

Abstract
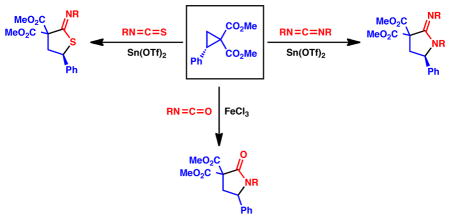
Isocyanates, isothiocyanates, and carbodiimides are effective substrates in (3 + 2) cycloadditions with donor–acceptor cyclopropanes for the synthesis of five-membered heterocycles. These reactions exhibit a broad substrate scope, high yields, and well-defined chemoselectivity. Discussed herein are the implications of Lewis acid choice on the stereochemical outcome and the reaction mechanism.
Donor-acceptor cyclopropanes are a useful class of building blocks for organic synthesis.1 Indeed, (3 + 2) cycloadditions of donor-acceptor cyclopropanes have proven to be a powerful strategy for the direct synthesis of 5-membered carbo- and heterocycles, and such methodologies have been applied toward natural product syntheses.2 Given our own interest in this field,2e we sought to examine heterocumulenes as potential dipolarophiles in stereoselective (3 + 2) cycloadditions to enable access to five-membered heterocycles. At the outset of this project, isocyanates3a,b and isothiocyanates3c,d had previously been shown to be reactive with alkoxy-substituted donor-acceptor cyclopropanes in only low to moderate yields, and stereocontrol of these reactions has relied on existing stereocenters remote to the site of reactivity. Based on the work of Johnson and others, we envisioned that the use of aryl substituents as the donor component would allow for an enantiospecific process by means of nucleophilic attack at the chiral benzylic center.4,5 The products formed could serve as useful building blocks toward optically active natural products and pharmaceutically relevant heterocyclic compounds.
We began by examining the reactivity of allyl isothiocyanate with aryl-substituted cyclopropanes (1). We found that a stoichiometric amount of tin(II) triflate enabled full conversion of the starting materials to thioimidates (2, Scheme 1). Notably, the chemoselectivity of this reaction is complementary with respect to the previous studies in which alkoxy-substituted donor-acceptor cyclopropanes are converted to thioamides.3,6
Scheme 1.

Substrate Scope of Isothiocyanate (3 + 2) Cycloaddition.
Conditions: cyclopropane 1 (0.4 mmol), isothiocyanate (0.8 mmol), Sn(OTf)2 (0.44 mmol), CH2Cl2 (1.3 mL). a Isolated yields.
We found that a variety of substituted aryl groups were tolerated in this reaction. Thioimidates with electron-rich aryl substituents (2b, 2c, 2m) were obtained with the shortest reaction times. Reactions leading to products with ortho or electron withdrawing arene substituents (2d, 2h, 2i) were the slowest. Cyclopropanes were not limited to those with aryl substituents; a vinyl group could also be used as an electron-donating substituent, offering 5-vinylthioimidate 2j in quantitative yield. In addition, cyclohexyl isothiocyanate was also compatible under these conditions, providing thioimidates 2k and 2l in 91% and 99% yield respectively.
Under tin(II) triflate mediated conditions, we found aryl isothiocyanates to be poorly reactive; however, concurrent with our studies, Li and coworkers disclosed an iron(III) chloride mediated (3 + 2) cycloaddition of aryl isothiocyanates with donor-acceptor cyclopropanes to form thiolactams (e.g. 4) rather than thioimidates (e.g. 5, Scheme 2a).7 We suspected that the products reported by Li may have been misassigned, and decided to investigate this further. Upon comparison of the 13C NMR spectra of our products (e.g. 2a–2m) to those reported by Li, we found similar shifts in the carbonyl range for both sets of spectra. In both cases, three signals are typically observed near 160 ppm: two correspond to the ester functionalities, and the third is consistent with a thioimidate; by contrast, a thioamide C=S 13C NMR signal is expected at approximately 200 ppm.8 Furthermore, our IR spectra consistently exhibited C=N stretches near 1650 cm−1, and C=S signals were not observed.9 Although no IR spectra were included in Li’s report, we reacted cyclopropane 3 with phenyl isothiocyanate under similar conditions to those reported by Li and obtained a compound with NMR spectra matching those reported (Scheme 2b). The product IR spectrum contained a C=N stretch at 1638 cm−1 and no C=S peak was observed. Finally, we found that the 5-mesityl substituted thioimidate (2h, Scheme 1) was crystalline and its structure was confirmed by single crystal X-ray diffraction (Scheme 2c).10a Combined, the IR, 13C NMR and X-ray crystallography data support our assignment of both the Li group’s and our products as thioimidates and not thioamides.
Scheme 2.

Structural Reassignment of Li’s Arylisothiocyanate (3 + 2) Products.
We then turned our attention to carbodiimide dipolarophiles and we observed that under tin(II) triflate mediated reaction conditions, the use of diisopropyl carbodiimide resulted in complete conversion of cyclopropane 3 to amidine 6a in only 80 minutes, and required only 1.1 equivalents of the dipolarophile.10b Electron rich 5-para-methoxyphenyl amidine 6b was formed almost quantitatively in less than 10 minutes (Scheme 3). Primary amidines (6e and 6f) could also be accessed using bis(trimethylsilyl)carbodiimide. Notably, (3 + 2) reactions were possible with cyclopropanes that are unreactive with isothiocyanates. For instance, a sterically congested 5,5-disubstituted amidine 6g could be generated in 58% yield. In addition, while aryl isothiocyanates were poorly reactive in the presence of tin(II) triflate, use of diphenylcarbodiimide resulted in the formation of the corresponding amidine (6h) in 79% yield. Overall, the shorter reaction times indicate that carbodiimides are considerably more reactive dipolarophiles than comparable isothiocyanates in these reactions.
Scheme 3.

Substrate Scope of Carbodiimide (3 + 2) Cycloaddition.
Conditions: cyclopropane 1 (0.4 mmol), carbodiimide (0.44 equiv), tin(II) triflate (0.44 mmol), CH2Cl2 (1.3 mL). a Isolated yields. b Bis(trimethylsilyl)carbodiimide is used as the dipolarophile.
We also examined isocyanates in (3 + 2) cycloadditions, however, they suffered from poor reactivity under tin mediated reaction conditions. Microwave heating resulted in shorter reaction times but an array of side products. Fortunately, we were able to obtain lactams in moderate yields using iron(III) chloride (Scheme 4).11 A variety of N-alkyl substituted lactams could be prepared, including isopropyl (7a), benzyl (7b and 7c), allyl (7d), and n-butyl (7e). In addition, a secondary lactam (7f) could be synthesized using trimethylsilylisocyanate as a dipolarophile. In general, these isocyanate reactions were considerably lower yielding than those with carbodiimides and isothiocyanates, nevertheless, they provide a meaningful entry into the important lactam series.
Scheme 4.
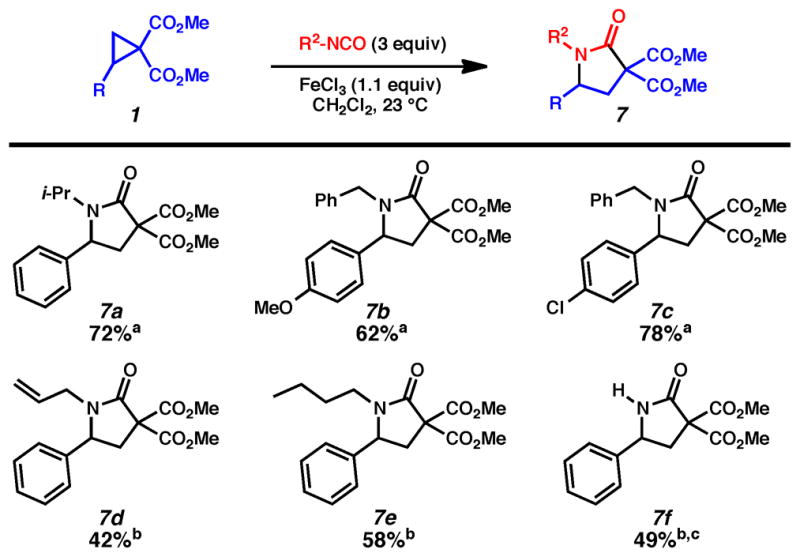
Isocyanate (3 + 2) Cycloaddition Substrate Scope.
Conditions: a Cyclopropane 1 (0.4 mmol), FeCl3 (glovebox, 0.44 mmol), isocyanate (1.2 mmol), CH2Cl2 (1.3 mL) or b cyclopropane (0.4 mmol), FeCl3 (benchtop, 0.44 mmol), isocyanate (1.2 mmol), MS 4 Å (50 mg), CH2Cl2 (1.3 mL). c Trimethylsilylisocyanate was used.
Finally, we sought to establish whether stereochemical information from the starting material is transferred to the product under the reaction conditions. We prepared enantioenriched cyclopropane (S)-3 according to literature methods and subjected it to our standard reaction conditions (Scheme 5).12 Although treatment of this substrate with an isocyanate in the presence of iron(III) chloride resulted in complete racemization of the benzylic stereocenter (Scheme 5a),13 we observed transfer of chirality in the case of tin(II) triflate mediated (3 + 2) cycloadditions (Schemes 5b and 5c). Notably substrates that required longer reaction times resulted in increased erosion of optical activity.14 We were able to confirm the absolute stereochemistry of the HBr salt of (R)-6a by single crystal X-ray diffraction, which revealed an inversion of configuration at the benzylic stereocenter through the course of the reaction (Figure 1).
Scheme 5.

Investigations into the Stereochemical Outcome of the (3 + 2) Reaction.
Figure 1.

Determination of Absolute Configuration.15
We propose that the mechanism of the isothiocyanate and carbodiimide reactions with tin(II) triflate involves a stereospecific intimate-ion pair mechanism analogous to that invoked by Johnson and coworkers for (3 + 2) cycloadditions of aldehydes and donor-acceptor cyclopropanes developed in their laboratories (Scheme 6).4c,d,16 Our observations including stereochemical inversion at the benzylic position, along with the greater reactivity of electron-rich dipolarophiles and of cyclopropanes with electron-rich aromatic substitutuents are all consistent with this mechanistic hypothesis.
Scheme 6.

Proposed Mechanism for (3 + 2) Cycloaddition.
In conclusion, we have disclosed an effective method for formation of pyrrolidinones, thioimidates and amidines from donor-acceptor cyclopropanes. Our data suggest that with comparable substituents, carbodiimides are more reactive than isothiocyanates, which are in turn more reactive than isocyanates. We have also disclosed a new mode of reactivity for isothiocyanates with donor-acceptor cyclopropanes. Furthermore, while iron(III) chloride caused racemization of the cyclopropane and formed racemic cycloadducts, tin(II) triflate mediated (3 + 2) reactions with isothiocyanates and carbodiimides were shown to proceed through an enantiospecific pathway, with inversion of configuration. Efforts to develop conditions catalytic in Lewis acid, as well as conditions for the enantioselective reactions of isocyanates with donor-acceptor cyclopropanes are currently underway. Applications of these methods for a range of purposes are also being investigated.
Supplementary Material
Acknowledgments
The authors wish to thank NIH-NIGMS (R01GM080269-01), Amgen, Abbott, Boehringer Ingelheim, and Caltech for financial support. A.G. gratefully acknowledges the Natural Sciences and Engineering Research Council (NSERC) of Canada for a PGS D scholarship. Dr. Michael R. Krout (Bucknell University) and Jonathan R. Gordon (Caltech) are thanked for helpful discussions. Lawrence Henling (Caltech) is gratefully acknowledged for X-ray crystallographic structural determination. The Bruker KAPPA APEXII X-ray diffractometer was purchased via an NSF CRIF:MU award to the California Institute of Technology, CHE-0639094. The Varian 400 MHz NMR spectrometer was purchased via an NIH grant (RR027690).
Footnotes
Supporting Information Available: Experimental details and NMR spectra of all intermediates. These materials are available free of charge via the Internet at http://pubs.acs.org.
References
- 1.(a) Mel’nikov MY, Budynina EM, Ivanova OA, Trushkov IV. Mendeleev Commun. 2011;21:293–301. [Google Scholar]; (b) Carson CA, Kerr MA. Chem Soc Rev. 2009;36:3051–3060. doi: 10.1039/b901245c. [DOI] [PubMed] [Google Scholar]; (c) De Simone F, Waser J. Synthesis. 2009:3353–3374. [Google Scholar]; (d) Rubin M, Rubina M, Gevorgyan V. Chem Rev. 2007;107:3117–3179. doi: 10.1021/cr050988l. [DOI] [PubMed] [Google Scholar]; (e) Yu M, Pagenkopf BL. Tetrahedron. 2005;61:321–347. [Google Scholar]; (f) Reissig H-U, Zimmer R. Chem Rev. 2003;103:1151–1196. doi: 10.1021/cr010016n. [DOI] [PubMed] [Google Scholar]; (g) Mochalov SS, Gazzaeva RA. Chem Heterocycl Compd. 2003;39:975–988. [Google Scholar]; (h) Reissig HU. Top Curr Chem. 1988;144:73–135. [Google Scholar]
- 2.Examples include: Karadeolian A, Kerr MA. Angew Chem, Int Ed. 2010;49:1133–1135. doi: 10.1002/anie.200906632.Campbell MJ, Johnson JS. J Am Chem Soc. 2009;131:10370–10371. doi: 10.1021/ja904136q.Zhang H, Curran DP. J Am Chem Soc. 2011;133:10376–10378. doi: 10.1021/ja2042854.Morales CL, Pagenkopf BL. Org Lett. 2008;10:157–159. doi: 10.1021/ol702376j.Goldberg AFG, Stoltz BM. Org Lett. 2011;13:4474–4476. doi: 10.1021/ol2017615.
- 3.(a) Brückner C, Suchland B, Reissig H-U. Liebigs Ann Chem. 1988:471–473. [Google Scholar]; (b) Graziano ML, Iesce MR. J Chem Res (S) 1987:362–363. [Google Scholar]; (c) Graziano ML, Cimminiello MR. J Chem Res (S) 1989:42–43. [Google Scholar]; (d) Graziano ML, Cimminiello G. J Chem Res (M) 1989:446–447. [Google Scholar]
- 4.Stereoselective (3 + 2) reactions of donor-acceptor cyclopropanes with aldimines: Parsons AT, Smith AG, Neel AJ, Johnson JS. J Am Chem Soc. 2010;132:9688–9692. doi: 10.1021/ja1032277.Aldehydes: Parsons AT, Johnson JS. J Am Chem Soc. 2009;131:3122–3123. doi: 10.1021/ja809873u.Pohlhaus PD, Sanders SD, Parsons AT, Li W, Johnson JS. J Am Chem Soc. 2008;130:8642–8650. doi: 10.1021/ja8015928.Pohlhaus PD, Johnson JS. J Am Chem Soc. 2005;127:16014–16015. doi: 10.1021/ja055777c.Vinylcyclopropanes with Azlactones: Trost BM, Morris PJ. Angew Chem, Int Ed. 2011;50:6167–6170. doi: 10.1002/anie.201101684.Alkynes: Lin M, Kang GY, Guo YA, Yu ZX. J Am Chem Soc. 2012;134:398–405. doi: 10.1021/ja2082119.
- 5.Additional examples of stereoselective cycloadditions of donor-acceptor cyclopropanes can be found in the review articles in ref 1.
- 6.Chemoselectivity comparable to that observed in our studies has been shown in a palladium-catalyzed (3 + 2) cycloaddition of isothiocyanates with aziridines. Baeg J-O, Bensimon C, Alper H. J Am Chem Soc. 1995;117:4700–4701.
- 7.Wang H, Yang W, Liu H, Wang W, Li H. Org Biomol Chem. 2012;10:5032–5035. doi: 10.1039/c2ob25682g. [DOI] [PubMed] [Google Scholar]
- 8.Pretsch E, Bühlmann P, Badertscher M. Structure Determination of Organic Compounds. 4. Springer-Verlag; Berlin: 2009. [Google Scholar]
- 9.A thioamide C=S IR stretch is expected as a strong band near 1140–1190 cm−1.8
- 10.(a) The assignment of Z-stereochemistry for thioimidate 2h was established via single crystal X-ray diffraction, and other thioimidates in Table 1 were assigned by analogy. (b) The assignment of E-stereochemistry for amidine 6a is based on the crystal structure of its HBr salt (see below), and other amidines in Table 2 were assigned by analogy.
- 11.Two methods were used to setup reactions with dry iron(III) chloride: (a) iron(III) chloride stored in a nitrogen-filled glovebox and dispensed into a flame-dried flask or (b) iron(III) chloride stored on a benchtop was dispensed into an oven-dried vial, and used in conjunction with 4 Å molecular sieves. Although (b) is more operationally convenient, method (a) appears to result in shorter reaction times and higher yields.
- 12.Davies HML, Bruzinski PR, Lake DH, Kong N, Fall MJ. J Am Chem Soc. 1996;118:6897–6907. [Google Scholar]
- 13.Treatment of cyclopropane (R)-3 with iron(III) chloride, in the absence of a dipolarophile, results in racemization of the material.
- 14.Prolonged exposure of cyclopropane (R)-3 to Sn(OTf)2 results in racemization. See ref 4c.
- 15.Methyl and phenyl hydrogen atoms and the bromide counterion are omitted for clarity.
- 16.Related mechanistic investigations were originally performed on cyclopropane nitrone cycloadditions: Karadeolian A, Kerr MA. J Org Chem. 2007;72:10251–10253. doi: 10.1021/jo702073w.Sapeta K, Kerr MA. J Org Chem. 2007;72:8597–8599. doi: 10.1021/jo701606u.Wanapun D, Van Gorp KA, Mosey NJ, Kerr MA, Woo TK. Can J Chem. 2005;83:1752–1767.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.