Nishikawa H, Qian F, Jungbluth AA, Frosina D, Gnjatic S, Ambrosone C, Kepner J, Odunsi T, Ritter G, Lele S, Chen YT, Ohtani H, Old LJ, Odunsi K. Intraepithelial CD8+ tumor-infiltrating lymphocytes and a high CD8+/regulatory T cell ratio are associated with favorable prognosis in ovarian cancer. Proc Natl Acad Sci U S A. 2005;102:18538–18543. doi: 10.1073/pnas.0509182102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Quezada SA, Simpson TR, Peggs KS, Merghoub T, Vider J, Fan X, Blasberg R, Yagita H, Muranski P, Antony PA, Restifo NP, Allison JP. Tumor-reactive CD4(+) T cells develop cytotoxic activity and eradicate large established melanoma after transfer into lymphopenic hosts. J Exp Med. 2010;207:637–650. doi: 10.1084/jem.20091918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Xie Y, Akpinarli A, Maris C, Hipkiss EL, Lane M, Kwon EK, Muranski P, Restifo NP, Antony PA. Naive tumor-specific CD4(+) T cells differentiated in vivo eradicate established melanoma. J Exp Med. 2010;207:651–667. doi: 10.1084/jem.20091921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ma Y, Aymeric L, Locher C, Mattarollo SR, Delahaye NF, Pereira P, Boucontet L, Apetoh L, Ghiringhelli F, Casares N, Lasarte JJ, Matsuzaki G, Ikuta K, Ryffel B, Benlagha K, Tesniere A, Ibrahim N, Dechanet-Merville J, Chaput N, Smyth MJ, Kroemer G, Zitvogel L. Contribution of IL-17-producing gamma delta T cells to the efficacy of anticancer chemotherapy. J Exp Med. 2011;208:491–503. doi: 10.1084/jem.20100269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nesbeth Y, Conejo-Garcia JR. Harnessing the effect of adoptively transferred tumor-reactive T cells on endogenous (host-derived) antitumor immunity. Clin Dev Immunol. 2010;2010:139304. doi: 10.1155/2010/139304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nesbeth Y, Scarlett U, Cubillos-Ruiz J, Martinez D, Engle X, Turk MJ, Conejo-Garcia JR. CCL5-mediated endogenous antitumor immunity elicited by adoptively transferred lymphocytes and dendritic cell depletion. Cancer Res. 2009;69:6331–6338. doi: 10.1158/0008-5472.CAN-08-4329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nesbeth YC, Martinez DG, Toraya S, Scarlett UK, Cubillos-Ruiz JR, Rutkowski MR, Conejo-Garcia JR. CD4+ T cells elicit host immune responses to MHC class II- ovarian cancer through CCL5 secretion and CD40-mediated licensing of dendritic cells. J Immunol. 2010;184:5654–5662. doi: 10.4049/jimmunol.0903247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hildner K, Edelson BT, Purtha WE, Diamond M, Matsushita H, Kohyama M, Calderon B, Schraml BU, Unanue ER, Diamond MS, Schreiber RD, Murphy TL, Murphy KM. Batf3 deficiency reveals a critical role for CD8alpha+ dendritic cells in cytotoxic T cell immunity. Science. 2008;322:1097–1100. doi: 10.1126/science.1164206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zitvogel L, Tesniere A, Kroemer G. Cancer despite immunosurveillance: immunoselection and immunosubversion. Nat Rev Immunol. 2006;6:715–727. doi: 10.1038/nri1936. [DOI] [PubMed] [Google Scholar]
- 32.Shankaran V, Ikeda H, Bruce AT, White JM, Swanson PE, Old LJ, Schreiber RD. IFNgamma and lymphocytes prevent primary tumour development and shape tumour immunogenicity. Nature. 2001;410:1107–1111. doi: 10.1038/35074122. [DOI] [PubMed] [Google Scholar]
- 33.Dunn GP, Old LJ, Schreiber RD. The three Es of cancer immunoediting. Annu Rev Immunol. 2004;22:329–360. doi: 10.1146/annurev.immunol.22.012703.104803. [DOI] [PubMed] [Google Scholar]
- 34.Dunn GP, Old LJ, Schreiber RD. The immunobiology of cancer immunosurveillance and immunoediting. Immunity. 2004;21:137–148. doi: 10.1016/j.immuni.2004.07.017. [DOI] [PubMed] [Google Scholar]
- 35.Schreiber RD, Old LJ, Smyth MJ. Cancer immunoediting: integrating immunity’s roles in cancer suppression and promotion. Science. 331:1565–1570. doi: 10.1126/science.1203486. [DOI] [PubMed] [Google Scholar]
- 36.DuPage M, Mazumdar C, Schmidt LM, Cheung AF, Jacks T. Expression of tumour-specific antigens underlies cancer immunoediting. Nature. 2012;482:405–409. doi: 10.1038/nature10803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Blankenstein T, Coulie PG, Gilboa E, Jaffee EM. The determinants of tumour immunogenicity. Nat Rev Cancer. 12:307–313. doi: 10.1038/nrc3246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schreiber RD, Old LJ, Smyth MJ. Cancer immunoediting: integrating immunity’s roles in cancer suppression and promotion. Science. 2011;331:1565–1570. doi: 10.1126/science.1203486. [DOI] [PubMed] [Google Scholar]
- 39.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 40.Wherry EJ. T cell exhaustion. Nat Immunol. 2011;12:492–499. doi: 10.1038/ni.2035. [DOI] [PubMed] [Google Scholar]
- 41.Baitsch L, Fuertes-Marraco SA, Legat A, Meyer C, Speiser DE. The three main stumbling blocks for anticancer T cells. Trends Immunol. 2012 doi: 10.1016/j.it.2012.02.006. [DOI] [PubMed] [Google Scholar]
- 42.Zheng Y, Zha Y, Gajewski TF. Molecular regulation of T-cell anergy. EMBO Rep. 2008;9:50–55. doi: 10.1038/sj.embor.7401138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Han S, Asoyan A, Rabenstein H, Nakano N, Obst R. Role of antigen persistence and dose for CD4+ T-cell exhaustion and recovery. Proc Natl Acad Sci U S A. 2010;107:20453–20458. doi: 10.1073/pnas.1008437107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Baitsch L, Baumgaertner P, Devevre E, Raghav SK, Legat A, Barba L, Wieckowski S, Bouzourene H, Deplancke B, Romero P, Rufer N, Speiser DE. Exhaustion of tumor-specific CD8(+) T cells in metastases from melanoma patients. J Clin Invest. 2011;121:2350–2360. doi: 10.1172/JCI46102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wang SF, Fouquet S, Chapon M, Salmon H, Regnier F, Labroquere K, Badoual C, Damotte D, Validire P, Maubec E, Delongchamps NB, Cazes A, Gibault L, Garcette M, Dieu-Nosjean MC, Zerbib M, Avril MF, Prevost-Blondel A, Randriamampita C, Trautmann A, Bercovici N. Early T cell signalling is reversibly altered in PD-1+ T lymphocytes infiltrating human tumors. PLoS One. 2011;6:e17621. doi: 10.1371/journal.pone.0017621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Beatty GL, Smith JS, Reshef R, Patel KP, Colligon TA, Vance BA, Frey NV, Johnson FB, Porter DL, Vonderheide RH. Functional unresponsiveness and replicative senescence of myeloid leukemia antigen-specific CD8+ T cells after allogeneic stem cell transplantation. Clin Cancer Res. 2009;15:4944–4953. doi: 10.1158/1078-0432.CCR-08-3332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Shay JW, Roninson IB. Hallmarks of senescence in carcinogenesis and cancer therapy. Oncogene. 2004;23:2919–2933. doi: 10.1038/sj.onc.1207518. [DOI] [PubMed] [Google Scholar]
- 48.Lechner MG, Liebertz DJ, Epstein AL. Characterization of cytokine-induced myeloid-derived suppressor cells from normal human peripheral blood mononuclear cells. J Immunol. 2010;185:2273–2284. doi: 10.4049/jimmunol.1000901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Norian LA, Rodriguez PC, O’Mara LA, Zabaleta J, Ochoa AC, Cella M, Allen PM. Tumor-infiltrating regulatory dendritic cells inhibit CD8+ T cell function via L-arginine metabolism. Cancer Res. 2009;69:3086–3094. doi: 10.1158/0008-5472.CAN-08-2826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Prendergast GC, Metz R, Muller AJ. Towards a genetic definition of cancer-associated inflammation: role of the IDO pathway. Am J Pathol. 2010;176:2082–2087. doi: 10.2353/ajpath.2010.091173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Stone TW, Darlington LG. Endogenous kynurenines as targets for drug discovery and development. Nat Rev Drug Discov. 2002;1:609–620. doi: 10.1038/nrd870. [DOI] [PubMed] [Google Scholar]
- 52.Ahmadzadeh M, Rosenberg SA. TGF-beta 1 attenuates the acquisition and expression of effector function by tumor antigen-specific human memory CD8 T cells. J Immunol. 2005;174:5215–5223. doi: 10.4049/jimmunol.174.9.5215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Torre-Amione G, Beauchamp RD, Koeppen H, Park BH, Schreiber H, Moses HL, Rowley DA. A highly immunogenic tumor transfected with a murine transforming growth factor type beta 1 cDNA escapes immune surveillance. Proc Natl Acad Sci U S A. 1990;87:1486–1490. doi: 10.1073/pnas.87.4.1486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sinha P, Okoro C, Foell D, Freeze HH, Ostrand-Rosenberg S, Srikrishna G. Proinflammatory S100 proteins regulate the accumulation of myeloid-derived suppressor cells. J Immunol. 2008;181:4666–4675. doi: 10.4049/jimmunol.181.7.4666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Meyer C, Sevko A, Ramacher M, Bazhin AV, Falk CS, Osen W, Borrello I, Kato M, Schadendorf D, Baniyash M, Umansky V. Chronic inflammation promotes myeloid-derived suppressor cell activation blocking antitumor immunity in transgenic mouse melanoma model. Proc Natl Acad Sci U S A. 2011;108:17111–17116. doi: 10.1073/pnas.1108121108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lu T, Ramakrishnan R, Altiok S, Youn JI, Cheng P, Celis E, Pisarev V, Sherman S, Sporn MB, Gabrilovich D. Tumor-infiltrating myeloid cells induce tumor cell resistance to cytotoxic T cells in mice. J Clin Invest. 2011;121:4015–4029. doi: 10.1172/JCI45862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Serafini P, Meckel K, Kelso M, Noonan K, Califano J, Koch W, Dolcetti L, Bronte V, Borrello I. Phosphodiesterase-5 inhibition augments endogenous antitumor immunity by reducing myeloid-derived suppressor cell function. J Exp Med. 2006;203:2691–2702. doi: 10.1084/jem.20061104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gabrilovich DI, Ostrand-Rosenberg S, Bronte V. Coordinated regulation of myeloid cells by tumours. Nat Rev Immunol. 2012;12:253–268. doi: 10.1038/nri3175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Nagaraj S, Gupta K, Pisarev V, Kinarsky L, Sherman S, Kang L, Herber DL, Schneck J, Gabrilovich DI. Altered recognition of antigen is a mechanism of CD8+ T cell tolerance in cancer. Nat Med. 2007;13:828–835. doi: 10.1038/nm1609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Corzo CA, Condamine T, Lu L, Cotter MJ, Youn JI, Cheng P, Cho HI, Celis E, Quiceno DG, Padhya T, McCaffrey TV, McCaffrey JC, Gabrilovich DI. HIF-1alpha regulates function and differentiation of myeloid-derived suppressor cells in the tumor microenvironment. J Exp Med. 2010;207:2439–2453. doi: 10.1084/jem.20100587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Conejo-Garcia JR, Benencia F, Courreges MC, Kang E, Mohamed-Hadley A, Buckanovich RJ, Holtz DO, Jenkins A, Na H, Zhang L, Wagner DS, Katsaros D, Caroll R, Coukos G. Tumor-infiltrating dendritic cell precursors recruited by a beta-defensin contribute to vasculogenesis under the influence of Vegf-A. Nat Med. 2004;10:950–958. doi: 10.1038/nm1097. [DOI] [PubMed] [Google Scholar]
- 62.Conejo-Garcia JR, Buckanovich RJ, Benencia F, Courreges MC, Rubin SC, Carroll RG, Coukos G. Vascular leukocytes contribute to tumor vascularization. Blood. 2005;105:679–681. doi: 10.1182/blood-2004-05-1906. [DOI] [PubMed] [Google Scholar]
- 63.Cubillos-Ruiz JR, Engle X, Scarlett UK, Martinez D, Barber A, Elgueta R, Wang L, Nesbeth Y, Durant Y, Gewirtz AT, Sentman CL, Kedl R, Conejo-Garcia JR. Polyethylenimine-based siRNA nanocomplexes reprogram tumor-associated dendritic cells via TLR5 to elicit therapeutic antitumor immunity. J Clin Invest. 2009;119:2231–2244. doi: 10.1172/JCI37716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Cubillos-Ruiz JR, Fiering S, Conejo-Garcia JR. Nanomolecular targeting of dendritic cells for ovarian cancer therapy. Future Oncol. 2009;5:1189–1192. doi: 10.2217/fon.09.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Cubillos-Ruiz JR, Rutkowski M, Conejo-Garcia JR. Blocking ovarian cancer progression by targeting tumor microenvironmental leukocytes. Cell Cycle. 9:260–268. doi: 10.4161/cc.9.2.10430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Huarte E, Cubillos-Ruiz JR, Nesbeth YC, Scarlett UK, Martinez DG, Buckanovich RJ, Benencia F, Stan RV, Keler T, Sarobe P, Sentman CL, Conejo-Garcia JR. Depletion of dendritic cells delays ovarian cancer progression by boosting antitumor immunity. Cancer Res. 2008;68:7684–7691. doi: 10.1158/0008-5472.CAN-08-1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Scarlett UK, Cubillos-Ruiz JR, Nesbeth YC, Martinez DG, Engle X, Gewirtz AT, Ahonen CL, Conejo-Garcia JR. In situ Stimulation of CD40 and Toll-like Receptor 3 Transforms Ovarian Cancer-Infiltrating Dendritic Cells from Immunosuppressive to Immunostimulatory Cells. Cancer Res. 2009;69:7329–7337. doi: 10.1158/0008-5472.CAN-09-0835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Curiel TJ, Wei S, Dong H, Alvarez X, Cheng P, Mottram P, Krzysiek R, Knutson KL, Daniel B, Zimmermann MC, David O, Burow M, Gordon A, Dhurandhar N, Myers L, Berggren R, Hemminki A, Alvarez RD, Emilie D, Curiel DT, Chen L, Zou W. Blockade of B7-H1 improves myeloid dendritic cell-mediated antitumor immunity. Nat Med. 2003;9:562–567. doi: 10.1038/nm863. [DOI] [PubMed] [Google Scholar]
- 69.Watkins SK, Zhu Z, Riboldi E, Shafer-Weaver KA, Stagliano KE, Sklavos MM, Ambs S, Yagita H, Hurwitz AA. FOXO3 programs tumor-associated DCs to become tolerogenic in human and murine prostate cancer. J Clin Invest. 121:1361–1372. doi: 10.1172/JCI44325. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 70.Cheong C, Matos I, Choi JH, Dandamudi DB, Shrestha E, Longhi MP, Jeffrey KL, Anthony RM, Kluger C, Nchinda G, Koh H, Rodriguez A, Idoyaga J, Pack M, Velinzon K, Park CG, Steinman RM. Microbial stimulation fully differentiates monocytes to DC-SIGN/CD209(+) dendritic cells for immune T cell areas. Cell. 143:416–429. doi: 10.1016/j.cell.2010.09.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Rivollier A, He J, Kole A, Valatas V, Kelsall BL. Inflammation switches the differentiation program of Ly6Chi monocytes from antiinflammatory macrophages to inflammatory dendritic cells in the colon. J Exp Med. 209:139–155. doi: 10.1084/jem.20101387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Diao J, Mikhailova A, Tang M, Gu H, Zhao J, Cattral MS. Immunostimulatory conventional dendritic cells evolve into regulatory macrophage-like cells. Blood. doi: 10.1182/blood-2011-11-392894. [DOI] [PubMed] [Google Scholar]
- 73.Scarlett UK, Rutkowski MR, Rauwerdink AM, Fields J, Escovar-Fadul X, Baird J, Cubillos-Ruiz JR, Jacobs AC, Gonzalez JL, Weaver J, Fiering S, Conejo-Garcia JR. Ovarian cancer progression is controlled by phenotypic changes in dendritic cells. J Exp Med. 209:495–506. doi: 10.1084/jem.20111413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Elia AR, Cappello P, Puppo M, Fraone T, Vanni C, Eva A, Musso T, Novelli F, Varesio L, Giovarelli M. Human dendritic cells differentiated in hypoxia down-modulate antigen uptake and change their chemokine expression profile. J Leukoc Biol. 2008;84:1472–1482. doi: 10.1189/jlb.0208082. [DOI] [PubMed] [Google Scholar]
- 75.Sica A, Larghi P, Mancino A, Rubino L, Porta C, Totaro MG, Rimoldi M, Biswas SK, Allavena P, Mantovani A. Macrophage polarization in tumour progression. Semin Cancer Biol. 2008;18:349–355. doi: 10.1016/j.semcancer.2008.03.004. [DOI] [PubMed] [Google Scholar]
- 76.Palucka K, Banchereau J. Cancer immunotherapy via dendritic cells. Nat Rev Cancer. 12:265–277. doi: 10.1038/nrc3258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Herber DL, Cao W, Nefedova Y, Novitskiy SV, Nagaraj S, Tyurin VA, Corzo A, Cho HI, Celis E, Lennox B, Knight SC, Padhya T, McCaffrey TV, McCaffrey JC, Antonia S, Fishman M, Ferris RL, Kagan VE, Gabrilovich DI. Lipid accumulation and dendritic cell dysfunction in cancer. Nat Med. 16:880–886. doi: 10.1038/nm.2172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Zhang Z, Liu Q, Che Y, Yuan X, Dai L, Zeng B, Jiao G, Zhang Y, Wu X, Yu Y, Yang R. Antigen presentation by dendritic cells in tumors is disrupted by altered metabolism that involves pyruvate kinase M2 and its interaction with SOCS3. Cancer Res. 70:89–98. doi: 10.1158/0008-5472.CAN-09-2970. [DOI] [PubMed] [Google Scholar]
- 79.Christofk HR, Vander Heiden MG, Harris MH, Ramanathan A, Gerszten RE, Wei R, Fleming MD, Schreiber SL, Cantley LC. The M2 splice isoform of pyruvate kinase is important for cancer metabolism and tumour growth. Nature. 2008;452:230–233. doi: 10.1038/nature06734. [DOI] [PubMed] [Google Scholar]
- 80.Grebhardt S, Veltkamp C, Strobel P, Mayer D. Hypoxia and HIF-1 increase S100A8 and S100A9 expression in prostate cancer. Int J Cancer. doi: 10.1002/ijc.27591. [DOI] [PubMed] [Google Scholar]
- 81.Ricciardi A, Elia AR, Cappello P, Puppo M, Vanni C, Fardin P, Eva A, Munroe D, Wu X, Giovarelli M, Varesio L. Transcriptome of hypoxic immature dendritic cells: modulation of chemokine/receptor expression. Mol Cancer Res. 2008;6:175–185. doi: 10.1158/1541-7786.MCR-07-0391. [DOI] [PubMed] [Google Scholar]
- 82.Cubillos-Ruiz JR, Baird JR, Tesone AJ, Rutkowski MR, Scarlett UK, Camposeco-Jacobs AL, Anadon-Arnillas J, Harwood NM, Korc M, Fiering SN, Sempere LF, Conejo-Garcia JR. Reprogramming tumor-associated dendritic cells in vivo using microRNA mimetics triggers protective immunity against ovarian cancer. Cancer Res. 2012;72:1683–1693. doi: 10.1158/0008-5472.CAN-11-3160. [DOI] [PMC free article] [PubMed] [Google Scholar]