

Abstract

The potential of a uniquely permissive engineered glycosyltransferase (OleD ASP) as a catalyst for steroid glycosylation is highlighted. The ability of OleD ASP to glucosylate a range of cardenolides and bufadienolides was assessed using a rapid LC-UV/MS-SPE-NMR analytical platform. While a bias toward OleD-catalyzed C3 mono-glucosylation was observed, subtle alterations of the steroidal architecture in some cases, invoked diglucosylation or, in one case (digoxigenin), C12 glucosylation. This latter case represents the first, and highly efficient, synthesis of digoxigenin 12-O-β-D-glucoside.
Steroidal glycosides such as digitoxin, digoxin and proscillaridin have been used for centuries to treat congestive heart failure and more recently noted to display highly potent anticancer activity.1 These ligands are known to bind the Na+,K+-ATPase alpha subunit with high affinity where they function to control both a range of intracellular signaling cascades critical to cell proliferation and regulate intracellular Na+ and K+ concentrations.2 C3 glycosylation of the steroidal core among this natural product class has a dramatic effect upon the delicate balance between signaling (antiproliferative) versus inotropic (cardiotoxicity) activities.3 Thus, there remains notable interest in the development of simple glycosylation platforms as a means to improve the putative therapeutic index of steroidal glycoside anticancer preclinical leads. Using a novel high throughput screen and LC-MS,4 representative steroidal aglycons were recently identified as substrates for a set of highly permissive glycosyltransferase variants derived from the macrolideinactivating glucosyltransferase OleD.5 Herein we extend this preliminary study through the application of a LC-UV/MS-SPE/NMR platform6 to rapidly probe the regio-/stereospecificity of OleD-catalyzed glycosylation of cardenolide and bufadienolide aglycons. While this study reveals a bias toward the desired C3 regiospecificity in the context of a range of non-native substrates for this enzyme, it also highlights how subtle modifications of the steroidal aglycon can dictate and/or prohibit glycosyltransfer.
To explore the feasibility of the LC-UV/MS-SPE-NMR platform for micro-scale structural elucidation of steroidal glycosides, an initial pilot study was conducted using digitoxigenin as the model (Scheme 1). For this study, the reaction contained 600 μg of OleD ASP, 2.5 mM UDP-Glc, 50 mM Tris-HCl (pH 8.0), 5 mM MgCl2, and 1 mM of digitoxigenin in a total volume of 1 mL. The reaction was allowed to proceed for 16 h at 25 °C and subsequently frozen, lyophilized, and the debris resuspended in 2 mL of ice cold MeOH, filtered and concentrated to 150 μl for LC-UV/MS-SPE-NMR analysis. The HPLC component of the subsequent analysis platform was accomplished using standard C18 reverse-phase chromatography with diode array detection wherein ~5 % of the flow was diverted to quadrupole time-of-flight mass (QTOF) detection. Fractions containing detected peaks were automatically diverted to pre-conditioned solid-phase extraction (SPE, C18) cartridges, which were subsequently dried via N2 gas and then eluted with 30 μl CD3CN into 1.7 mm NMR tubes for direct analysis via a Bruker Avance III 600 MHz spectrometer with a 1.7 mm 1H{13C/15N} cryogenic probe.
Scheme 1.
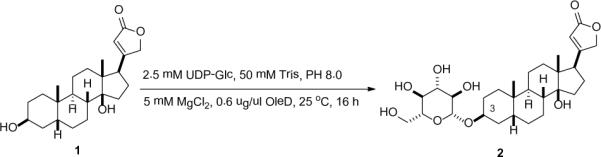
OleD catalyzed glucosylation of digitoxigenin.
LC-MS analysis of the pilot reaction described above revealed the formation of a single glycoside in 20% yield. Structure elucidation of this glycoside, based upon 1H NMR, 1H-1H COSY, 1H-13C HSQC, and 1H-13C HMBC, was consistent with the 3-O-β-D-glucoside 2 (Scheme 1).7 Key support for the regiospecificity assignment derived from an observed HMBC correlation between the sugar anomeric C1' proton and the C3 of digitoxigenin. An observed large anomeric proton coupling constant (δH 4.32, doublet, 8.0 Hz) supported the formation of the β-anomer, consistent with an established inverting mechanism for OleD variants studied to date8 C3 glucosylation also led to a notably consistent large downfield 13C shift (~7 ppm) of the C3 carbon (`glycosylation shift'),9 which served as a convenient indicator in probing OleD-catalyzed glycosylation of alternative steroidal aglycons (Table 1).
Table 1.
C3 13C chemical shifts of substrates and products
| compound | C3 shift (ppm) | compound | C3 shift (ppm) |
|---|---|---|---|
| 1 | 66.7 | 2 | 73.5 |
| 3 | 66.3a | 10 | 66.0b |
| 4 | 66.810 | 11 | 73.6 |
| 7 | 67.73(c) | 12 | 75.6 |
| 8 | 66.711 | 13 | 73.7 |
| 14 | 74.8 | ||
| 9 | 66.612 | 15 | 73.2 |
| 16 | 74.1 |
C12 13C chemical shift 73.7 ppm;
C12 13C chemical shift 82.1 ppm
Given the success of the digitoxigenin pilot study, steroidal aglycons 3–9 (Figure 1) were subsequently treated in an identical manner. Five (aglycon 3, 4, 7, 8, and 9) out of the seven putative aglycons led to glucosylated products, three (3, 4, and 7) of which led to mono-glucosides (10,13 (11,14 and 1215) and two (8 and 9) of which provided both mono- (13, and 1516) and di-glucosides (14, and 16).17 A bias toward C3 glycosylation was observed with one surprising exception – digoxigenin, which led to C12 glucosylation exclusively in high efficiency.
Figure 1.

Potential substrates and determined products of OleD-catalyzed test reactions.
The docking and molecular dynamics (MD) simulation of OleD complexed with 1 or 3 are consistent with our experimental data (Figure 2). In comparison to the binding mode of 1, the binding model of 3 invokes an `inverse binding mode' due to electrostatic and steric repulsions between the steroid C12-OH and the Tyr140 side chain-OH (Figure S3). Similar docking and MD simulations with ligands 4–9, 13 and 15 revealed the following key observations. The predicted binding of ouabaingenin (5) and strophanthidin (6) are misaligned and distant (~4.7 Å, Fig. S4, panels b and c) with the active-site base, the His19 side chain imidazole, due in part to the electrostatic attraction between the Asn80 side-chain carboxamide and functional groups at C19 in 5 or 6. In contrast, the predicted binding modes of 8 and 9 highlight favorable dipole-quadrupole stabilization between the substrate C16 methyl ester and the Tyr140 (Figure S4), also prevalent in the binding models for the corresponding monoglucosides forms 13 and 15, which may be important to the observed iterative (steroid C3 and subsequent glucoside C2') glucosylation of 8 and 9.
Figure 2.

Simulated ligand-bound models for OleD-1 (a) and OleD-3 (b). Ligands are represented in cyan, OleD in green, key active-site residues in purple and H-bonds as dashed lines. The calculated binding energy of the compounds 1 and 3 with OleD are −12.1 and −11.9 kcal/mol.
While the impact of C3 glycosylation upon the anticancer activity of steroidal glycosides has been previously reported,3a,18 the influence of C12 glycosylation upon bioactivity of this natural product class had not. Thus, the enzymatic synthesis of 10 was subsequently scaled to enable bioactivity assessment. Specifically, digoxigenin 3 (10 mg, 25.6 μmol) was dissolved in 0.65 mL DMSO, transferred to 25 mL assay buffer solution (50 mM Tris HCl, 5 mM MgCl2, pH 8.0) and the reaction initiated via addition of UDP-Glc (62 mg, 0.124 mmol) and 16 mg of OleD ASP. After 24 hr of agitation at room temperature, the reaction was quenched and flushed though a 12 mL HLB column, which was subsequently dried with N2 gas and then eluted with methanol. The methanolic eluent was concentrated and the collected residue subjected to flash chromatography using CH2Cl2/MeOH to afford digoxigenin 12-O-β-D-Glc 10 (13 mg, 23.5 μmol, 92%)-highlighting the first synthesis of digoxigenin 12-O-β-D-Glc. Consistent with the belief that steroidal glycosides bind the Na+, K+-ATPase alpha subunit with the lactone (and presumably C12) buried deep within the ligand binding pocket,19 the C12 glucoside was found to be ~5–10-fold less active than the corresponding aglycon based upon in vitro cytotoxicity against two lung cancer cell lines (Table 2). The rather surprising fact that the activity of the C12 glycoside was not further diminished suggests notable flexibility in the Na+, K+-ATPase alpha subunit ligand-binding pocket.
Table 2.
In vitro cytotoxicity (μM)
H460, large cell lung carcinoma;
A549, small cell lung carcinoma
In summary, this study highlights the capabilities of OleD ASP in the context of steroidal glycoside construction and reveals how subtle alterations of the steroidal architecture can dramatically influence regioselectivity of the reaction. As such, this study serves to provide additional information for understanding substrate specificity of the uniquely permissive OleD ASP variant and also sets the stage for rapid enzymatic glycorandomization of this class of natural product through the use of alternative sugar donors.20
Supplementary Material
Acknowledgment
This work was supported by NIH AI52218 (JST) and also made use of the National Magnetic Resonance Facility at Madison, which is supported by NIH grants P41RR02301 (BRTP/ NCRR) and P41GM66326 (NIGMS). Additional equipment was purchased with funds from the University of Wisconsin, the NIH (RR02781, RR08438), the NSF (DMB-8415048, OIA-9977486, BIR-9214394), and the USDA.
Footnotes
Supporting Information Available: Experimental and computational procedures, characterization, and biological testing. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- (1).(a) Newman RA, Yang P, Pawlus AD, Block KI. Mol. Interv. 2008;8:36–49. doi: 10.1124/mi.8.1.8. [DOI] [PubMed] [Google Scholar]; (b) Prassas I, Diamandis EP. Nat. Rev. Drug Discov. 2008;7:926–935. doi: 10.1038/nrd2682. [DOI] [PubMed] [Google Scholar]; (c) Vaklavas C, Chatzizisis YS, Tsimberidou AM. Pharmacol. Ther. 2011;130:177–190. doi: 10.1016/j.pharmthera.2011.01.009. [DOI] [PubMed] [Google Scholar]; (d) Gao H, Popescu R, Kopp B, Wang Z. Nat. Prod. Rep. 2011;28:953–969. doi: 10.1039/c0np00032a. [DOI] [PubMed] [Google Scholar]; (e) Agrawal AA, Petschenka G, Bingham RA, Weber MG, Rasmann S. New Phytol. 2012;194:28–45. doi: 10.1111/j.1469-8137.2011.04049.x. [DOI] [PubMed] [Google Scholar]; (f) Yang EH, Shah S, Criley JM. Am. J. Med. 2012;125:337–343. doi: 10.1016/j.amjmed.2011.09.019. [DOI] [PubMed] [Google Scholar]
- (2).(a) Schoner W, Scheiner-Bobis G. Am. J. Physiol. Cell Physiol. 2007;293:C509–36. doi: 10.1152/ajpcell.00098.2007. [DOI] [PubMed] [Google Scholar]; (b) Suhail M. J. Clin. Med. Res. 2010;2:1–17. doi: 10.4021/jocmr2010.02.263w. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Mijatovic T, Dufrasne F, Kiss R. Curr. Med. Chem. 2012;19:627–646. doi: 10.2174/092986712798992075. [DOI] [PubMed] [Google Scholar]; (d) Hauck C, Frishman WH. Cardiol. Rev. 2012;20:130–138. doi: 10.1097/CRD.0b013e31823c835c. [DOI] [PubMed] [Google Scholar]
- (3).(a) Langenhan JM, Peters NR, Guzei IA, Hoffmann FM, Thorson JS. Proc. Natl. Acad. Sci. 2005;102:12305–12310. doi: 10.1073/pnas.0503270102. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Langenhan JM, Engle JM, Slevin LK, Fay LR, Lucker RW, Smith KR, Endo MM. Bioorg. Med. Chem. Lett. 2008;18:670–673. doi: 10.1016/j.bmcl.2007.11.058. [DOI] [PubMed] [Google Scholar]; (c) Hutchinson CR, Shekhani MS, Prudent JR. US patent. 2011 20110319350.
- (4).(a) Gantt RW, Goff RD, Williams GJ, Thorson JS. Angew. Chem. Int. Ed. 2008;47:8889–8892. doi: 10.1002/anie.200803508. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Gantt RW, Peltier-Pain P, Cournoyer WJ, Thorson JS. Nat. Chem. Biol. 2011;7:685–691. doi: 10.1038/nchembio.638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).(a) Yang M, Proctor MR, Bolam DN, Errey JC, Field RA, Gilbert HJ, Davis BG. J. Am. Chem. Soc. 2005;127:9336–9337. doi: 10.1021/ja051482n. [DOI] [PubMed] [Google Scholar]; (b) Williams GJ, Zhang C, Thorson JS. Nat. Chem. Biol. 2007;3:657–662. doi: 10.1038/nchembio.2007.28. [DOI] [PubMed] [Google Scholar]; (c) Williams GJ, Goff RD, Zhang C, Thorson JS. Chem Biol. 2008;15:393–401. doi: 10.1016/j.chembiol.2008.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).(a) Seger C, Godejohann M, Tseng L, Spraul M, Girtler A, Sturm S, Stuppner H. Anal. Chem. 2005;77:878–885. doi: 10.1021/ac048772r. [DOI] [PubMed] [Google Scholar]; (b) Motti CA, Freckelton ML, Tapiolas DM, Willis RH. J. Nat. Prod. 2009;72:290–294. doi: 10.1021/np800562m. [DOI] [PubMed] [Google Scholar]; (c) Castro A, Moco S, Coll J, Vervoort J. J. Nat. Prod. 2010;73:962–965. doi: 10.1021/np9005025. [DOI] [PubMed] [Google Scholar]
- (7).Synthesis of digitoxigenin Glc: Elderfield RC, Uhle FC, Fried J. J. Am. Chem. Soc. 1947;69:2235–2236. doi: 10.1021/ja01201a506.. Kihara M, Yoshioka K, Kitatsuji E, Hashimoto T, Fullerton D.t S., Rohrer DC. Steroids. 1983;42:37–54. doi: 10.1016/s0039-128x(83)90072-7.. Ooi Y, Hasimoto T, Mitsuo N, Satoh T. Tetrahedron Lett. 1984;25:2241–2244.
- (8).(a) Quirós LM, Carbajo RJ, Salas JA. FEBS Lett. 2000;476:186–189. doi: 10.1016/s0014-5793(00)01721-x. [DOI] [PubMed] [Google Scholar]; (b) Quirós LM, Carbajo RJ, Braña AF, Salas JA. J. Biol. Chem. 2000;275:11713–11720. doi: 10.1074/jbc.275.16.11713. [DOI] [PubMed] [Google Scholar]; (c) Bolam DN, Roberts S, Proctor MR, Turkenburg JP, Dodson EJ, Martinez-Fleites C, Yang M, Davis BG, Davies GJ, Gilbert H. Proc. Natl. Acad. Sci. USA. 2007;104:5336–5341. doi: 10.1073/pnas.0607897104. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Zhou M, Thorson JS. Org. Lett. 2011;13:2786–2788. doi: 10.1021/ol200977u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).(a) Kasai R, Suzuo M, Asakawa J, Tanaka O. Tetrahedron Lett. 1977;2:175–178. [Google Scholar]; (b) Taki T, Kuroyanagi M, Yoshioka H, Handa S. J. Biochem. 1992;111:614–619. doi: 10.1093/oxfordjournals.jbchem.a123806. [DOI] [PubMed] [Google Scholar]
- (10).Tori K, Ishii H, Wolkowski ZW, Chachaty C, Sangaré M, Piriou F, Lukacs G. Tetrahedron Lett. 1973;14:1077–1080. [Google Scholar]
- (11).Yoshiaki K, Ayano K. Collect. Czech. Chem. Commun. 1998;63:1663–1670. [Google Scholar]
- (12).Ye M, Han J, Guo H, Guo D. Magn. Reson. Chem. 2002;40:786–788. [Google Scholar]
- (13).For the synthesis of digoxigenin 3-Gc, 7 Brown L, Boutagy J, Thomas R. Arzneimittel Forschung. 1981;31:1059–10604.. For the chemical synthesis of the 3,12-diglucoside of digoxigenin: Makarevich IF. Pharm. Chem. J. 1969;5:266–269.
- (14).Makarevich IF. Khimiya Prirodnykh Soedinenii. 1971;7:45–46. [Google Scholar]
- (15).Krenn L, Kopp B, Deim A, Robien W, Kubelka W. Planta Med. 1994;60:63–69. doi: 10.1055/s-2006-959410. [DOI] [PubMed] [Google Scholar]
- (16).(a) Ye M, Dai J.i, Guo H, Cui Y, Guo D. Tetrahedron Lett. 2002;43:8535–8538. [Google Scholar]; (b) Ye M, Han J, Guo H, Guo D. Magn. Reson. Chem. 2002;40:786–788. [Google Scholar]
- (17).The diglucosylation was evidenced by the HMBC coupling of the anomeric proton of the second sugar with C2 carbon of the first sugar
- (18).(a) Wang HY, Xin W, Zhou M, Stueckle TA, Rojanasakul Y, O'Doherty GA. ACS Med. Chem. Lett. 2011;2:73–78. doi: 10.1021/ml100219d. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Rashan LJ, Franke K, Khine MM, Kelter G, Fiebig HH, Neumann J, Wessjohann LA. J. Ethnopharmacol. 2011;134:781–8. doi: 10.1016/j.jep.2011.01.038. [DOI] [PubMed] [Google Scholar]
- (19).(a) Ogawa H, Shinoda T, Cornelius F, Toyoshima C. Proc. Natl. Acad. Sci. USA. 2009;106:13742–13747. doi: 10.1073/pnas.0907054106. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Katz A, Lifshitz Y, Bab-Dinitz E, Kapri-Pardes E, Goldshleger R, Tal DM, Karlish SJ. J. Biol. Chem. 2010;285:19582–19592. doi: 10.1074/jbc.M110.119248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).(a) Williams GJ, Thorson JS. Adv. Enzymol. Relat. Areas. Mol. Biol. 2009;76:55–119. doi: 10.1002/9780470392881.ch2. [DOI] [PubMed] [Google Scholar]; (b) Gantt RW, Peltier-Pain P, Thorson JS. Nat. Prod. Rep. 2011;28:1811–1853. doi: 10.1039/c1np00045d. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.