

Abstract
Background
Atypical variants of Alzheimer’s disease (AD) have been pathologically defined based on the distribution of neurofibrillary tangles; hippocampal sparing (HpSp) AD shows minimal involvement of the hippocampus and limbic predominant (LP) AD shows neurofibrillary tangles restricted to the medial temporal lobe. We aimed to determine whether MRI patterns of atrophy differ across HpSp AD, LP AD and typical AD, and whether imaging could be a useful predictor of pathological subtype during life.
Methods
In this case-control study, we identified 177 patients who had been prospectively followed in the Mayo Clinic Alzheimer’s Disease Research Center, were demented during life, had AD pathology at autopsy (Braak stage ≥ IV, intermediate-high probability AD) and an antemortem MRI. Cases were assigned to one of three pathological subtypes (HpSp n=19, typical n=125, or LP AD n=33) based on neurofibrillary tangle counts and their ratio in association cortices to hippocampus, without reference to neuronal loss. Voxel-based morphometry and atlas-based parcellation were used to compare patterns of grey matter loss across groups, and to controls.
Findings
The severity of medial temporal and cortical grey matter atrophy differed across subtypes. The most severe medial temporal atrophy was observed in LP AD, followed by typical AD, and then HpSp AD. Conversely, the most severe cortical atrophy was observed in HpSp AD, followed by typical AD, and then LP AD. A ratio of hippocampal-to-cortical volume provided the best discrimination across all three AD subtypes. The majority of typical AD (98/125;78%) and LP AD (31/33;94%) subjects, but only 8/19 (42%) of the HpSp AD subjects, presented with a dominant amnestic syndrome.
Interpretation
Patterns of atrophy on MRI differ across the pathological subtypes of AD, suggesting that MR regional volumetrics reliably track the distribution of neurofibrillary tangle pathology and can predict pathological subtype during life.
Funding
US National Institutes of Health (National Institute on Aging)
INTRODUCTION
Structural MRI measurements of atrophy are important biomarkers to track disease progression in Alzheimer’s disease (AD). Progression of atrophy has been shown to match the stereotypic pattern of progression of neurofibrillary tangles (NFT) described by the Braak staging scheme1, 2; with early changes observed in medial temporal lobe, particularly hippocampus, and then later spreading to involve association cortex. The density of NFT in the hippocampus correlates well with the degree of atrophy on MRI3-5, showing that atrophy is an antemortem correlate of NFT pathology in the elderly.
However, it has recently been shown that 25% of AD cases do not show the typical distribution of NFTs in the brain6. Hippocampal sparing (HpSp) cases showed a relative sparing of the hippocampus with higher NFT counts observed in cortex, and limbic predominant (LP) cases showed unusually high NFT counts in the hippocampus, with a relative sparing of cortex6. Clinical presentation, age at onset, disease duration and rate of cognitive decline differed across these groups, and compared to cases that showed a typical distribution of NFTs (typical AD), demonstrating that these represent different clinicopathological subtypes of AD. The presence of atypical subtypes could be an important confounder in MRI biomarker studies of AD. It is currently unknown whether patterns of atrophy observed on antemortem MRI differ across these subtypes.
The aim of our study was to determine whether antemortem patterns of volume loss on MRI differ between HpSp, LP, and typical AD, particularly in limbic and association cortices. We also aimed to determine whether a ratio of hippocampal-to-cortical volume on antemortem MRI could accurately predict these pathological subtypes at autopsy.
METHODS
Subject selection
A total of 198 subjects were identified from the Mayo Clinic Rochester, MN, neuropathological database that fulfilled the following inclusion criteria: Braak stage IV or greater1, intermediate-high probability of AD on NIA-Reagan criteria7, and had an antemortem volumetric MRI. Of these subjects, 125 were classified pathologically as typical AD (70%), 33 as LP AD (19%), and 19 as HpSp AD (11%). Subjects with hippocampal sclerosis (n=21) were excluded from the study to avoid contamination by cases with low NFT counts due to neuronal loss6.
All subjects had been prospectively recruited and followed longitudinally in the Mayo Clinic Alzheimer’s Disease Research Center (ADRC) or Alzheimer’s Disease Patient Registry (ADPR)8 between 1992 and 2005. The ADRC recruits subjects referred to the Department of Neurology with cognitive difficulties and the ADPR recruits subjects from the local community. Autopsy is performed on all subjects that provide consent. All subjects underwent clinical evaluations, cognitive testing and apolipoprotein E genotyping. Diagnosis of dementia was made based on the Diagnostic and Statistical Manual of Mental Disorders, Fourth Edition9. The medical records of all cases were reviewed by an experienced behavioral neurologist (K.A.J) blinded to pathological subtype in order to determine whether the presenting syndrome of each subject was dominated by deficits in memory, or another cognitive domain. This approach was used since it was not possible to retrospectively fit current diagnostic criteria for all subjects in this cohort. Each of the 177 subjects were age and gender-matched to a normal control subject, resulting in a total of 177 controls. Controls were cognitively normal individuals that had been seen in internal medicine for routine examinations and asked to enroll in the ADRC/ADPR. All controls were evaluated by a neurologist to ensure they did not have active neurologic or psychiatric conditions, had normal neurological and neurocognitive examinations, and were not taking any medications that would affect cognition.
This study was approved by the Mayo Clinic IRB. All subjects and/or their proxies provided written informed consent before participating in any research activity.
Pathological procedures
Neuropathologic examinations were performed according to the recommendations of the Consortium to Establish a Registry for Alzheimer’s disease (CERAD)10 by one of two neuropathologists (DWD/JEP), as described11. Each specimen was assigned a Braak stage1 using Bielschowsky silver stain based on the earliest appearance of NFT pathology, and AD was diagnosed based on NIA–Reagan criteria as low, intermediate, or high probability AD7. The presence of Lewy bodies and cerebrovascular disease was noted in all cases, and amyloid burden was measured using CERAD. The presence of cerebrovascular disease was considered if on H&E white matter rarefaction and/or cribriform change of the basal ganglia was seen in association with marked arteriosclerosis or atherosclerosis; in addition to the presence of multiple microinfarcts, lacunar infarcts, or large infarcts.
For this study, thioflavin-S fluorescence microscopy was utilized to quantitatively measure NFT densities with an Olympus BH2 fluorescence microscope (Center Valley, PA, USA). NFT counts, without separation of intracellular and extracellular tangles, were performed in hippocampus (CA1 and subiculum) and three association cortices (mid-frontal, inferior parietal, and superior temporal). Final NFT density for each region was based on the average NFT density determined from the area with the highest density of pathology. An algorithm was utilized to classify subjects into HpSp and LP AD using the hippocampal and cortical NFT counts, and the ratio of hippocampal-to-cortical counts, as previously described6 and detailed in Table 1. The algorithm was developed using an independent cohort of 889 AD cases6. Cases not designated as HpSp or LP AD were classified as typical AD.
TABLE 1.
Pathological classification algorithm used to define HpSp and LP AD subtypes
| Criteria | HpSp AD | LP AD |
|---|---|---|
| Ratio of average hippocampal-to-cortical neurofibrillary tangle count | < 25th percentile of all AD cases | > 75th percentile of all AD cases |
| Hippocampal neurofibrillary tangle counts (CA1, subiculum, CA1-subiculum average) | All counts < median* | All counts > median* |
| Cortical neurofibrillary tangle counts (superior temporal, inferior parietal, middle frontal, cortical average) | At least 3 cortical regions ≥ median† | At least 3 cortical regions ≤ median† |
Median neurofibrillary tangle counts were calculated in a cohort of 889 AD cases (CA1=12, subiculum=20 and CA1-subiculum average=17)
Median cortical neurofibrillary tangle counts were calculated in a cohort of 889 AD cases (superior temporal=10, inferior parietal=8, middle frontal=5 and cortical average=8)
MRI analysis
All subjects had undergone a standardized protocol head MRI that included a T1-weighted 3-dimensional SPGR sequence (22×16.5cm or 24×18.5cm FOV, 25° flip angle, 124 contiguous 1.6mm thick coronal slices). Scans were performed on a total of 16 different scanners. All scanners undergo a standardized quality control calibration procedure daily, which monitors geometric fidelity over a 200mm volume along all three cardinal axes, signal-to-noise, and transmit gain, and maintains the scanner within a tight calibration range. All images underwent pre-processing correction for gradient non-linearity and intensity non-uniformity12. The earliest MRI was used for each subject in order to determine whether imaging could be predictive of pathological subtype at an early stage of the disease. If a subject had been scanned whilst clinically normal then we used the first MRI after the subject became demented.
Grey matter atrophy was assessed at the voxel-level using voxel-based morphometry (VBM)13 and SPM5. Standard preprocessing steps were employed, including normalization to a customized template14, unified segmentation15, modulation and smoothing at 8mm full-width at half-maximum. A full-factorial (ANCOVA) model was used to compare each AD group (HpSp, typical, and LP AD) to their own specific age-matched control group (i.e. 19 HpSp subjects were compared to 19 matched controls (median [range] age=66yrs [51-86]), 125 typical subjects were compared to 125 matched controls (77yrs [43-94]), and 33 LP AD subjects were compared to 33 matched controls (82yrs [71-92]). In addition, each AD group was compared to the entire cohort of 177 controls, including age and gender as covariates. Direct comparisons were also performed between AD groups, including age and gender as covariates. Comparisons were assessed at p<0.05 using family wise error (FWE) correction and uncorrected at p<0.001. Effect size maps were also assessed.
Grey matter volumes of specific regions-of-interest were also calculated using atlas-based parcellation in SPM5 and the automated anatomic labeling (AAL) atlas16, as previously described17. Grey matter volumes were calculated for hippocampus and three association cortices (lateral frontal, temporal and parietal lobe) matching the NFT density assessments. In addition, volumes were calculated for regions considered to be part of the limbic system (amygdala, entorhinal cortex, parahippocampal gyrus, cingulate gyrus, orbitofrontal cortex, substantia innominata and thalamus) and striatum and precuneus since these regions are implicated in AD. Left and right volumes were averaged for each region. Total intracranial volume (TIV) was calculated by propagating a template-drawn TIV mask to subject space using a discrete-cosine transform registration model in SPM5. The propagated mask was eroded by one pass of a 3×3×3 cube structuring element to remove border voxels, and the volume estimate was obtained by multiplying the number of voxels in the mask by the voxel size. Regional volumes were scaled by TIV to correct for head size.
Statistical analysis
We compared demographic characteristics across groups using one-way ANOVA followed by pair-wise contrasts from the ANOVA model or chi-squared tests. For the chi-squared test comparing clinical symptoms we report a p-value calculated using Monte Carlo simulation due to low cell counts. We compared gray matter volumes across groups using ANOVA and pair-wise contrasts. In these models the response was the natural log transform of grey matter volume divided by TIV. In this way the response is adjusted by head size and the contrast coefficients can be interpreted as the approximate percentage difference, i.e. a relative difference, between groups due to the properties of logs18.
To summarize pair-wise group differences, we report the area under the receiver operator characteristic curve (AUROC). This two-sample measure of effect size and group-wise separation (i.e. discrimination) can be generalized to three or more groups using estimates of the probability a subject belongs to a given group19. We estimated these probabilities using a multi-nominal model although a linear discriminant analysis provided very similar group membership probabilities. What we term the three-way AUROC has an interpretation of how often three randomly selected subjects from the three AD groups can correctly be classified taking into account their grey matter volume scaled by TIV. With three groups there are 3×2×1=6 possible classifications thus chance alone would suggest a success rate of 1/6 (~17%). Therefore, we also report the observed three-way AUROC divided by 1/6 to estimate the relative improvement in classification. For both the three-way AUROC and the ratio we provide 95% bootstrap confidence intervals using the BCa method and 1000 replicates. We note that like the AUROC, the three-way AUROC is not sensitive to the relative sizes of the groups, i.e. group prevalence rates. In contrast, a measure of discrimination such as the percent correctly classified is highly sensitive to the relative sizes of the groups and could be increased simply by adding more subjects in whichever group is most common.
Since the estimated three-way AUROCs are used to infer how well grey matter volumes can discriminate among subjects in general (and not just our particular sample) we performed a bootstrap validation analysis to assess the extent to which our estimates were a result of over-fitting to the sample at hand and as such overly optimistic. We did this as follows: (a) take 100 bootstrap samples; (b) fit a multinomial model to each bootstrap sample, and (c) use the predictions from the bootstrap model to calculate the three-way AUROC based on the entire sample. The mean difference between the values obtained in step (c) and the original three-way AUROC is an estimate of the “over-optimism” of the reported statistics.
We also performed a number of sensitivity analyses to evaluate the robustness of our findings. The models were evaluated adjusted for age as a main effect and as an interaction term and found no appreciable differences in our results. Nor did we find differences when adjusting our models for pathology covariates including Braak stage, CERAD score, or presence of Lewy body or vascular pathology. In further sensitivity analyses, we modeled the response on the untransformed scale, as well as with TIV as a covariate rather than a scaling factor, and found no notable differences in our results. Lastly, since the MRI studies were performed across 16 different scanners, we performed a variance components analysis using mixed effects linear models with random group and random scanner effects. For each of the ROIs we found that variance due to scanner constituted less than 0.5% of the total variability.
We report p-values obtained from the individual models and did not adjust them to account for examining 14 ROIs in total20, 21. A central motivation for this approach is that each ROI represents a distinct anatomic region which was of interest and specified a priori. We note that in the VBM analysis described above we utilize FWE correction due to the massive scale of the hypothesis testing in that context. All analyses were performed using R statistical software version 2.14.1 (R Foundation for Statistical Computing, Vienna, http://www-R-project.org).
RESULTS
Clinical findings across groups
The three pathologically-defined AD groups did not differ in gender, education, APOE genotype, or disease duration (Table 2). However, the groups did differ in age at scan, onset and death (all p<0.001), with youngest age observed in HpSp AD and oldest age observed in LP AD. Time from scan to death was shorter in HpSp AD, although, importantly, time from onset-to-scan and cognitive performance at scan did not differ across groups suggesting they are well matched for disease stage. Memory was the most commonly affected domain across all three AD groups, although the proportion of subjects that presented with an amnestic syndrome was lowest in HpSp AD (42%) and highest in LP AD (94%).
TABLE 2.
Subject demographics, clinical and pathological features
| Control (n =177) | HpSp AD (n = 19) | Typical AD (n = 125) | LP AD (n = 33) | ANOVA P-value
|
||
|---|---|---|---|---|---|---|
| Comparing AD groups | Comparing all four groups | |||||
| Female, n (%). | 94 (53) | 7 (37) | 70 (56) | 22 (67) | 0.11* | 0.20* |
| Education, years | 14 (8, 20) | 15 (8, 20) | 13 (8, 20) | 14 (8, 20) | 0.22 | 0.24 |
| Short Test of Mental Status | 35 (28, 38) | 24 (11, 36) | 26 (7, 37) | 27 (16, 35) | 0.39 | <0.001† |
| CDR Sum of Boxes. | 0 (0, 0.5) | 5.0 (0.5, 12) | 3.5 (0.0, 18) | 3 (0.5, 12) | 0.55 | <0.001† |
| APOE ε4 carrier, n (%) | 30 (17) | 12 (63) | 73 (58) | 23 (70) | 0.49* | <0.001*† |
| Age at scan, years | 77 (43, 95) | 66 (51, 82) | 76 (47, 97) | 81 (69, 95) | <0.001‡ | <0.001§ |
| Age at onset, years | — | 63 (49, 79) | 73 (42, 96) | 79 (68, 94) | <0.001‡ | — |
| Age at death, years | — | 70 (55, 88) | 84 (51, 104) | 89 (76, 100) | <0.001‡ | — |
| Time from onset to scan, years | — | 3 (0, 9) | 3 (0, 11) | 2 (1, 9) | 0.90 | — |
| Time from scan to death, years | — | 4 (2, 9) | 6 (0, 16) | 7 (1, 14) | 0.04¶ | — |
| Disease duration, years | — | 7 (5, 13) | 9 (2, 27) | 11 (3, 19) | 0.12 | — |
| Total intracranial volume, L | 1.46 (1.16,1.85) | 1.46 (1.25,1.96) | 1.45 (1.13,2.07) | 1.45 (1.10,1.77) | 0.19 | — |
| Dominant cognitive domain affected at presentation | ||||||
| Memory | — | 8 (42) | 98 (78) | 31 (94) | 0.01** | — |
| Language | — | 3 (16) | 8 (6) | 0 (0) | — | |
| Executive | — | 2 (11) | 3 (2) | 0 (0) | — | |
| Spatial/perceptual | — | 2 (11) | 1 (1) | 0 (0) | — | |
| Praxis | — | 2 (11) | 4 (3) | 0 (0) | — | |
| Acalculia | — | 0 (0) | 1 (1) | 0 (0) | — | |
| Mixed | — | 2 (11) | 8 (6) | 1 (3) | — | |
| Unable to discern | — | 0 (0) | 2 (2) | 1 (3) | — | |
| Neurofibrillary tangle count | ||||||
| Mid-frontal | — | 14 (2, 35) | 5 (0, 25) | 1.0 (0, 10) | <0.001‡ | — |
| Superior temporal | — | 12 (7, 30) | 8 (0, 33) | 3.0 (1, 15) | <0.001‡ | — |
| Inferior parietal | — | 13 (6, 23) | 6 (0, 31) | 2 (1, 7) | <0.001‡ | — |
| Hippocampus CA1 | — | 4.0 (2, 10) | 12 (1, 45) | 20 (7, 38) | <0.001‡ | — |
| Hippocampus subiculum | — | 9.0 (1, 17) | 16 (0, 66) | 41 (18, 90) | <0.001‡ | — |
| Braak stage | – | 6 (5, 6) | 6 (4, 6) | 6 (4, 6) | 0.06 | – |
| CERAD score, n (%) | 0.42* | – | ||||
| Sparse | – | 1 (5) | 6 (5) | 1 (3) | ||
| Moderate | – | 3 (16) | 23 (18) | 11 (33) | ||
| Frequent | – | 15 (79) | 97 (77) | 21 (64) | ||
| Lewy bodies, n (%) | – | 8 (42) | 37 (30) | 10 (30) | 0.54* | – |
| Vascular disease, n (%) | – | 4 (21) | 31 (25) | 10 (30) | 0.73* | – |
Note: Unless otherwise indicated values shown are median (minimum, maximum)
Chi-square test
Chi-square test based on 5 domains by 3 groups cross-classification table evaluating differences in domains of impairment across the three AD subtypes.
Controls significantly different from each AD group (p < 0.01)
All pair-wise comparisons were significant (p ≤ 0.01)
Control group significantly younger than LP AD (p = 0.001) and significantly older than HpSp AD (p < 0.001)
HpSp AD significantly different from LP AD (p = 0.006) and Typical AD (p = 0.03) : LP AD, limbic AD; HpSp AD, hippocampal sparing AD; CDR, Clinical Dementia Rating; APOE, apolipoprotein E; CERAD = Consortium to Establish a Registry for Alzheimer’s disease
Voxel-level findings across groups
Patterns of grey matter loss in each AD group compared to specific age-matched control groups are shown in Figure 1. Typical AD showed grey matter loss throughout medial temporal lobe and lateral temporoparietal cortex, with additional involvement of precuneus, insula, medial and lateral frontal lobe, and caudate nucleus, compared to controls. In contrast, HpSp AD showed a relative sparing of the medial temporal lobe, with loss instead observed predominantly in the temporoparietal cortex, insula and precuneus. Mild loss was observed in frontal lobe and caudate nucleus. In contrast to both other groups, grey matter loss in LP AD was focused on medial temporal lobe, particularly involving hippocampus, amygdala, and entorhinal cortex, with no loss observed in lateral temporoparietal cortex, compared to controls. Patterns were very similar when each group was compared to the entire cohort of 177 controls (Supplemental Figure 1), except some mild lateral temporal loss was then observed in the LP AD group.
Figure 1.
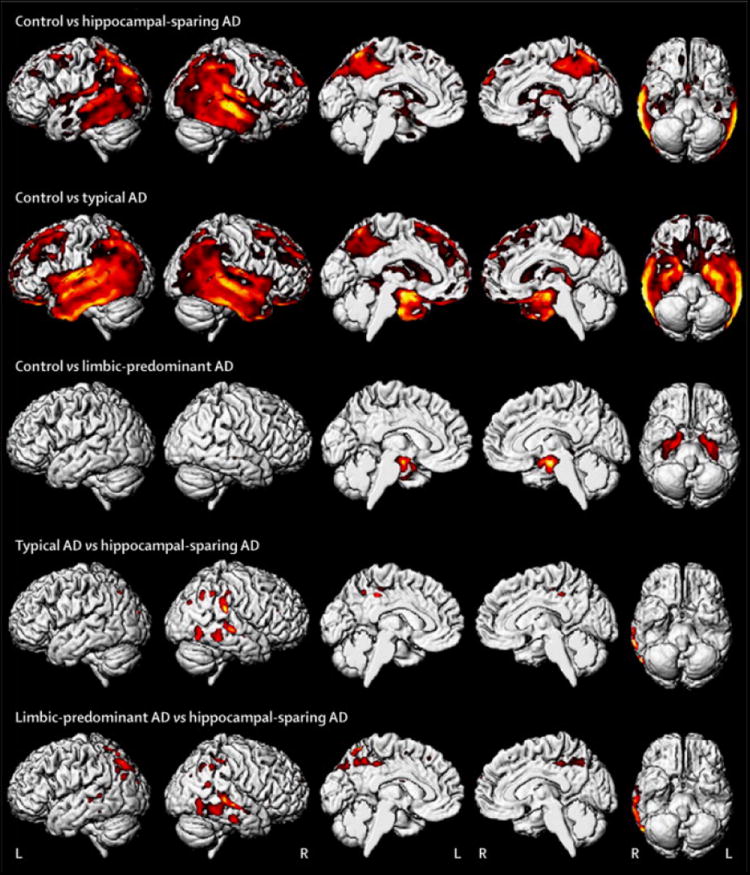
Group comparisons of grey matter volume using voxel-based morphometry. Rows 1-3 show patterns of loss in each pathological subtype compared to specific age-matched control groups. Rows 4-5 show differences between the pathological subtypes. Results are shown on three dimensional renderings of the brain after correction for multiple comparisons using the family wise error correction at p<0.05.
HpSp AD showed greater cortical loss than both typical and LP AD, predominantly involving posterior temporal lobe, inferior parietal lobe, and precuneus; with greatest loss observed in the right hemisphere (Figure 1). No other differences were observed between any of the AD groups at FWE correction of p<0.05. However, typical AD showed greater loss than LP AD in lateral temporal and frontal lobe uncorrected at p<0.001. Effect size maps for all group comparisons are shown in Figure 2.
Figure 2.
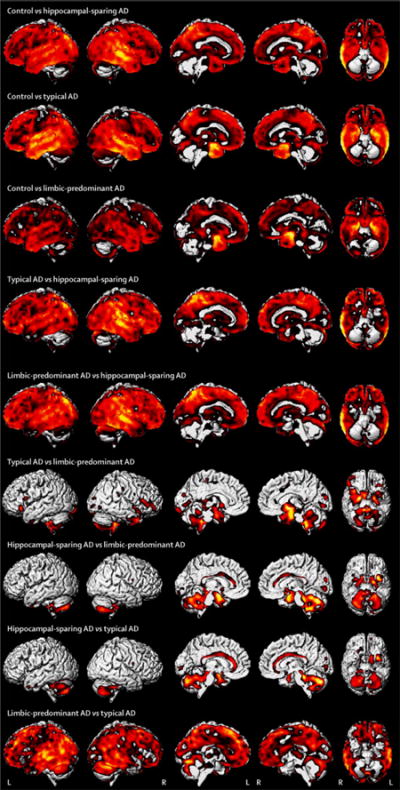
Unthresholded effect size maps showing all group comparisons of grey matter volume using voxel-based morphometry.
ROI-level findings across groups
The ROI volumes for each group are shown in Figure 3, with differences across groups highlighted in Figure 4. Amygdala (p=0.003), hippocampus (p=0.006), and entorhinal cortex (p=0.003) volumes were smaller in LP compared to HpSp AD, with the hippocampus (p=0.006) and amygdala (p=0.04) also smaller in LP compared to typical AD, and the entorhinal cortex (p=0.04) smaller in typical compared to HpSp AD. Conversely, lateral temporal, frontal and parietal lobe and precuneus volumes were all smaller in HpSp compared to typical (p=0.003, p=0.02, p<0.001, p=0.002 respectively) and LP AD (p<0.001, p=0.01, p<0.001, p<0.001 respectively). Striatal volumes were smaller in typical (p=0.02) and HpSp (p=0.004) compared to LP AD. The ratio of hippocampal-to-cortical volume gave the most significant differences across groups (HpSp= median 0.049, (range 0.039-0.060), typical = 0.042 (0.031-0.059), LP = 0.039 (0.022-0.050), p<0.001 for all comparisons), and allowed the best discrimination across all three groups; with the three-way AUROC of 0.52 (95% confidence interval 0.47, 0.52) more than three times higher than the chance classification rate (0.17). In fact, this ratio significantly improves classification beyond what is obtained for age and clinical presentation alone (p<0.001). The bootstrap validation analysis found no evidence that this three-way AUROC was overly optimistic and biased high due to overfitting.
Figure 3.
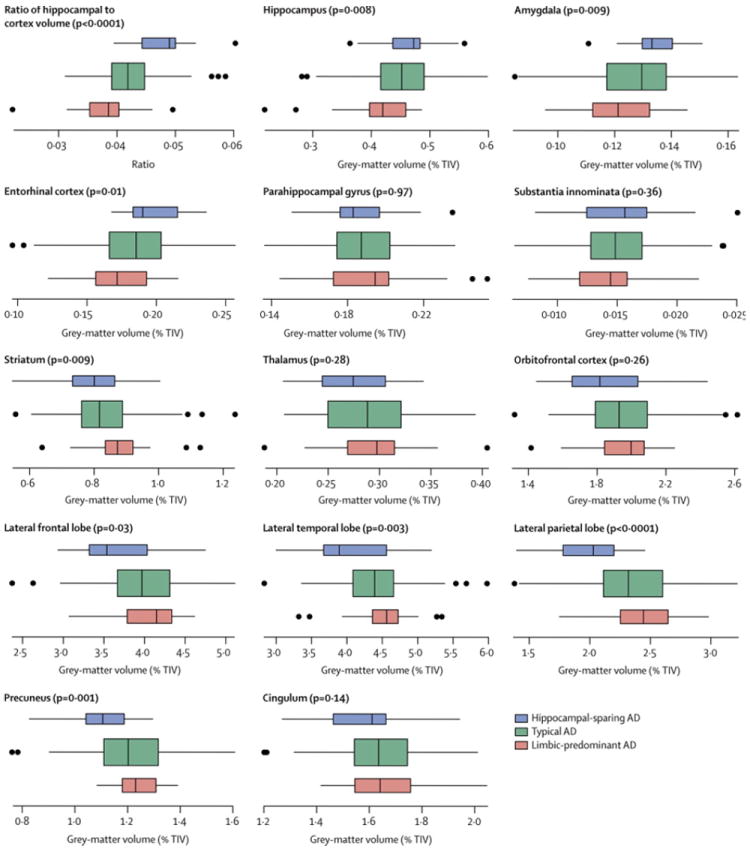
Box plots of gray matter findings by AD dementia group for 14 regions of interest. The set of box plots at the top left show the hippocampus-to-cortex volume ratio. Otherwise the value shown is the gray matter volume expressed as a percentage of TIV. Each box indicates the lower quartile (25th percentile), the median, and the upper quartile (75th percentile) of the distribution. By convention the “whiskers” of the box extend to the point furthest from the box yet still within 1.5 times the width of the box (the inter-quartile range). Points beyond the whiskers are individually indicated. As indicated in the key in the lower-right corner of the figure, the boxes are color coded. Box height on the vertical axis is related to sample size. The p-value for the one-way ANOVA test is shown in parenthesis at the top of each plot.
Figure 4.
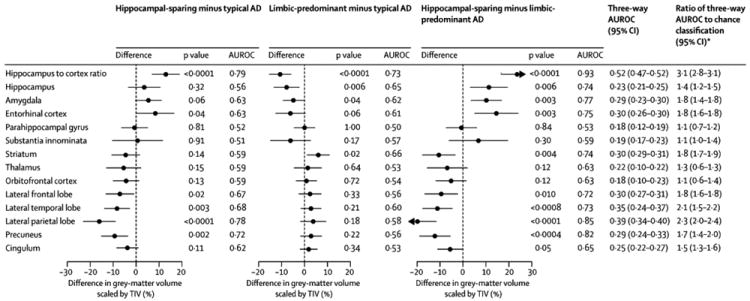
Estimates and 95% confidence intervals for the difference in gray matter volume scaled by TIV expressed as an approximate percentage difference. The numbers to the right of each confidence interval indicate the P-value for the group-wise comparison from the ANOVA model placed above the estimated AUROC for the two groups. At the right of the figure is the three-way AUROC, with a 95% confidence interval, placed above the ratio of the three-way AUROC to chance classification of 1/6 (95% confidence interval).
Correlates of clinical syndrome within typical AD
In order to understand the neuroanatomical basis of non-amnestic syndromes in typical AD, regional comparisons were performed between typical AD subjects with and without a dominant memory presentation. Subjects with non-memory presentations showed a significantly greater ratio of hippocampal-to-cortical volume (Figure 5) compared to those with memory presentations (non-memory= 0.045 (0.035-0.056), memory= 0.041 (0.031-0.057), p=0.001). In addition, those with non-memory presentations showed smaller lateral temporal, striatal and substantia innominata volumes, and greater hippocampal, amygdala and entorhinal cortex volumes, compared to those with dominant memory impairment (Supplementary Table 1).
Figure 5.
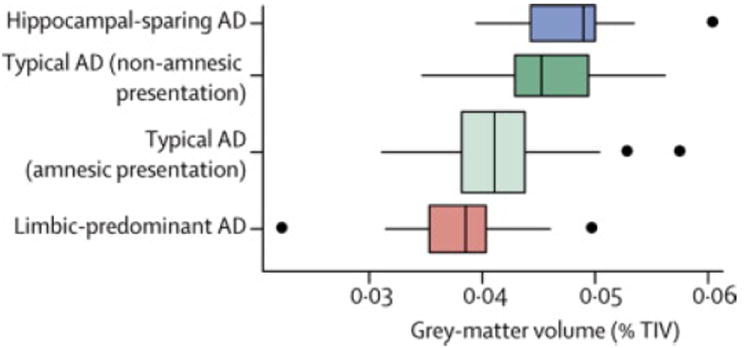
Box plots of the hippocampus-to-cortex gray matter volume for each AD dementia group. The typical AD group has been divided into those with a predominantly memory domain of impairment versus others. Two subjects from the typical AD group are not shown due to insufficient clinical information. Each box indicates the lower quartile (25th percentile), the median, and the upper quartile (75th percentile) of the distribution. By convention the “whiskers” of the box extend to the point furthest from the box yet still within 1.5 times the width of the box (the inter-quartile range). Points beyond the whiskers are individually indicated. Box height on the vertical axis is related to sample size. At the top left is the p-value for the one-way ANOVA test.
DISCUSSION
This study demonstrates striking differences in patterns of atrophy on MRI across the pathological subtypes of AD, supporting the pathological classification and suggesting that patterns of atrophy can predict pathological subtype during life.
A ratio of hippocampal-to-cortical volume provided the best discrimination across all three AD subtypes, with HpSp AD showing the least hippocampal atrophy, yet the most cortical atrophy, and at the other extreme, LP AD showing the most hippocampal atrophy, yet the least cortical atrophy. These patterns further support a relationship between NFT deposition and atrophy2-5. The ratio of hippocampal-to-cortical NFT density was, of course, critical to the classification of these subjects6. However, we now demonstrate that differences in this ratio can be detected using MRI, and, importantly, that differences across groups can be identified on the earliest MRI obtained for each subject, an average 6-8 years before death. Discrimination was greatest between HpSp and LP AD since these types represent the two extremes of the spectrum; however, discrimination across all three groups was also excellent suggesting that this simple imaging ratio could be useful to differentiate pathological subtypes at the individual level.
Although hippocampal involvement was the most obvious element of LP AD, other limbic structures, such as amygdala and entorhinal cortex, were also involved. Conversely, these limbic structures were relatively preserved in HpSp AD. However, other regions that are often considered part of the limbic system, such as parahippocampal gyrus, orbitofrontal cortex, thalamus and cingulate gyrus, did not differ across groups. If anything, they were involved to a greater degree in HpSp AD. The striatum and precuneus also showed greatest involvement in HpSp AD. It appears as though LP AD specifically targets medial temporal lobe structures that are usually involved earliest in AD1, while HpSp AD targets cortical and subcortical regions that are usually involved later in the typical AD pathological process1.
The profile of cognitive impairment differed strikingly across AD subtypes. Memory impairment was the presenting symptom in the majority of typical and LP AD subjects, but was less common in HpSp AD. The majority of HpSp AD subjects, instead, presented with non-amnestic symptoms, such as language impairment, spatial/perceptual impairment, and praxis, or a mixture of these symptoms. Atypical clinical syndromes defined by these non-memory impairments have indeed previously been associated with AD pathology22-25. We, and others, have demonstrated that atrophy patterns in these non-memory presentations of AD typically differ from those observed in the amnestic form of Alzheimer’s dementia, with involvement of the cortex but often relative sparing of the hippocampus17, 24, 26, 27. It is likely that these findings are driven by the presence of HpSp AD cases in the non-amnestic groups. Our findings suggest that non-amnestic presentations of AD arise not only as a result of hippocampal sparing, but also due to proportionally greater involvement of the cortex compared to the hippocampus. Some of the subjects with typical AD did, however, also present with non-amnestic impairments, and interestingly, these subjects showed higher hippocampal-to-cortical ratios than the typical AD subjects with memory impairments. These subjects therefore appear to be closer on the neurodegenerative continuum to HpSp AD. In contrast, almost every LP AD subject presented with memory complaints, which is unsurprising given the severe hippocampal loss in this group.
Although non-amnestic syndromes were common in HpSp AD, some subjects did present with memory impairment. A subject presenting with typical Alzheimer’s dementia, dominated by memory impairment, could therefore have HpSp, typical or LP AD. This has important implications for clinical trials that recruit patients with typical Alzheimer’s dementia, especially since hippocampal atrophy has been used as a disease biomarker. The presence of different pathological subtypes will introduce variability into the study, inflating or decreasing hippocampal estimates, and influencing observed treatment effects on hippocampal volume. It is likely that rates of hippocampal and cortical atrophy will also differ across pathological types, although longitudinal studies will be needed to test this hypothesis.
Age also differed across pathological subtypes, with youngest age observed in HpSp AD, and oldest age observed in LP AD, as previously reported6. Consistent with our clinical findings, early age-at-onset has previously been associated with the presence of atypical clinical syndromes in AD28. Our findings also likely explain why subjects with early-onset Alzheimer’s dementia have been observed to have more widespread cortical involvement than late-onset cases29, 30.
Although we studied a large cohort of pathologically-confirmed subjects, the power of the analyses involving HpSp and LP AD subjects could have been limited by the small number of subjects in these groups. In addition, the subjects were recruited from a specialized tertiary care facility which may limit the generalizability of the findings. However, the proportion of different AD subtypes, and the clinical/demographic differences across subtypes, are almost identical to those observed in a larger (n=889) cohort recruited from a more culturally and economically diverse population in Florida6, suggesting our findings may generalize to more diverse healthcare settings.
This study demonstrates striking differences in patterns of atrophy across AD pathological subtypes showing that the pathological classification is clinically meaningful and highlighting the importance of recognizing atypical subtypes in imaging studies of AD. A ratio of hippocampal-to-cortical volume could help predict pathological subtype during life. Identifying the pathological subtypes of AD will be important clinically to help predict progression, since cognitive decline is more rapid in HpSp AD6. In addition, recognizing the fact that AD pathology may not always be associated with prominent hippocampal atrophy will be important to help target treatment for these atypical patients.
RESEARCH IN CONTEXT
Systematic review
We performed a pubmed search for articles published in English between Jan 1 1984 and May 1 2012, with the search terms “atypical Alzheimer’s disease”, “early onset Alzheimer’s disease”, “hippocampal sparing Alzheimer’s disease”, “neurofibrillary tangles”, “hippocampus”, and “imaging biomarkers”. We specifically selected studies that assessed imaging in patients with autopsy-confirmed Alzheimer’s disease (AD) that had atypical clinical presentations, and studies that had assessed correlations between imaging biomarkers and pathology in AD.
Interpretation
We demonstrate that patterns of grey matter loss on antemortem MRI differ between recently identified atypical pathological subtypes of AD (hippocampal sparing AD and limbic predominant AD) and typical AD, with a ratio of hippocampal-to-cortical volume providing good separation across AD subtypes. Hippocampal sparing AD was associated with sparing of the hippocampus, atypical non-amnestic clinical presentations and younger age, and is therefore the most likely cause of non-amnestic presentations of AD reported in previous clinical studies. The pathological grouping was based solely on neurofibrillary tangle densities, but we demonstrate clinical and imaging differences across subtypes suggesting these groupings are clinically meaningful and useful. Imaging has the potential to help predict these pathological subtypes during life.
Supplementary Material
Acknowledgments
This study was funded by NIH grants R21-AG038736 (PI Whitwell), R01-AG037491 (PI Josephs), R01-AG011378 (PI Jack), and P50-AG016574 (PI Petersen). The authors would like to acknowledge Dr.’s Emre Kokmen and Daniel A. Drubach for performing clinical assessments of some of the ADRC subjects.
Role of the funding source
The funding sources had no role in the study design, data collection, data analysis, data interpretation, writing of the report, or decision to submit for publication.
Footnotes
CONTRIBUTORS
JLW, KAJ and DWD were responsible for study concept and design and data interpretation. JLW performed all voxel-based morphometry processing and analysis and created Figure 1. KAJ was responsible for acquisition and interpretation of clinical data and for study supervision. DWD and MEM were responsible for neurofibrillary tangle data acquisition and pathological classification. Pathological examinations were performed by DWD and JEP. SDW and NT were responsible for statistical analysis and for creating Figures 2-4. MLS was responsible for acquisition of regional MRI data. DSK, BFB and RCP were responsible for acquisition of clinical data. CRJ was responsible for acquisition of MRI. JLW drafted the original report which was reviewed and revised by KAJ, DWD, MEM, SDW, NT, MLS, DSK, BFB, JEP, RCP and CRJ. Funding was obtained by JLW, KAJ, RCP and CRJ.
CONFLICTS OF INTEREST
The authors have no conflicts of interest relevant to this manuscript
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Braak H, Braak E. Neuropathological stageing of Alzheimer-related changes. Acta neuropathologica. 1991;82:239–259. doi: 10.1007/BF00308809. [DOI] [PubMed] [Google Scholar]
- 2.Whitwell JL, Josephs KA, Murray ME, et al. MRI correlates of neurofibrillary tangle pathology at autopsy: a voxel-based morphometry study. Neurology. 2008;71:743–749. doi: 10.1212/01.wnl.0000324924.91351.7d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jack CR, Jr, Dickson DW, Parisi JE, et al. Antemortem MRI findings correlate with hippocampal neuropathology in typical aging and dementia. Neurology. 2002;58:750–757. doi: 10.1212/wnl.58.5.750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gosche KM, Mortimer JA, Smith CD, et al. Hippocampal volume as an index of Alzheimer neuropathology: findings from the Nun Study. Neurology. 2002;58:1476–1482. doi: 10.1212/wnl.58.10.1476. [DOI] [PubMed] [Google Scholar]
- 5.Csernansky JG, Hamstra J, Wang L, et al. Correlations between antemortem hippocampal volume and postmortem neuropathology in AD subjects. Alzheimer disease and associated disorders. 2004;18:190–195. [PubMed] [Google Scholar]
- 6.Murray ME, Graff-Radford NR, Ross OA, et al. Neuropathologically defined subtypes of Alzheimer’s disease with distinct clinical characteristics: a retrospective study. Lancet neurology. 2011;10:785–796. doi: 10.1016/S1474-4422(11)70156-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hyman BT, Trojanowski JQ. Consensus recommendations for the postmortem diagnosis of Alzheimer disease from the National Institute on Aging and the Reagan Institute Working Group on diagnostic criteria for the neuropathological assessment of Alzheimer disease. Journal of neuropathology and experimental neurology. 1997;56:1095–1097. doi: 10.1097/00005072-199710000-00002. [DOI] [PubMed] [Google Scholar]
- 8.Petersen RC, Kokmen E, Tangalos E, et al. Mayo Clinic Alzheimer’s Disease Patient Registry. Aging (Milan, Italy) 1990;2:408–415. doi: 10.1007/BF03323961. [DOI] [PubMed] [Google Scholar]
- 9.American Pyschiatric Association. Diagnostic and Statistical Manual of Mental Disorders. 4. Washington DC: American Psychiatric Association; 1994. DSM IV. [Google Scholar]
- 10.Mirra SS, Heyman A, McKeel D, et al. The Consortium to Establish a Registry for Alzheimer’s Disease (CERAD). Part II. Standardization of the neuropathologic assessment of Alzheimer’s disease. Neurology. 1991;41:479–486. doi: 10.1212/wnl.41.4.479. [DOI] [PubMed] [Google Scholar]
- 11.Knopman DS, Parisi JE, Salviati A, et al. Neuropathology of cognitively normal elderly. Journal of neuropathology and experimental neurology. 2003;62:1087–1095. doi: 10.1093/jnen/62.11.1087. [DOI] [PubMed] [Google Scholar]
- 12.Sled JG, Zijdenbos AP, Evans AC. A nonparametric method for automatic correction of intensity nonuniformity in MRI data. IEEE transactions on medical imaging. 1998;17:87–97. doi: 10.1109/42.668698. [DOI] [PubMed] [Google Scholar]
- 13.Ashburner J, Friston KJ. Voxel-based morphometry--the methods. NeuroImage. 2000;11:805–821. doi: 10.1006/nimg.2000.0582. [DOI] [PubMed] [Google Scholar]
- 14.Vemuri P, Gunter JL, Senjem ML, et al. Alzheimer’s disease diagnosis in individual subjects using structural MR images: validation studies. NeuroImage. 2008;39:1186–1197. doi: 10.1016/j.neuroimage.2007.09.073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ashburner J, Friston KJ. Unified segmentation. NeuroImage. 2005;26:839–851. doi: 10.1016/j.neuroimage.2005.02.018. [DOI] [PubMed] [Google Scholar]
- 16.Tzourio-Mazoyer N, Landeau B, Papathanassiou D, et al. Automated anatomical labeling of activations in SPM using a macroscopic anatomical parcellation of the MNI MRI single-subject brain. NeuroImage. 2002;15:273–289. doi: 10.1006/nimg.2001.0978. [DOI] [PubMed] [Google Scholar]
- 17.Whitwell JL, Jack CR, Jr, Przybelski SA, et al. Temporoparietal atrophy: A marker of AD pathology independent of clinical diagnosis. Neurobiology of aging. 2009 doi: 10.1016/j.neurobiolaging.2009.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gelman A, Hill J. Data analysis using regression and multilevel/hierarchical models. New York: Cambridge University Press; 2007. [Google Scholar]
- 19.Li J, Fine JP. ROC analysis with multiple classes and multiple tests: methodology and its application in microarray studies. Biostatistics. 2008;9:566–576. doi: 10.1093/biostatistics/kxm050. [DOI] [PubMed] [Google Scholar]
- 20.Perneger TV. What’s wrong with Bonferroni adjustments. BMJ (Clinical Research Ed) 1998;316:1236–1238. doi: 10.1136/bmj.316.7139.1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rothman KJ. No adjustments are needed for multiple comparisons. Epidemiology (Cambridge, Mass) 1990;1:43–46. [PubMed] [Google Scholar]
- 22.Alladi S, Xuereb J, Bak T, et al. Focal cortical presentations of Alzheimer’s disease. Brain. 2007;130:2636–2645. doi: 10.1093/brain/awm213. [DOI] [PubMed] [Google Scholar]
- 23.Galton CJ, Patterson K, Xuereb JH, Hodges JR. Atypical and typical presentations of Alzheimer’s disease: a clinical, neuropsychological, neuroimaging and pathological study of 13 cases. Brain. 2000;123(Pt 3):484–498. doi: 10.1093/brain/123.3.484. [DOI] [PubMed] [Google Scholar]
- 24.Josephs KA, Whitwell JL, Duffy JR, et al. Progressive aphasia secondary to Alzheimer disease vs FTLD pathology. Neurology. 2008;70:25–34. doi: 10.1212/01.wnl.0000287073.12737.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mesulam M, Wicklund A, Johnson N, et al. Alzheimer and frontotemporal pathology in subsets of primary progressive aphasia. Annals of neurology. 2008;63:709–719. doi: 10.1002/ana.21388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lehmann M, Rohrer JD, Clarkson MJ, et al. Reduced cortical thickness in the posterior cingulate gyrus is characteristic of both typical and atypical Alzheimer’s disease. J Alzheimers Dis. 2010;20:587–598. doi: 10.3233/JAD-2010-1401. [DOI] [PubMed] [Google Scholar]
- 27.Migliaccio R, Agosta F, Rascovsky K, et al. Clinical syndromes associated with posterior atrophy: early age at onset AD spectrum. Neurology. 2009;73:1571–1578. doi: 10.1212/WNL.0b013e3181c0d427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Licht EA, McMurtray AM, Saul RE, Mendez MF. Cognitive differences between early- and late-onset Alzheimer’s disease. American journal of Alzheimer’s disease and other dementias. 2007;22:218–222. doi: 10.1177/1533317506299156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Frisoni GB, Testa C, Sabattoli F, et al. Structural correlates of early and late onset Alzheimer’s disease: voxel based morphometric study. Journal of neurology, neurosurgery, and psychiatry. 2005;76:112–114. doi: 10.1136/jnnp.2003.029876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ishii K, Kawachi T, Sasaki H, et al. Voxel-based morphometric comparison between early- and late-onset mild Alzheimer’s disease and assessment of diagnostic performance of z score images. Ajnr. 2005;26:333–340. [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.