

Abstract
Various binuclear metal ion clusters and complexes have been reconstituted in crystalline human arginase I by removing the Mn2+2-cluster of the wild-type enzyme with metal chelators and subsequently soaking the crystalline apoenzyme in buffer solutions containing NiCl2 or ZnCl2. X-ray crystal structures of these metal ion variants are correlated with catalytic activity measurements that reveal differences resulting from metal ion substitution. Additionally, treatment of crystalline Mn2+2-human arginase I with Zn2+ reveals for the first time the structural basis for inhibition by Zn2+, which forms a carboxylate-histidine-Zn2+ triad with H141 and E277. The imidazole side chain of H141 is known to be hyper-reactive and its chemical modification or mutagenesis is known to similarly compromise catalysis. The reactive substrate analogue 2(S)-amino-6-boronohexanoic acid (ABH) binds as a tetrahedral boronate anion to Mn2+2, Co2+2, Ni2+2, and Zn2+2 clusters in human arginase I, and it can be stabilized by a third inhibitory Zn2+ ion coordinated by H141. Since ABH binds as an analogue of the tetrahedral intermediate and its flanking transition states in catalysis, this implies that the various metallosubstituted enzymes are capable of some level of catalysis with an actual substrate. Accordingly, we establish the following trend for turnover number (kcat) and catalytic efficiency (kcat/KM): Mn2+ > Ni2+ ≈ Co2+ ≫ Zn2+. Therefore, Mn2+ is required for optimal catalysis by human arginase I.
Human arginase I is a binuclear manganese metalloenzyme that catalyzes the hydrolysis of L-arginine (L-Arg) to form products L-ornithine (L-Orn) and urea during the final cytosolic step of the urea cycle.1–3 As demonstrated for rat arginase I, a complete Mn2+2 cluster is required for maximal catalytic activity in vivo.4,5 The same holds true for human arginase I; however, this enzyme can be reconstituted with a Co2+2 cluster that is said to yield 10-fold higher catalytic activity in vitro.6 Early studies demonstrated that rat arginase I treated with metal ion chelators can be reconstituted with Co2+, Ni2+, and Cd2+ to yield enzymes with 25–35% activity compared with Mn2+2-rat arginase I.7 However, since chelators extract only one metal ion from the binuclear manganese cluster of rat arginase I,8 the residual activity observed in early metal reconstitution experiments with this enzyme7 most likely derived from mixed metal clusters. Significantly, metal binding properties and catalytic metal requirements can vary for arginases from different species. Indeed, certain bacterial arginases utilize binuclear metal clusters other than Mn2+2, such as the Co2+2 preference observed for arginase from Helicobacter pylori,9 or the Ni2+2 preference for arginase from Bacillus anthracis. 10
Metal coordination geometry in the active site of human arginase I is distorted octahedral for both metal ions in the native, unliganded enzyme,11 and in the enzyme complex with the tetrahedral transition state analogue inhibitor 2(S)-amino-6-boronohexanoic acid (ABH),12 which binds as the tetrahedral boronate anion (Figure 1).13 Octahedral metal coordination polyhedra may account for the relative ease with which the Mn2+2 cluster is substituted with alternative metal ions that readily adopt octahedral metal coordination, such as Co2+ and Ni2+. Our recent crystal structure determinations of Co2+2-human arginase I and its complexes with ABH and the product L-Orn reveal identical metal coordination geometries compared to those observed in the corresponding complexes with Mn2+2-human arginase I.14 Presuming identical metal coordination geometries, then, activity differences among Mn2+2-, Co2+2-, and Ni2+2-substituted arginases may result in part from the differing abilities of each particular metal ion cluster in activating the catalytic nucleophile, believed to be a metal-bridging hydroxide ion,1–3 and in stabilizing the tetrahedral intermediate and its flanking transition states in catalysis. For example, the 10-fold activity enhancement observed for L-Arg hydrolysis by Co2+2-human arginase I in comparison with Mn2+2-human arginase I is believed to result from a pKa for the Co2+-bridging water molecule that is approximately one unit lower than that of the Mn2+-bound water molecule, thereby resulting in a higher concentration of the nucleophilic metal-bridging hydroxide ion at pH 8.5.6
Figure 1.
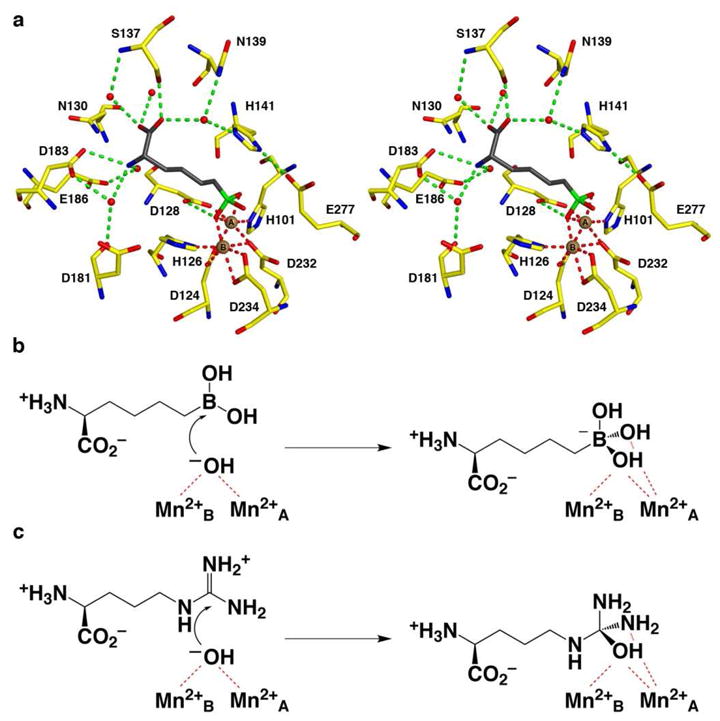
(a) Binding of the reactive substrate analogue inhibitor ABH in the active site of Mn2+2-human arginase I (PDB entry 2AEB), which mimics the binding of the tetrahedral intermediate and its flanking transition states in catalysis. Atoms are color-coded as follows: C = yellow, N = blue, O = red, B = green, Mn2+ = brown spheres, solvent = red spheres. (b) The nucleophilic metal-bridging hydroxide ion of Mn2+2-human arginase I attacks the sp2-hybridized boron atom in the trigonal planar boronic acid moiety of ABH to form a stable tetrahedral boronate anion. (c) The nucleophilic metal-bridging hydroxide ion of Mn2+2- human arginase I attacks the sp2-hybridized carbon atom in the trigonal planar guanidinium group of substrate L-arginine to form a metastable tetrahedral intermediate during catalysis.
It is currently unknown as to whether arginase reconstituted with a Zn2+2 cluster exhibits catalytic activity, despite the fact that Zn2+ is ubiquitous in hydrolytic metalloenzymes, e.g., such as the prototypical metalloproteases carboxypeptidase A and thermolysin.15,16 Although octahedral Zn2+ coordination geometry in proteins is occasionally observed,17,18 tetrahedral coordination geometry is more typically preferred because the Zn2+ ion is not subject to ligand field stabilization energy.19 Accordingly, the octahedral metal binding sites of arginase may not be ideal for Zn2+ binding. Moreover, Zn2+ binding to native arginase enzymes from various species is inhibitory.20–23 This inhibition could result from Zn2+ association with an adventitious tetrahedral binding site formed by catalytic residues and metal-bound solvent molecules, as observed for the inhibition of carboxypeptidase A and thermolysin by excess Zn2+.24,25
To understand the structural basis for activity differences in human arginase I containing Mn2+2, Co2+2, Ni2+2, and Zn2+2 clusters, and to understand the structural basis for the inhibitory activity of Zn2+ against Mn2+2-human arginase I, we now report X-ray crystal structures of human arginase I substituted with Ni2+ or Zn2+ in the presence and absence of the inhibitor ABH. Additionally, we show that the inhibitory effect of Zn2+ against Mn2+2-human arginase I results from the formation of a novel trinuclear Mn2+2Zn2+ cluster, in which the inhibitory Zn2+ ion is liganded by active site residue H141 in alternate binding modes depending on whether or not ABH is also bound.
EXPERIMENTAL PROCEDURES
Materials
Dipicolinic acid, manganese(II) chloride tetrahydrate (≥99%), cobalt chloride hexahydrate (>99%) and nickel chloride hexahydrate were purchased from Sigma. Tris(2-carboxyethyl)phosphine hydrochloride (98%, TCEP) was purchased from Alfa Aesar. 2(S)-Amino-6-boronohexanoic acid (ABH) ammonium salt was purchased from Enzo Life Sciences (Farmingdale, NY). Prepared solutions of 50% (w/v) Jeffamine ED-2001, 100 mM HEPES (pH 7.0) and 30% (v/v) Jeffamine ED-2001, 100 mM HEPES (pH 7.0) were purchased from Hampton Research (Aliso Viejo, CA). Ethylenediaminetetraacetic acid tetrasodium salt (EDTA, 99.5%), zinc chloride (99.1%), and all other chemicals were purchased from Fisher Scientific.
Expression and Purification of Metallosubstituted Human Arginase I
Human arginase I was expressed in Escherichia coli (E. coli) strain BL21-Gold (DE3) cells (Agilent Technologies) by transformation of the pBS(KS) plasmid.13 A 2 mL starter culture containing Luria-Bertani media and ampicillin at a final concentration of 100 μg/mL was grown for 8 hr in a shaker at 37 °C and 250 rpm. A second starter culture (250 mL) containing a minimal media recipe [minimal media M9 salts (Sigma), casamino acids, 18 mM D-(+)-glucose (Acros, ACS reagent), 1.8 mM MgSO4, 89 μM CaCl2, and 100 μg/mL ampicillin] was inoculated with 500 μL of the 2 mL starter culture and incubated at 37 °C and 220 rpm for 14 hours. Subsequently, 500 mL cultures in the minimal media recipe were inoculated with 25 mL of the second starter culture and incubated at 37 °C and 220 rpm until the OD600 reached 0.8 – 1.0. The incubating temperature was then lowered to 25 °C, and each culture was supplemented with 500 μL of 1M IPTG and 500 μL of a 0.1 M solution of either MnCl2, CoCl2, NiCl2, or ZnCl2. Cultures were incubated at 25 °C and 220 rpm for 12 hours. These cells were pelleted by centrifugation and stored at −80 °C.
Metal-substituted arginases were purified as described for the native enzyme13 with slight modification. Briefly, the pelleted cells were thawed to room temperature and resuspended in a lysis buffer [10 mM Tris (pH 8.0), 150 mM NaCl, 1 mM EDTA, 5 mM of either MnCl2, CoCl2, NiCl2, or ZnCl2]. Cells were sonicated on ice using a Sonifier 450 (Branson) [33% duty cycle with maximum tip setting (#7)] and sonications were performed in 3 cycles (one cycle = 3 min of sonication followed by a 2 min rest period). The cell lysate was centrifuged at 15,000 rpm for 45 min, after which the supernatant was recovered. In a round-bottom flask, the supernatant was heated and stirred gently at 50 °C for 20 min, then cooled on ice for 10 min, and finally centrifuged at 15,000 rpm for 5 min. The supernatant was then heated again in a second round-bottom flask at 60 °C for 20 min, cooled on ice for 10 min, centrifuged at 15,000 rpm for 15 min, and then dialyzed against 20 mM Tris (pH 7.5), 2 mM TCEP, 1 mM EDTA; the preparation was subsequently equilibrated with 2 mM of either MnCl2, CoCl2, NiCl2, or ZnCl2. A carboxymethyl cellulose (CM-52) resin (Whatman) was sequentially washed with (1) 500 mL of deionized water, (2) 500 mL of a metal chelation buffer [100 mM Tris (pH 7.5), 150 mM NaCl, 10 mM EDTA], and (3) 500 mL of deionized water over a sintered funnel (frit pore diameter = 10 – 20 μM) under vacuum. The washed resin, which was almost dry, was suspended in 80 mL of the CM-52 mobile phase A [20 mM Tris (pH 7.5), 2 mM TCEP, 1 mM EDTA, and 2 mM of either MnCl2, CoCl2, NiCl2, or ZnCl2]. A column was packed with CM-52 resin (3 cm I.D. × 7 cm resin bed height) and equilibrated with mobile phase A. The dialysate was loaded onto the CM-52 column and was subject to step-elution with 100% mobilie phase B [20 mM Tris (pH 7.5), 2 mM TCEP, 300 mM NaCl, 1 mM EDTA, and 2 mM of either MnCl2, CoCl2, NiCl2, or ZnCl2]. The eluted enzyme was >98% pure as judged by SDS-PAGE analysis. Finally, purified enzyme was exchanged into a final buffer of 50 mM bicine (pH 8.5) and 100 μM of either MnCl2, CoCl2, NiCl2, or ZnCl2 using a PD-10 desalting column (GE Healthcare).
Activity Assays
Arginase activity was monitored spectrophotometrically based on the formation of urea, using the colorimetric assay developed by Archibald.26 All measurements were performed in quadruplicate at 22 °C. A stock solution of 100 mM L-Arg in 50 mM HEPPS (pH 8.5), 100 μM MCl2 (M = Mn2+, Co2+, or Ni2+) was prepared. From this stock solution, 200 μL samples were prepared with a range of L-Arg concentrations (0.20 – 18.0 mM for Mn2+2-human arginase I, 0.06 – 9.1 mM for Co2+2-human arginase I, and 0.20 – 18.0 mM for Ni2+2- human arginase I) in 50 mM HEPPS (pH 8.5). Each reaction was initiated by adding 20 μL of 0.073 μM trimeric Mn2+2-human arginase I (ε280 = 22,990 M−1 cm−1 for the monomer) and terminated after 90 seconds (time optimum) using 30 μL of a 3:1 (v/v) concentrated acid/chromophore dye solution [H2SO4:H3PO4:H2O (1:3:1 v/v/v)/245 mM α-isonitrosopropiophenone in ethanol]. Samples were heated to 90 °C for 1 hour in a thermocycler to ensure complete reaction of urea with the dye. For urea quantification, 150 μL of each sample was transferred into a flatbottom 96-well plate (Fisher) and spectrophotometric absorbance readings were recorded at λ = 550 nm with an Envision plate reader (Perkin Elmer) immediately after solutions were heated. A urea standard curve was made every time the plate reader was used and the concentrations of the urea standards ranged 0 – 2.0 mM.
To confirm catalytic activity measurements for Mn2+2-human arginase I and Co2+2-human arginase I, arginase activity was also measured by a sensitive carbon-14 radioactive assay as described by Rüegg and Russell.27 All measurements were performed in triplicate at 22 °C. Briefly, a typical reaction mixture contained 40 μL of assay buffer [50 mM HEPPS (pH 8.5), 0.02275 μM L-[guanidino-14C]arginine, and 100 μM of either MnCl2 or CoCl2] in each reaction tube. The L-[guanidino-14C]arginine (specific activity 55 mCi mmol−1) was purchased from American Radiolabeled Chemicals. Typically, 5 μL of increasing concentrations of unlabeled LArg were added to each 40 μL reaction mixture such that final L-Arg concentrations ranged as follows: 0.070 – 40 mM for Mn2+2-human arginase I and 0.070 – 30 mM for Co2+2-human arginase I. Reactions were initiated by the addition of 5 μL of assay buffer containing the metal-reconstituted arginase (0.081 μM Mn2+2-human arginase I or 0.174 μM Co2+2-human arginase I). After 90 seconds, reactions were terminated by the addition of 200 μL of stop solution [0.25 M acetic acid (pH 4.5), 7 M urea, 0.010 M unlabeled L-Arg], immediately followed by the addition of 200 μL of Dowex solution [1:1 (v/v) slurry of Dowex 50 W-X8 (Acros) in deionized water], and vortexed. Reaction samples were gently mixed for 10 min and centrifuged at 2,000 rpm for 2 min. Supernatant from each reaction (200 μL) was added to 3.0 mL of Ecoscint solution (National Diagnostics) for liquid scintillation counting, which was performed using a Beckman model LS5000E counter.
Inductively Coupled Plasma-Mass Spectrometry (ICP-MS) Analysis
Purified samples of Mn2+2-, Co2+2-, and Ni2+2-human arginase I were subject to ICP-MS for the quantitative analysis of metal ion content and stoichiometry at the University of Georgia, Center for Applied Isotope Studies, Athens, GA. Each sample, which was initially solubilized in 50 mM bicine (pH 8.5) and 100 μM of either MnCl2, CoCl2, or NiCl2, was subject to a buffer exchange to minimize the free metal ion concentration in solution. A separate PD-10 column (GE Healthcare) was used for each sample. These columns were first washed with (1) 25 mL of deionized water, (2) 25 mL of a metal chelation solution [10 mM bicine (pH 8.5), 5 mM EDTA], and then pre-equilibrated with 25 mL of 10 mM bicine (pH 8.5) prior to running each sample. Samples were dialyzed using dialysis cassettes (Pierce, 10 kDa molecular weight cut-off) against a total of 6 L of 10 mM bicine (pH 8.5) at 4 °C, with three buffer exchanges. All dialysis glassware was pre-washed with the same metal chelation solution followed by rinsing with deionized water. Each sample was concentrated using an Amicon Ultra-4 (10 kDa molecular weight cut-off) centrifugal concentrator; the final volume was adjusted to 1.0 mL and the protein concentration was determined by amino acid analysis at the W.M. Keck Biotechnology Resource Laboratory, Yale University School of Medicine, New Haven, CT.
Preparation of Metallosubstituted Crystalline Human Arginase I
While the preparation of metal-substituted arginases for activity assays in solution is described above, we opted to prepare metal-substituted arginases in the crystal using the more rapid and efficient protocol that we previously developed for our X-ray crystallographic studies of Co2+2-human arginase I.14 Briefly, human arginase I was expressed in Escherichia coli, purified, and crystallized as previously described.11,13 A 3 μL drop of enzyme solution [14.7 mg/mL Mn2+2-human arginase I (ε280 = 22,900 M−1 cm−1 for the monomer), 1.4 mM thymine, 50 mM bicine (pH 8.5), 100 μM MnCl2] and a 3 μL drop of precipitant solution [30% (v/v) Jeffamine ED-2001, 100 mM HEPES (pH 7.0)] were combined on a siliconized cover slide and equilibrated against 500 μL of precipitant solution. Crystals of the unliganded enzyme grew within 5 days at room temperature. These crystals were soaked for 14 days in a buffer solution of 15 mM EDTA, 15 mM dipicolinic acid, 100 mM HEPES (pH 7.0), 30% (v/v) Jeffamine ED-2001, and solutions were replenished daily. Both chelators (EDTA and dipicolinic acid) were required to ensure complete removal of Mn2+ ions, as we previously demonstrated in the preparation of metal-free human arginase I.14 Crystals of the metal-free enzyme were reconstituted with a Ni2+2 cluster at pH 7.0 by soaking in a buffer solution containing 5 mM NiCl2, 100 mM HEPES (pH 7.0), 30% (v/v) Jeffamine ED-2001 for 26 hours. A crystal of the Ni2+2-human arginase I complex with ABH was prepared by soaking a crystal of the metal-free enzyme in 5 mM NiCl2, 5 mM ABH, 100 mM HEPES (pH 7.0), and 32% (w/v) Jeffamine ED-2001 for 26 hours. A crystal of the Zn2+5-human arginase I-ABH complex was prepared by soaking a crystal of the metal-free enzyme in 5 mM ZnCl2, 5 mM ABH, 100 mM HEPES (pH 7.0), and 30% (v/v) Jeffamine ED-2001 for 18 hours. A crystal of the Mn2+2Zn2+-human arginase I complex with ABH was prepared by soaking a crystal of Mn2+2-human arginase I in 5 mM ZnCl2, 5 mM ABH, 100 mM HEPES (pH 7.0), and 32% (w/v) Jeffamine ED-2001 for 26 hours. The unliganded Mn2+2Zn2+-human arginase I complex was prepared by soaking a Mn2+2-HAI crystal in 5 mM ZnCl2, 50 mM boric acid, 100 mM HEPES (pH 7.0), and 30% (v/v) Jeffamine ED-2001 for 26 hours. All crystals were flash-cooled in liquid nitrogen with their respective mother liquor solutions serving as cryoprotectants.
X-ray Crystal Structure Determinations
X-ray diffraction data from single crystals of Ni2+2-HAI, the Ni2+2-HAI-ABH complex, the Zn2+2,Zn2+CD-HAI-ABH complex, the Mn2+2,Zn2+CD-HAI-ABH complex, the inhibitor-free Mn2+2,Zn2+CE-HAI complex, and the Mn2+2-HAI-ABH complex were collected on beamline X29 (λ = 1.075 Å) of the National Synchrotron Light Source at Brookhaven National Laboratory (Upton, NY). Diffraction data were indexed, integrated, and scaled using the HKL-2000 suite of programs.28 All crystals were hemihedrally twinned, as initially reported for the Mn2+2-human arginase I-ABH complex,13 and belonged to apparent space group P3 with similar unit cell dimensions (Table 1). Structures were determined by molecular replacement using the program Phaser as implemented in the CCP4 suite of programs.29,30 The search model used for rotation and translation function calculations was the A-chain structure of Mn2+2-HAI (PDB entry 2PHA11) less metal ions and solvent atoms (for structures with intact binuclear manganese clusters, only solvent atoms were omitted).
Table 1.
Data Collection and Refinement Statistics.
| metallosubstituted human arginase I | Ni2+2 | Ni2+2, ABH complex | Zn2+5, ABH complex | Mn2+2Zn2+ | Mn2+2Zn2+, ABH complex |
|---|---|---|---|---|---|
| Data Collection | |||||
| resolution limits (Å) | 50.0 – 1.70 | 50.0 – 1.48 | 50.0 – 2.20 | 50.0 – 1.90 | 50.0 – 1.53 |
| total/unique reflections measured | 795439/70032 | 498285/106524 | 147051/32953 | 579461/51066 | 272261/97388 |
| space group symmetry unit cell dimensions | P3 | P3 | P3 | P3 | P3 |
| a, b, c (Å) | 90.35, 90.35, 69.81 | 90.71, 90.71, 69.66 | 91.12, 91.12, 69.77 | 90.83,90.83, 70.07 | 91.08, 91.08, 69.90 |
| α, β,γ (deg) | 90, 90, 120 | 90, 90, 120 | 90, 90, 120 | 90, 90, 120 | 90, 90, 120 |
| R mergea,b | 0.093 (0.432) | 0.067 (0.354) | 0.096 (0.698) | 0.101 (0.978) | 0.069 (0.341) |
| I/σ (I )a | 26.53 (7.07) | 21.59 (4.36) | 14.20 (2.19) | 26.39 (2.67) | 15.50 (3.21) |
| completeness (%)a | 99.9 (100) | 99.8 (100) | 99.9 (100) | 99.9 (100) | 99.5 (98.2) |
| Refinement | |||||
| reflections used in refinement/test set | 69431/3213 | 105002/5116 | 29796/1457 | 49137/2368 | 94853/4626 |
| twinning fraction | 0.50 | 0.45 | 0.21 | 0.50 | 0.47 |
| R twinc | 0.1542 | 0.1531 | 0.1723 | 0.1540 | 0.1598 |
| R twin/freed | 0.1889 | 0.1925 | 0.2328 | 0.2211 | 0.2153 |
| solvent moleculese | 163 | 198 | 106 | 79 | 175 |
| ligand moleculese | 0 | 2 | 2 | 0 | 2 |
| metal ionse | 4 | 4 | 9 | 6 | 6 |
| Root Mean Square Deviation | |||||
| bonds (Å) | 0.008 | 0.007 | 0.008 | 0.008 | 0.008 |
| angles (deg) | 1.5 | 1.5 | 1.6 | 1.5 | 1.5 |
| Average B-factors ( Å 2) | |||||
| main chain | 26 | 22 | 46 | 38 | 24 |
| side chain | 28 | 24 | 48 | 39 | 26 |
| solvent | 27 | 23 | 38 | 31 | 24 |
| ligand | -- | 23 | 40 | -- | 21 |
| metal ionsf | 20 (4) | 17 (4) | 37 (9) | 30 (6) | 19 (6) |
| Ramachandran Plot (%) | |||||
| allowed | 85.8 | 88.8 | 85.8 | 85.4 | 88.6 |
| additionally allowed | 13.8 | 10.8 | 13.4 | 13.8 | 11.0 |
| generously allowed | 0.2 | 0.0 | 0.6 | 0.6 | 0.0 |
| disallowed (Q65) | 0.2 | 0.4 | 0.2 | 0.2 | 0.4 |
| PDB accession code | 4GSM | 4GSV | 4GSZ | 4GWC | 4GWD |
Values in parenthesis are for the highest resolution shell.
Rmerge = Σ |I − 〈I〉|/Σ I, where I is the observed intensity, 〈I〉 is the average intensity calculated from replicate data.
Rtwin = Σ ||Fo|−|Fc||/Σ |Fo| for reflections contained in the working set.
Rtwin/free = Σ ||Fo|−|Fc||/Σ |Fo| for 5% of reflections contained in the test set held aside during refinement. |Fo| and |Fc|are the observed and calculated structure factor amplitudes, respectively.
Per asymmetric unit cell.
Values in parentheses indicate the total number of metal ions in the unit cell for each structure from which the average B-factor was calculated.
Refinement was performed with CNS (version 1.2)31 using the hemihedral twinning operation parameters -h, -k, and l; the twinning fraction varied slightly from one structure to another (Table 1). Model building was performed using Coot (version 0.6.1).32 Atomic coordinates of the bound inhibitor ABH and water molecules were included in the later stages of refinement. For the Ni2+2-human arginase I-ABH, Zn2+5-human arginase I-ABH, and the Mn2+2Zn2+-human arginase I-ABH complexes, gradient omit maps showed ABH bound in the active site of each monomer in the asymmetric unit, and all ABH atoms were refined with full occupancy. In addition, average B-factors for ABH in these complexes were similar to the main chain average B-factors for the entire protein (Table 1).
In all the crystal structures reported herein, metal ion sites A and B were refined with full occupancy. For the Zn2+5-human arginase I-ABH complex, metal ion sites CD, D, and E were refined with occupancy values of 0.55, 0.35, and 0.35, respectively (reported occupancy values are averaged over all monomers in the asymmetric unit). For the Mn2+2Zn2+-human arginase I structure, metal ion site CE was refined with an occupancy of 0.48. For the Mn2+2Zn2+-human arginase I-ABH complex, metal ion site CD was refined to full occupancy. The average B-factors for metal ions were slightly lower than the average B-factors for main chain atoms in each structure.
Disordered segments at the N-terminus (residues M1 – S5) and at the C-terminus (residues P320 – K322) are excluded from all final models. For unliganded Ni2+2-human arginase I, unliganded Mn2+2Zn2+-human arginase I, and the Zn2+5-human arginase I-ABH complex, Q65 in monomer A of the asymmetric unit adopts a disallowed conformation based on the Ramachandran plot (data not shown). For the Ni2+2-human arginase I-ABH complex and the Mn2+2Zn2+-human arginase I-ABH complex, Q65 adopts a disallowed conformation in both monomers A and B of the asymmetric unit. Since the main chain atoms of Q65 are characterized by clear and unambiguous electron density, this is not likely to be an artifact. Ramachandran statistics were calculated with the program PROCHECK,33 and average B-factors were calculated with the program MOLEMAN.34 Data collection and refinement statistics for all structure determinations are recorded in Table 1.
RESULTS
Catalytic Activity Measurements
Using the colorimetric assay developed by Beale and Croft,35 Stone and colleagues6 report that Co2+2-human arginase I at 37 °C exhibits kcat = 240 ± 14 s−1, KM = 0.19 ± 0.04 mM, and kcat/KM = 1,270 ± 330 mM−1 s−1 at pH 7.4; notably, the catalytic efficiency (kcat/KM) is said to be 10-fold higher than that measured for Mn2+2-human arginase I (kcat = 300 ± 12 s−1, KM = 2.33 ± 0.26 mM, and kcat/KM = 129 ± 20 mM−1 s−1 at pH 7.4). However, we obtain slightly different results using the colorimetric assay developed by Archibald26 and the radioactive L-[guanidino-14C]arginine assay of Rüegg and Russell27 (Table 2). A possible reason for these differences is the two different oxime reagents used for urea quantification in the colorimetric assays. The assay pH may also contribute to the observed activity differences – Stone and colleagues6 report measurements at pH 7.4, whereas our measurements are made at pH 8.5, close to the pH optimum for catalytic activity. We were not able to prepare Zn2+2-human arginase I due to protein precipitation. For each soluble metallosubstituted arginase, ICP-MS measurements confirm the incorporation of the desired metal ions without contamination by other adventitious metal ions (Table 3). The trend in turnover number (kcat) and catalytic efficiency (kcat/KM) for metallosubstituted human arginase I is as follows: Mn2+ > Ni2+ ≈ Co2+ ≫ Zn2+ (Zn2+ is always inhibitory).
Table 2.
Catalytic Activities of Metallosubstituted Enzymes.
| enzyme | KM (mM) | kcat (s−1) | kcat/KM × 104 (M−1s−1) |
|---|---|---|---|
| Mn2+2-human arginase Ia | 3±1 | 340±160 | 11±2 |
| Co2+2-human arginase Ia | 1.7±0.3 | 90±60 | 6±4 |
| Ni2+2-human arginase Ia | 4±1 | 110±70 | 3±1 |
| Mn2+2-human arginase Ib | 8±3 | 410±120 | 5.4±0.6 |
| Co2+2-human arginase Ib | 3.7±0.3 | 100±10 | 2.6±0.3 |
Colorimetric assay, measurements in quadruplicate.
Radioactive L-[guanidino-14C]arginine assay, measurements in triplicate.
Table 3.
ICP-MS Analysis.
| enzyme | molar ratio of metal : proteina,b | |||||
|---|---|---|---|---|---|---|
|
| ||||||
| Fe | Mn | Co | Ni | Cu | Zn | |
| Mn2+2-human arginase I | 0.01 | 1.76 | 0.00 | 0.00 | 0.00 | 0.02 |
| Co2+2-human arginase I | 0.08 | 0.04 | 1.82 | 0.00 | 0.00 | 0.03 |
| Ni2+2-human arginase I | 0.02 | 0.03 | 0.00 | 1.47 | 0.00 | 0.04 |
The metal ion content of metallosubstituted human arginase I was determined by ICP-MS and the protein concentration was determined by amino acid analysis.
The ratio of metal:protein was determined by dividing the metal content (determined by ICP-MS) by the protein concentration (i.e., the concentration of arginase monomer).
Ni2+2-Human Arginase I
The overall structure of unliganded Ni2+2-human arginase I is essentially identical to that of Mn2+2-human arginase I (PDB entry 2PHA11), with an r.m.s. deviation of 0.24 Å for 313 Cα atoms. The coordination numbers and geometries of Ni2+A and Ni2+B are essentially identical to those observed for the corresponding metal ions in unliganded Mn2+2-human arginase I and Co2+2-human arginase I.11,14 However, the average metal-metal separation in Ni2+2-human arginase I is 3.2 Å, which is slightly shorter than that observed in Mn2+2- and Co2+2-human arginase I (3.4 Å each). Furthermore, the average metal coordination distances are generally shorter in Ni2+2-human arginase I: the average Ni2+-ligand separation is 2.2 Å in Ni2+2-human arginase I at pH 7.0, whereas the average Mn2+-ligand separation is 2.4 Å in Mn2+2-human arginase I at pH 7.5 (PDB entry 2PHA11). For comparison, the average Co2+-ligand separation in Co2+2-human arginase I is 2.2 Å at pH 7.0 and pH 8.5 (PDB entries 3TH7 and 3THE, respectively).14 It is interesting to note that the trend in average metal-ligand separations is consistent with the Irving-Williams series, which suggests generally weaker metal-ligand coordination interactions for Mn2+ compared with Co2+ or Ni2+.
Ni2+2-Human Arginase I-ABH Complex
The overall fold of the Ni2+2-human arginase I-ABH complex is essentially identical to that of the Mn2+2-human arginase I-ABH complex (PDB entry 2AEB13), with an r.m.s. deviation of 0.21 Å for 313 Cα atoms. The average metal-metal separation in Ni2+2-human arginase I is 3.2 Å, which is close to that measured in the Mn2+2-human arginase I-ABH complex (3.3 Å) and equal to that measured in the Co2+2-human arginase I-ABH complex (3.2 Å) (PDB entries 2AEB and 3THH, respectively).13,14 Interestingly, ABH binding to Ni2+2-human arginase I does not change the average metal-metal separation, but ABH binding to Mn2+2- and Co2+2-human arginase I slightly decreases the average metal-metal separation.
Upon binding to the enzyme, the boronic acid moiety of ABH undergoes nucleophilic attack by the metal-bridging hydroxide ion of the native Ni2+2-substituted enzyme to yield a tetrahedral boronate anion that mimics the tetrahedral intermediate and its flanking transition states in catalysis. That the metal-bridging hydroxide ion in Ni2+2-human arginase I is sufficiently nucleophilic to react with ABH suggests that it is sufficiently nucleophilic to react with the actual substrate L-arginine, thereby accounting for the significant catalytic activity measured for Ni2+2-human arginase I (Table 2).6 A simulated annealing omit map showing the bound inhibitor is found in Figure 2.
Figure 2.
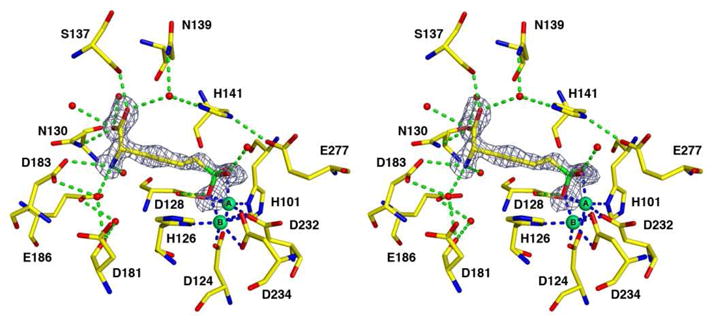
Omit map of the inhibitor ABH bound in the active site of Ni2+2-human arginase I, contoured at 3.4σ. Atoms are color-coded as follows: C = yellow, N = blue, O = red, B = green, Ni2+ = light green spheres, solvent = red spheres. Metal coordination and hydrogen bond interactions are represented by blue and green dashed lines, respectively.
In the enzyme-inhibitor complex, boronate oxygen atom O1 coordinates to the Ni2+A and Ni2+B ions with an average separation of 2.3 Å; this atom corresponds to the metal-bridging hydroxide ion observed in the unliganded enzyme. Boronate oxygen atom O2 coordinates to Ni2+A with an average interatomic separation of 2.4 Å. Boronate oxygen O3 hydrogen bonds with a water molecule that in turn is hydrogen bonded with the side chain of T246. Interestingly, the average metal-ligand separations are nearly equal in the Ni2+2-, Co2+2-, and Mn2+2-human arginase I-ABH complexes (2.3 Å, 2.2 Å, and 2.2 Å, respectively), in contrast with the trend observed for the unliganded metal-substituted enzymes. Finally, the α-carboxylate and α-amino groups of ABH make the same array of hydrogen bond interactions observed in the Mn2+2- and Co2+2-human arginase I-ABH complexes.13,14
Zn2+5-Human Arginase I-ABH Complex
When crystalline metal-free human arginase I is reconstituted with Zn2+ and ABH, a total of five Zn2+ ions are observed to bind, three of which are located in the active site. The overall structure of the Zn2+5-human arginase I-ABH complex at pH 7.0 is very similar to that of the Mn2+2-human arginase I-ABH complex (PDB entry 2AEB13), with an r.m.s. deviation of 0.29 Å for 313 Cα atoms. In the active site, metal ion binding sites A and B have been successfully reconstituted with Zn2+ ions, and ABH binds as the tetrahedral boronate anion in identical fashion to that observed in the Mn2+2-, Co2+2-, and Ni2+2-human arginase I-ABH complexes.13,14 Strikingly, a third Zn2+ ion binds in the active site and is coordinated by the Nd atom of the H141 imidazole group, the Oe1 atom of E277, boronate hydroxyl groups O2 and O3 of ABH, and a water molecule; the zinc-bound water molecule donates a hydrogen bond to the Oε2 atom of E277. However, the geometry of the latter interaction is poor, so it is likely to be a weak hydrogen bond at best. This new metal binding site is designated as site CD. Average metal-metal separations in the trinuclear zinc cluster are as follows: Zn2+A-Zn2+B, 3.4 Å; Zn2+B-Zn2+CD, 5.2 Å; Zn2+A-Zn2+CD, 4.7 Å. A simulated annealing omit map showing the bound inhibitor and active site Zn2+ ions is found in Figure 3a.
Figure 3.
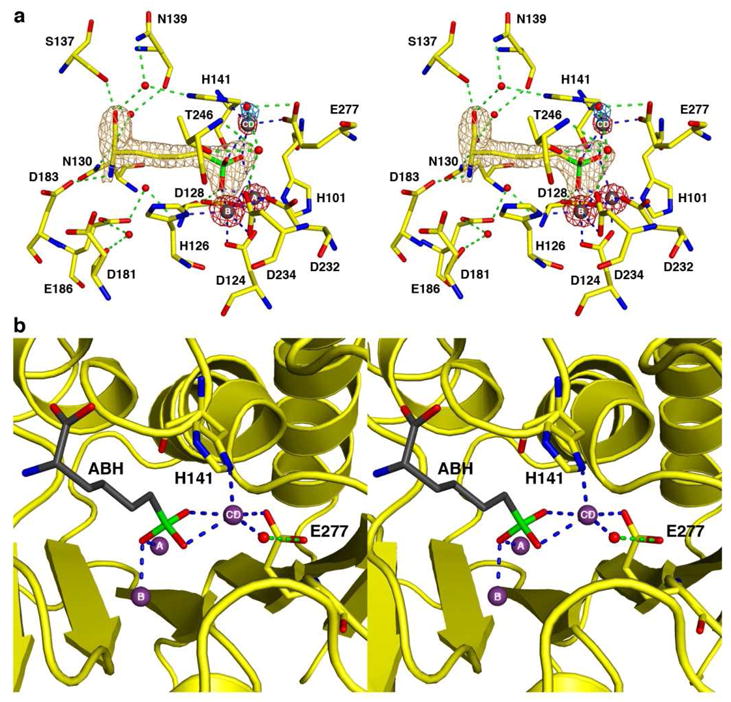
(a) Simulated annealing omit maps of the inhibitor ABH (tan, contoured at 5.5s), three Zn2+ ions (red, contoured at 14σ), and the Zn2+CD-bound water molecule (blue, contoured at 6.5s) in the active site of the Zn2+5-human arginase I-ABH complex. Atoms are color-coded as follows: C = yellow, N = blue, O = red, B = green, Zn2+ = dark gray spheres, solvent = red spheres. Metal coordination and hydrogen bond interactions are represented by blue and green dashed lines, respectively. (b) Another orientation of the complex shown in (a) highlights the square pyramidal coordination geometry of the Zn2+CD ion.
That ABH binds to Zn2+-substituted human arginase I as a tetrahedral boronate anion indicates that a possible metal-bridging hydroxide ion in native Zn2+2-human arginase I is sufficiently nucleophilic to attack the electrophilic boronic acid moiety of ABH, suggesting that nucleophilic attack at the guanidinium group of the actual substrate L-arginine could be possible. However, the binding of the Zn2+CD ion further stabilizes the tetrahedral boronate anion, so it is possible that the binding of this tetrahedral species is mainly observed due to the additional metal coordination interactions with Zn2+CD, assuming that free Zn2+ in solution does not affect the equilibrium between the boronic acid and boronate anion forms of ABH. We were unable to prepare protein or protein crystals containing solely a Zn2+2 cluster in the active site, so we are unable to draw any conclusions regarding the possible catalytic activity of Zn2+2-human arginase I. Presumably, however, catalytic activity would be minimal due to facile inhibition by excess Zn2+.
The Zn2+CD ion has a coordination number of 5 and adopts approximately square pyramidal coordination geometry (Figure 3b), with four oxygen atoms in equatorial positions and a nitrogen atom in the axial position. Notably, while Zn2+C coordination by E277 is characterized by an average separation of 2.2 Å, indicative of a strong electrostatic interaction, the Zn2+CD ion is located perpendicular to the plane of the E277 carboxylate, which constitutes poor geometry for a carboxylate-metal ion coordination interaction.36
Parenthetically, we note that two additional Zn2+ ions are observed in this structure. First, the side chain of H312 near the C-terminus coordinates to a Zn2+ ion at the contact surface between two monomers, approximately 15 Å away from the active site (data not shown). Metal ion binding causes some minor conformational changes in the polypeptide segment flanking H312. This Zn2+ ion, designated Zn2+D, is refined with an occupancy of 0.35. Finally, an additional metal ion, designated Zn2+E, is found on the protein surface coordinated by side chains of H115, D117, and a water molecule. The Zn2+E ion is refined with an occupancy of 0.35 in chains A and B (data not shown).
Mn2+2Zn2+-Human Arginase I
The overall structure of unliganded Mn2+2Zn2+-human arginase I is very similar to that of unliganded Mn2+2-human arginase I (PDB entry 2PHA11), with an r.m.s. deviation of 0.31 Å for 313 Cα atoms. Metal coordination interactions in the binuclear manganese cluster are similar to those observed in native unliganded Mn2+2-human arginase I and the average Mn2+A-Mn2+B separation is 3.2 Å. However, a Zn2+ ion is coordinated by the Nε atom and not the Nδ atom of the H141 imidazole group (Figure 4), so the position of this Zn2+ ion differs from that of the Zn2+CD ion described above. Accordingly, we designate this new position as metal binding site CE. The H141 Nd-H group donates a hydrogen bond to E277 with an average N---O separation of 2.9 Å. This interaction appears to be stronger than that in the Zn2+-free enzyme, in which the corresponding average separation is 3.5 Å. It is likely that metal coordination by the Nε atom of H141 increases the partial positive charge on the H141 Nδ-H group, thereby enhancing the favorable electrostatic interaction with E277.
Figure 4.
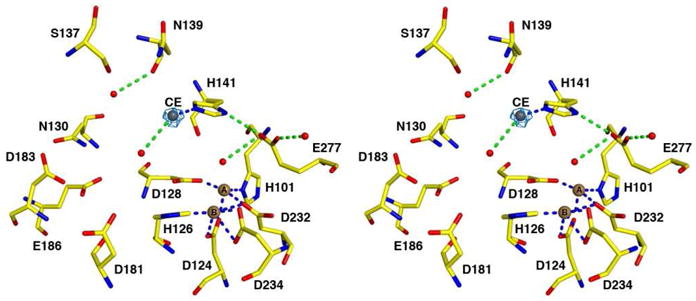
Simulated annealing omit map (blue) of the Zn2+CE ion in the active site of monomer B of Mn2+2Zn2+-human arginase I (contoured at 5.2σ), where it is coordinated by the Ne atom of the H141 imidazole group. Formation of the carboxylate-histidine-zinc interaction with E277 and H141 is the structural basis for inhibition of arginase by zinc. Atoms are color-coded as follows: C = yellow, N = blue, O = red, Mn2+ = brown spheres, Zn2+ = dark gray sphere, solvent = red spheres. Metal coordination and hydrogen bond interactions are represented by blue and green dashed lines, respectively. The solvent molecule near the Mn2+ ions is too distant to be considered an inner-sphere ligand (Mn2+A-O and Mn2+B-O separations are 2.9 Å and 3.4 Å, respectively).
The Zn2+CE ion has only two ordered ligands, H141 and a water molecule; in chain A, this water molecule receives a hydrogen bond from R21. Parenthetically, we note that this complex does not contain Zn2+D or Zn2+E ions bound to the surface sites observed in the Zn2+5-human arginase I-ABH complex. This crystal structure shows that the inhibition20-23 of arginase by Zn2+ results from the coordination of H141 to Zn2+. This is consistent with mutagenesis studies of H141 in rat liver arginase demonstrating the catalytic importance of this residue.37,38
Mn2+2Zn2+-Human Arginase I-ABH Complex
The structure of the Mn2+2Zn2+-human arginase I-ABH complex is essentially identical to that of the Mn2+2-human arginase I-ABH complex (PDB entry 2AEB13), with an r.m.s. deviation of 0.26 Å for 313 Cα atoms. This complex contains the Zn2+CD ion coordinated by the Nd atom of H141, the Oε1 atom of E277, boronate hydrogen groups O1 and O2 of ABH, and a water molecule (Figure 5a). This structure lacks the Zn2+D and Zn2+E ions as observed in the Zn2+5-human arginase I-ABH complex. Average metal-metal separations in the trinuclear metal cluster are as follows: Mn2+A-Mn2+B, 3.3 Å; Mn2+B-Zn2+CD, 4.9 Å; Mn2+A-Zn2+CD, 4.5 Å. Notably, the binding of Zn2+CD does not perturb the average Mn2+A-Mn2+B separation, which is also 3.3 Å in the Mn2+2-human arginase I-ABH complex. Metal ions Mn2+A, Mn2+B, and Zn2+CD are coordinated with identical geometries to those observed for Zn2+A, Zn2+B, and Zn2+CD in the structure of the Zn2+5-human arginase I-ABH complex. A superposition of the structure of the Mn2+2Zn2+-human arginase I-ABH complex with that of unliganded Mn2+2Zn2+-human arginase I shows the change of H141 coordination to the inhibitory Zn2+ ion, i.e., the Zn2+CE→Zn2+CD transition, triggered by the binding of ABH (Figure 5b).
Figure 5.
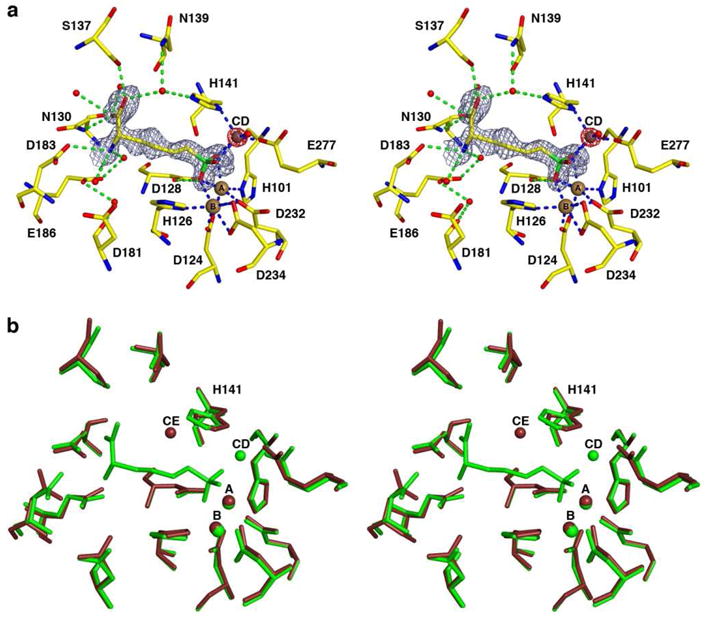
(a) Simulated annealing omit map of the inhibitor ABH (gray, contoured at 2.2σ) and the Zn2+CD ion (red, contoured at 12s) bound in the active site of the Mn2+2Zn2+-human arginase I-ABH complex. Atoms are color-coded as follows: C = yellow, N = blue, O = red, Mn2+ = brown spheres, Zn2+ = dark gray sphere, solvent = red spheres. Metal coordination and hydrogen bond interactions are represented by blue and green dashed lines, respectively. (b) Superposition of the Mn2+2Zn2+-human arginase I-ABH complex (chain A, all atoms green) and Mn2+2Zn2+-human arginase I (chain A, all atoms brown) highlights the different binding modes of the inhibitory zinc ion to H141 in the unliganded and liganded forms of the enzyme.
DISCUSSION
The X-ray crystal structure of unliganded Mn2+2Zn2+-human arginase I is the first to reveal the structural basis for inhibition by Zn2+. In this structure, a carboxylate-histidine-metal triad is fully formed that involves E277, H141, and Zn2+CE. In such a triad, the basicity of the histidine imidazole for metal ion coordination is enhanced by the carboxylate group, and the acidity of the histidine imidazole for hydrogen bonding to the carboxylate is enhanced.39 In Mn2+2Zn2+-human arginase I, the binding of Zn2+CE significantly strengthens the hydrogen bond interaction between E277 and H141, as reflected by the shorter E277-H141 separation of 2.8 Å in the fully formed triad compared with 3.5 Å in the native, unliganded enzyme.11 Formation of the triad renders H141 nonfunctional in catalysis, where it is believed to act as a proton shuttle. In accord with these results, mutation or chemical modification of H141 results in significant but usually not complete loss of catalytic activity in rat arginase I37,38,40 and in human arginase I.41,42 For example, H141N and H141A rat arginases I exhibit 11% and 0.2% residual catalytic activity, respectively, compared to the wild-type enzyme based on kcat.37,38 These activity losses are consistent with the loss of an active site general base/general acid for which less efficient rescue pathways might be available, e.g., involving buffer and/or solvent molecules. Accordingly, compromising the function of a general base/general acid histidine imidazole group by coordination of an inhibitory Zn2+ ion is particularly effective.
Metal ion sites A and B have distorted octahedral geometry in Mn2+2-human arginase I, and it is notable that this geometry is preserved regardless of what metal ions are substituted in or added to the enzyme active site (Figure 6). Curiously, however, we were unable to prepare crystals of unliganded Zn2+2-human arginase I. Our attempts to prepare crystals of this particular unliganded metalloenzyme resulted in substantial physical deterioration of the crystals during soaking experiments in buffer solutions containing 5 mM ZnCl2, making the crystals unsuitable for X-ray diffraction experiments. Even so, we were able to reconstitute the enzyme with a Zn2+3 cluster in the active site, but only in the presence of the inhibitor ABH. Possibly, the binding of Zn2+ ions to the distorted octahedral ligand fields in sites A and B is only sufficiently stabilized when ABH is bound, such that additional stabilization is derived from ABH coordination interactions with Zn2+CD.
Figure 6.
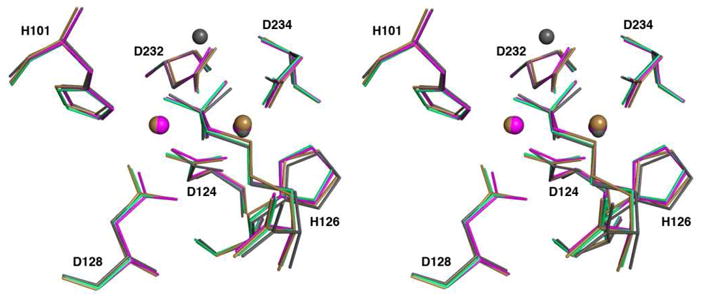
(a) Superposition of the A chains of the Mn2+2-human arginase I-ABH complex (PDB entry 2AEB, brown), the Co2+2-human arginase I-ABH complex (PDB entry 3THH, magenta), the Ni2+2-human arginase I-ABH complex (light green), and the Zn2+5-human arginase I-ABH complex (gray).
Even so, it is conceivable that Zn2+2-human arginase I could catalyze the hydrolysis of L-Arg to form products L-Orn and urea because the crystal structure of the Zn2+5-human arginase I-ABH complex reveals that ABH also binds as the tetrahedral boronate anion (Figure 3a). As mentioned previously, the binding of ABH as a boronate anion requires a chemically reactive metal-bridging hydroxide ion that would be similarly reactive against the L-Arg substrate. However, since the third Zn2+ ion binds readily in the active site as Zn2+CD or Zn2+CE, catalytic turnover with the L-Arg substrate is prevented.
Finally, it is notable that human arginase I is capable of metal ion dissociation from sites A and B when the crystalline enzyme is dialyzed for one week against the metal ion chelators EDTA and dipicolinic acid at pH 7.0.14 This feature enables structure-function studies of multiple metal-substituted arginases for the first time ever. While rat arginase I was the first arginase to yield a crystal structure,43 complete metal ion extraction could not be achieved with this enzyme: only the Mn2+A ion could be removed,8 and the protein precipitated if more aggressive dialysis conditions were utilized. Nevertheless, reconstitution of metal-depleted Mn2+B-rat arginase I with different metal ions resulted in the formation of hetereonuclear metal clusters,5 some of which appeared to be functional. For example, Mn2+Co2+ and Mn2+Cd2+ clusters in H101N rat arginase I restore some, but not all, of the catalytic function of Mn2+2-H101N rat arginase I, which itself exhibits ~50% activity compared with the wild-type enzyme.8,37 We conclude that rat and human arginase I are versatile systems for the preparation and study of homonuclear and heteronuclear metal clusters in a metalloprotein, and these systems will prove quite useful as we continue to probe the metal-dependent catalytic versatility of these enzymes.
Acknowledgments
Funding
This work was supported by National Institutes of Health Grant GM49758.
We thank the National Synchrotron Light Source at Brookhaven National Laboratory (beamline X29) for access to X-ray crystallographic data collection facilities. Additionally, we thank Dr. Mustafa Köksal for helpful discussions.
ABBREVIATIONS
- L-Arg
L-arginine
- L-Orn
L-ornithine
- ABH
2(S)-amino-6-boronohexanoic acid
- EDTA
ethylenediaminetetraacetic acid
- HEPES
N-(2-hydroxyethyl)piperazine-N′-(2-ethanesulfonic acid)
- HEPPS
N-(2-hydroxyethyl)piperazine-N′-(3-propanesulfonic acid)
- TCEP
Tris(2-carboxyethyl)phosphine hydrochloride
- BME
mercaptoethanol
- CM-52
carboxymethyl cellullose
- ICP-MS
inductively coupled plasma-mass spectrometry
- PDB
Protein Data Bank
Footnotes
Accession Codes
The atomic coordinates and structure factors of Ni2+2-human arginase I, the Ni2+2-human arginase I-ABH complex, the Zn2+5-human arginase I-ABH complex, Mn2+2Zn2+-human arginase I, and the Mn2+2Zn2+-human arginase I-ABH complex have been deposited in the Protein Data Bank (www.rcsb.org) with accession codes 4GSM, 4GSV, 4GSZ, 4GWC, and 4GWD, respectively.
References
- 1.Ash DE, Cox JD, Christianson DW. Arginase: a binuclear manganese metalloenzyme. In: Sigel A, Sigel H, editors. Manganese and Its Role in Biological Processes, Vol 37 of Metal Ions in Biological Systems. New York: M. Dekker; 1999. pp. 407–428. [PubMed] [Google Scholar]
- 2.Christianson DW, Cox JD. Catalysis by metal-activated hydroxide in zinc and manganese metalloenzymes. Annu Rev Biochem. 1999;68:33–57. doi: 10.1146/annurev.biochem.68.1.33. [DOI] [PubMed] [Google Scholar]
- 3.Christianson DW. Arginase: structure, mechanism, and physiological role in male and female sexual arousal. Acc Chem Res. 2005;38:191–201. doi: 10.1021/ar040183k. [DOI] [PubMed] [Google Scholar]
- 4.Hirsch-Kolb H, Kolb HJ, Greenberg DM. Nuclear magnetic resonance studies of manganese binding of rat liver arginase. J Biol Chem. 1971;246:395–401. [PubMed] [Google Scholar]
- 5.Reczkowski RS, Ash DE. EPR evidence for binuclear manganese(II) centers in rat liver arginase. J Am Chem Soc. 1992;114:10992–10994. [Google Scholar]
- 6.Stone EM, Glazer ES, Chantranupong L, Cherukuri P, Breece RM, Tierney DL, Curley SA, Iverson BL, Georgiou G. Replacing Mn2+ with Co2+ in human arginase I enhances cytotoxicity toward L-arginine auxotrophic cancer cell lines. ACS Chem Biol. 2010;5:333–342. doi: 10.1021/cb900267j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dahlig E, Porembska Z. Reactivation of the EDTA-treated arginase from rat and calf liver. Acta Biochim Pol. 1977;24:187–196. [PubMed] [Google Scholar]
- 8.Scolnick LR, Kanyo ZF, Cavalli RC, Ash DE, Christianson DW. Altering the binuclear manganese cluster of arginase diminishes thermostability and catalytic function. Biochemistry. 1997;36:10558–10565. doi: 10.1021/bi970800v. [DOI] [PubMed] [Google Scholar]
- 9.McGee DJ, Zabaleta J, Viator RJ, Testerman TL, Ochoa AC, Mendz GL. Purification and characterization of Helicobacter pylori arginase, RocF: unique features among the arginase superfamily. Eur J Biochem. 2004;271:1952–1962. doi: 10.1111/j.1432-1033.2004.04105.x. [DOI] [PubMed] [Google Scholar]
- 10.Viator RJ, Rest RF, Hildebrandt E, McGee DJ. Characterization of Bacillus anthracis arginase: effects of pH, temperature, and cell viability on metal preference. BMC Biochemistry. 2008;9:15. doi: 10.1186/1471-2091-9-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Di Costanzo L, Pique ME, Christianson DW. Crystal structure of human arginase I complexed with thiosemicarbazide reveals an unusual thiocarbonyl μ-sulfide ligand in the binuclear manganese cluster. J Am Chem Soc. 2007;129:6388–6389. doi: 10.1021/ja071567j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Baggio R, Elbaum D, Kanyo ZF, Carroll PJ, Cavalli RC, Ash DE, Christianson DW. Inhibition of Mn2+2-arginase by borate leads to the design of atransition state analogue inhibitor, 2(S)-amino-6-boronohexanoic acid. J Am Chem Soc. 1997;119:8107–8108. [Google Scholar]
- 13.Di Costanzo L, Sabio G, Mora A, Rodriguez PC, Ochoa AC, Centeno F, Christianson DW. Crystal structure of human arginase I at 1.29-Å resolution and exploration of inhibition in the immune response. Proc Natl Acad Sci U S A. 2005;102:13058–13063. doi: 10.1073/pnas.0504027102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.D’Antonio EL, Christianson DW. Crystal structures of complexes with cobalt-reconstituted human arginase I. Biochemistry. 2011;50:8018–8027. doi: 10.1021/bi201101t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Christianson DW, Lipscomb WN. Carboxypeptidase A. Acc Chem Res. 1989;22:62–69. [Google Scholar]
- 16.Matthews BW. Structural basis of the action of thermolysin and related zinc peptidases. Acc Chem Res. 1988;21:333–340. [Google Scholar]
- 17.Christianson DW. Structural biology of zinc. Adv Prot Chem. 1991;42:281–355. doi: 10.1016/s0065-3233(08)60538-0. [DOI] [PubMed] [Google Scholar]
- 18.Alberts IL, Nadassy K, Wodak SJ. Analysis of zinc binding sites in protein crystal structures. Protein Sci. 1998;7:1700–1716. doi: 10.1002/pro.5560070805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Berg JM, Merkle DL. On the metal ion specificity of zinc finger proteins. J Am Chem Soc. 1989;111:3759–3761. [Google Scholar]
- 20.Greenberg DM, Bagot AE, Roholt OA. Liver arginase. III. Properties of highly purified arginase. Arch Biochem Biophys. 1956;62:446–453. doi: 10.1016/0003-9861(56)90143-6. [DOI] [PubMed] [Google Scholar]
- 21.Tormanen CD. Inhibition of rat and soybean arginase by zinc ion. Abstracts, 236th Annual ACS Meeting; Philadelphia. August 17–21, 2008; 2008. p. 35. BIOL. [Google Scholar]
- 22.Issaly IM, Issaly AS. Control of ornithine carbamoyltransferase activity by arginase in Bacillus subtilis. Eur J Biochem. 1974;49:485–495. doi: 10.1111/j.1432-1033.1974.tb03853.x. [DOI] [PubMed] [Google Scholar]
- 23.Moreno-Vivian C, Soler G, Castillo F. Arginine catabolism in the phototrophic bacterium Rhodobacter capsulatus E1F1: Purification and properties of arginase. Eur J Biochem. 1992;204:531–537. doi: 10.1111/j.1432-1033.1992.tb16664.x. [DOI] [PubMed] [Google Scholar]
- 24.Gomez-Ortiz M, Gomis-Ruth FX, Huber R, Aviles FX. Inhibition of carboxypeptidase A by excess zinc: analysis of the structural determinants by X-ray crystallography. FEBS Lett. 1997;400:336–340. doi: 10.1016/s0014-5793(96)01412-3. [DOI] [PubMed] [Google Scholar]
- 25.Holland DR, Hausrath AC, Juers D, Matthews BW. Structural analysis of zinc substitutions in the active site of thermolysin. Protein Sci. 1995;4:1955–1965. doi: 10.1002/pro.5560041001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Archibald RM. Colorimetric determination of urea. J Biol Chem. 1945;157:507–518. [Google Scholar]
- 27.Rüegg UT, Russell AS. A rapid and sensitive assay for arginase. Anal Biochem. 1980;102:206–212. doi: 10.1016/0003-2697(80)90340-1. [DOI] [PubMed] [Google Scholar]
- 28.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 29.McCoy AJ, Grosse-Kunstleve RW, Storoni LC, Read RJ. Likelihood-enhanced fast translation functions. Acta Crystallogr, Sect D: Biol Crystallogr. 2005;61:458–464. doi: 10.1107/S0907444905001617. [DOI] [PubMed] [Google Scholar]
- 30.Collaborative Computational Project Number 4. The CCP4 suite: programs for protein crystallography. Acta Crystallogr, Sect D: Biol Crystallogr. 1994;50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 31.Brünger AT, Adams PD, Clore GM, Delano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges N, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL. Crystallography and NMR system (CNS): A new software system for macromolecular structure determination. Acta Crystallogr, Sect D: Biol Crystallogr. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 32.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr, Sect D: Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 33.Laskowski RA, MacArthur MW, Moss DS, Thornton JM. PROCHECK: a program to check the stereochemical quality of protein structures. J Appl Crystallogr. 1993;26:283–291. [Google Scholar]
- 34.Kleywegt GJ, Zou JY, Kjeldgaard M, Jones TA. Around O. In: Rossmann MG, Arnold E, editors. International Tables of Crystallography. Kluwer Academic Publishers; Dordrecht, The Netherlands: 2001. pp. 353–356.pp. 366–367. [Google Scholar]
- 35.Beale RN, Croft D. A sensitive method for the colorimetric determination of urea. J Clin Pathol. 1961;14:418–424. doi: 10.1136/jcp.14.4.418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Carrell CJ, Carrell HL, Erlebacher J, Glusker JP. Structural aspects of metal ion-carboxylate interactions. J Am Chem Soc. 1988;110:8651–8656. [Google Scholar]
- 37.Cavalli RC, Burke CJ, Soprano DR, Kawamoto S, Ash DE. Mutagenesis of rat liver arginase expressed in Escherichia coli: Role of conserved histidines. Biochemistry. 1994;33:10652–10657. doi: 10.1021/bi00201a012. [DOI] [PubMed] [Google Scholar]
- 38.Colleluori DM, Reczkowski RS, Emig FA, Cama E, Cox JD, Scolnick LR, Compher K, Jude K, Han S, Viola RE, Christianson DW, Ash DE. Probing the role of the hyper-reactive histidine residue of arginase. Arch Biochem Biophys. 2005;444:15–26. doi: 10.1016/j.abb.2005.09.009. [DOI] [PubMed] [Google Scholar]
- 39.Christianson DW, Alexander RS. Carboxylate-histidine-zinc interactions in protein structure and function. J Am Chem Soc. 1989;111:6412–6419. [Google Scholar]
- 40.Daghigh F, Cavalli RC, Soprano DR, Ash DE. Chemical modification and inactivation of rat liver arginase by N-bromosuccinimide: Reaction with His141. Arch Biochem Biophys. 1996;327:107–112. doi: 10.1006/abbi.1996.0098. [DOI] [PubMed] [Google Scholar]
- 41.Carvajal N, Olate J, Salas M, Uribe E, López V, Herrera P, Cerpa J. Chemical modification and site-directed mutagenesis of human liver arginase: Evidence that the imidazole group of histidine-141 is not involved in substrate binding. Arch Biochem Biophys. 1999;371:202–206. doi: 10.1006/abbi.1999.1421. [DOI] [PubMed] [Google Scholar]
- 42.Vockley JG, Tabor DE, Kern RM, Goodman BK, Wissmann PB, Kang DS, Grody WW, Cederbaum SD. Identification of mutations (D128G, H141L) in the liver arginase gene of patients with hyperargininemia. Hum Mutat. 1994;4:150–154. doi: 10.1002/humu.1380040210. [DOI] [PubMed] [Google Scholar]
- 43.Kanyo ZF, Scolnick LR, Ash DE, Christianson DW. Structure of a unique binuclear manganese cluster in arginase. Nature. 1996;383:554–557. doi: 10.1038/383554a0. [DOI] [PubMed] [Google Scholar]