

Abstract
Commissural dI1 neurons have been extensively studied to elucidate the mechanisms underlying axon guidance during development1,2. These neurons are located in the dorsal spinal cord and send their axons along stereotyped trajectories. Commissural axons initially project ventrally towards and then across the floorplate. After crossing the midline, these axons make a sharp rostral turn and project longitudinally towards the brain. Each of these steps is regulated by the coordinated activities of attractive and repulsive guidance cues. The correct interpretation of these cues is crucial to the guidance of axons along their demarcated pathway. Thus, the physiological contribution of a particular molecule to commissural axon guidance is ideally investigated in the context of the living embryo. Accordingly, gene knockdown in vivo must be precisely controlled in order to carefully distinguish axon guidance activities of genes that may play multiple roles during development.
Here, we describe a method to knockdown gene expression in the chicken neural tube in a cell type-specific, traceable manner. We use novel plasmid vectors3 harboring cell type-specific promoters/enhancers that drive the expression of a fluorescent protein marker, followed directly by a miR30-RNAi transcript4 (located within the 3'-UTR of the cDNA encoding the fluorescent protein) (Figure 1). When electroporated into the developing neural tube, these vectors elicit efficient downregulation of gene expression and express bright fluorescent marker proteins to enable direct tracing of the cells experiencing knockdown3. Mixing different RNAi vectors prior to electroporation allows the simultaneous knockdown of two or more genes in independent regions of the spinal cord. This permits complex cellular and molecular interactions to be examined during development, in a manner that is fast, simple, precise and inexpensive. In combination with DiI tracing of commissural axon trajectories in open-book preparations5, this method is a useful tool for in vivo studies of the cellular and molecular mechanisms of commissural axon growth and guidance. In principle, any promoter/enhancer could be used, potentially making the technique more widely applicable for in vivo studies of gene function during development6.
This video first demonstrates how to handle and window eggs, the injection of DNA plasmids into the neural tube and the electroporation procedure. To investigate commissural axon guidance, the spinal cord is removed from the embryo as an open-book preparation, fixed, and injected with DiI to enable axon pathways to be traced. The spinal cord is mounted between coverslips and visualized using confocal microscopy.
Keywords: Neuroscience, Issue 68, Developmental Biology, Molecular Biology, Genetics, Spinal cord, neural development, microRNA, chicken, in ovo electroporation, RNA interference, knock down, neural circuit, dissection, open-book preparation
Protocol
1. Preparation of RNAi Plasmid DNA for Cell Type-specific Gene Silencing
Plasmids (Figure 1) are synthesized using standard molecular cloning techniques, as previously described in detail3,4.
1.1 Cloning into the vectors: oligonucelotide design
- We use the same universal oligonucleotides and cloning protocols that are described in the product information provided with the pRFPRNAiC vector4 (ARK-Genomics).
- For cloning into the first hairpin site: 5' primer HP1: 5'-GGCGGGGCTAGCTGGAGAAGATGCCTTCCGGAGAGGTGCTGCTGAGCG 3' primer HP1: 5'-GGGTGGACGCGTAAGAGGGGAAGAAAGCTTCTAACCCCGCTATTCACCACCACTAGGCA
- For cloning into the second hairpin site: 5' primer HP2: 5'-GGCGGGACGCGTGCTGTGAAGATCCGAAGATGCCTTGCGCTGGTTCCTCCGTGAGCG 3' primer HP2:
- 5'-CGCCGCGCATGCACCAAGCAGAGCAGCCTGAAGACCAGTAGGCA
We use Genscript's siRNA Target Finder to select gene-specific target sequences: https://www.genscript.com/ssl-bin/app/rnai.
Primers for cloning a gene-specific miRNA into the first hairpin site:
An example for silencing GFP is shown below. Target sequence (22nt): 5'-GGCACAAGCTGGAGTACAACTA GFP Forward HP1 = 59mer ![]() GFP Reverse HP1 = 58mer
GFP Reverse HP1 = 58mer ![]() There are common sequences in these oligos that form part of the miRNA flanking sequences (chicken-specific), and common loop/stem sequences (from human miRNA30). The gene-specific target sequences are underlined. Note that there is a mismatch at the 5' base of the forward strand (shown in bold; G→A in this example) to mimic the natural mismatch in miRNA30 at this position.
There are common sequences in these oligos that form part of the miRNA flanking sequences (chicken-specific), and common loop/stem sequences (from human miRNA30). The gene-specific target sequences are underlined. Note that there is a mismatch at the 5' base of the forward strand (shown in bold; G→A in this example) to mimic the natural mismatch in miRNA30 at this position.
Primers for cloning a gene-specific miRNA into the second hairpin site:
An example for silencing LacZ is shown below. Target sequence (22nt): 5'-CGCGCTGTATCGCTGGATCAAA LacZ Forward HP2: ![]() LacZ Reverse HP2:
LacZ Reverse HP2: ![]() Note that again, the 5' base of the target sequence in the forward strand has been changed (shown in bold; C→A in this example) so that it mismatches the antisense sequence, mimicking miRNA30.
Note that again, the 5' base of the target sequence in the forward strand has been changed (shown in bold; C→A in this example) so that it mismatches the antisense sequence, mimicking miRNA30.
1.2 PCR Reaction and Subcloning
The gene-specific oligos are used together with the universal oligos in a PCR reaction to generate the miRNA30-like hairpin with chicken miRNA flanking sequences.
| Cloning into first hairpin site: 1 μl - 10 ng GFP Forward primer HP1 1 μl - 100 ng 5' primer HP1 1 μl - 100 ng 3' primer HP1 1 μl dNTPs (10 mM) 5 μl 10x Pfu reaction buffer 1 μl Pfu DNA Polymerase (Promega) 39 μl PCR-Grade water | OR | Cloning into second hairpin site: 1 μl - 10 ng LacZ Forward primer HP2 1 μl - 100 ng 5' primer HP2 1 μl - 100 ng 3' primer HP2 1 μl dNTPs (10 mM) 5 μl 10x Pfu reaction buffer 1 μl Pfu DNA Polymerase (Promega) 39 μl PCR-Grade water |
Cycles:
| 94 °C | 94 °C | 55 °C | 72 °C | 72 °C | 4 °C |
| 1 min | 30 sec | 30 sec | 1 min | 9 min | hold |
| 30 cycles |
- Purify the PCR product, digest with restriction enzymes and subclone into the vector:
- First hairpin site: use NheI and MluI
- Second hairpin site: use MluI and SphI
Follow standard techniques for transformation of competent bacterial cells. Plate cells on LB agar (containing ampicillin) and harvest DNA by plasmid midipreparation (e.g. Nucleobond Xtra Midi kit, Machery-Nagel).
Suspend concentrated plasmid DNA in sterile ddH20, measure concentration by spectrophotometry and store at -20 °C.
1.3 Sequencing miRNA plasmids
Under standard conditions the sequencing reaction often fails due to strong secondary structure of the hairpins. To improve this7:
Perform sequencing reaction in 10 mM Tris-Cl with 0.01 mM EDTA (pH 8.0) instead of water. This increases conversion of supercoiled DNA to ssDNA, which is more amenable for sequencing.
Add a heat denaturation step (98 °C, 5 min) prior to sequencing. This converts supercoiled plasmid DNA to ssDNA.
2. Electroporation
2.1 Egg handling
Place the eggs into an incubator set to 38.5 °C and ~45% humidity.
Incubate the eggs until the embryos have reached the desired stage of development. To study axon guidance of commissural neurons, we typically electroporate embryos when they have reached Hamburger & Hamilton (HH) stage 17-18 (after approximately 3 days of incubation)8. However, injection and electroporation need to be done before the protein of interest has accumulated, to make knockdown efficient.
Remove the eggs from the incubator and put them in a stable horizontal position for 20 min, to reposition the embryo on top of the yolk at the upper side of the egg. Return the eggs to the incubator during this period.
2.2 Preparation of reagents and equipment
Prepare phosphate buffered saline (PBS) and sterilize by autoclaving or passing the solution through a 0.2 μm filter.
Make glass micropipettes by pulling capillaries (World Precision Instruments 1B120F-4; 1.2 / 0.68 OD/ID (mm)) in a suitable pulling device (eg. Narishige PC-10). Break off the tip of a micropipette to obtain a tip diameter of ~5 μm and connect it to a piece of tubing with the appropriate diameter.
Mount the platinum electrodes (0.5 cm long) firmly in a hand-held frame, with an inter-electrode distance of 0.5 cm. Connect the electrodes to a square wave pulse generator (BTX ECM 830).
- Set the pulse generator with the following parameters:
- For unilateral electroporation: voltage: 25 V, number of pulses: 5, length of pulse: 50 msec, interpulse interval: 1 sec
- For bilateral electroporation: voltage: 18 V, number of pulses: 5, length of pulse: 50 msec, interpulse interval: 1 sec
Prepare a syringe with 18 G needle, a scalpel, a spritz bottle of 70% ethanol and tape.
Melt paraffin wax in a beaker on a heating plate at 80 °C.
2.3 Windowing
Wipe the eggs using a tissue soaked in 70% ethanol.
Place a strip of tape along the long axis of the egg.
Make two small holes in the eggshell using a scalpel; one at the blunt end of the egg and the other at the corner of the area to be windowed.
Using a syringe, remove approx. 3 ml of albumen from each egg by inserting the needle at an angle of 45° into the hole at the blunt end of the egg. Carefully avoid damaging the yolk.
Cut a window into the eggshell using small scissors, held horizontally to avoid damaging the embryo. If required, the position of the embryo can be verified and marked with a pencil by holding a strong light against the blunt end of the egg. A window of 1.5 to 2 cm square is useful for most applications.
Seal the hole at the blunt end of the egg and any cracks in the eggshell with melted paraffin wax, applied with a brush.
Seal the window using transparent tape (e.g. Scotch Magic).
Return the egg to the incubator.
2.4 Electroporation
Prepare sterile tools and wipe the work area with 70% ethanol.
Prepare the injection solution. The appropriate DNA concentration must be determined by the user and will vary according to the enhancer/promoter used to drive expression. As a guide, we typically use 0.2-2.0 μg/μl (see Discussion). In a total of 20 μl, the injection mixture should contain:
| RNAi plasmid DNA (in H20) | X μl |
| 20x PBS | 1 μl |
| 0.4% trypan blue | 2 μl |
| sterile ddH20, to a final volume of | 20 μl |
Use gentle suction to load the DNA mixture into the glass microcapillary attached to the tubing.
Remove the tape from the windowed egg and stage the embryo according to Hamburger and Hamilton8.
Use forceps and spring scissors to carefully remove the extraembryonic membranes from the caudal half of the embryo. The membranes can be easily lifted away from the embryo in the region where the major right and left vitelline veins enter the trunk. Gently tear or cut the membranes and pull them towards the tail. If required, the embryo can be visualized better by injecting a solution of India ink or Fast Green between embryonic disc and egg yolk as shown in a video by Boulland and colleagues9.
Inject the DNA solution into the central canal of the neural tube, just above the hindlimbs. Control the injection volume by mouth. The blue dye should spread from the tip of the tail up to the ventricle of the developing brain.
Add a few drops of sterile PBS on top of the embryo.
Place the electrodes parallel to the anterior-posterior axis of the embryo. Avoid touching the embryo or blood vessels.
Hold the electrodes steady, and electroporate.
- Carefully remove the electrodes and rinse them with sterile water to remove denatured proteins from the egg white.
- For bilateral electroporation, switch the polarity of the electrodes, reposition the electrodes parallel to the embryo and repeat the electroporation. Rinse the electrodes in sterile water.
Drop some more sterile PBS onto the embryo. Reseal the egg with tape and return it to the incubator until the desired developmental stage is reached. Proper sealing is crucial to avoid dehydration of the embryo.
3. Spinal Cord Preparations
3.1 Dissection of embryos
At HH25-26 (day 5 of incubation), remove the embryo from the egg using forceps or a small spoon. Place it in PBS in a Petri dish coated with silicone (Sylgard elastomer).
- Remove the extraembryonic membranes and lay the embryo on its back. Stabilize it on the plate by pinning it through the neck and tail (using 0.20 mm insect pins), with gentle stretching.
- Whole embryos (for sectioning later) can be fixed here, by replacing the PBS with 4% paraformaldehyde (PFA) and incubating for 1 hr at room temperature. To make open-book preparations, continue the protocol with unfixed tissue as described below.
3.2 Isolation of spinal cords from embryos
Pin the limbs, ensuring that the pins are inserted at an angle away from the embryo so that they do not interfere with the dissection. The embryo should be illuminated from below to enable tissue density to be perceived in the following steps.
Remove the heart and internal organs using spring scissors and gently scraping with forceps. The segmented vertebrae and spinal cord should be visible if all the organs have been removed completely.
Using spring scissors, make a shallow cut through the vertebrae overlying the spinal cord at the neck. Rotate the embryo 180° and make two short longitudinal cuts through the vertebrae on either side of the spinal cord, from the neck towards the tail. Use forceps to lift the flap of vertebrae away from the spinal cord and peel the tissue (containing all the vertebrae) in a single strip towards the tail.
Gently stretch and re-pin the embryo through the tail and limbs.
Use a fine microsurgical scalpel (e.g. Grieshaber 68101) or a tungsten needle to cut away the meninges overlying the spinal cord. Look for a dark, dense line of tissue between the neural tube and the dorsal root ganglia. Adjust the illumination angle, if necessary. Gently cut longitudinally along this line on each side of the spinal cord, from the neck to the tail. The meninges should separate from the spinal cord due to the gentle stretching of the pinned embryo.
Cut through the spinal cord at the level of the wing bud and caudal to the limb bud, and lift the whole spinal cord out of the embryo in one smooth rostral to caudal motion, using forceps. The isolated spinal cord should be kept immersed in PBS during this step. Do not lift it out of the dish.
3.3 Fixation of open-books
Spread the isolated spinal cord onto a spatula and transfer it to a new silicone-lined Petri dish containing 4% paraformaldehyde in PBS.
Produce a flat-mount preparation by carefully pinning the spinal cord in six positions (rostrally, medially and caudally on each side, using 0.10 mm insect pins). We label each preparation by a little 'flag' that not only identifies the embryo but also indicates the anterior end of the open-book preparation.
Incubate at room temperature for 30 min to 1 hr. Open-books should not be over-fixed as this reduces the efficiency of DiI diffusion10 and increases background.
Carefully pour off the 4% PFA and replace it with PBS. Keep the dishes at 4 °C until ready to inject with DiI or mount.
3.4 DiI injection into commissural neurons
Observe the open-books under fluorescence miscroscopy and select the appropriate side of the open-book to inject with DiI.
Prepare Fast DiI (5 mg/ml in ethanol) and draw the solution into a glass micropipette attached to plastic tubing. Break off the end of the micropipette as finely as possible to obtain a very small diameter tip. Insert the needle into a dish of PBS and check that the DiI does not leak from the needle. If the DiI leaks, the needle diameter is too big. In that case, prepare a new needle.
Illuminate the open-book preparations from below. Look for a denser longitudinal stripe of tissue, located approximately 1/5 of the width of a hemibook from the lateral edge of the preparation. This corresponds to the cell bodies of the commissural neurons, which are found just ventral to the roof plate. Starting at one end of the open-book, insert the glass needle into the tissue and, as the needle is withdrawn, puff in a small amount of DiI using a mouth pipette.
Work quickly, making several injections along the length of the open-book at regular intervals of approximately 0.5 mm. If the needle becomes clogged, carefully clear it with forceps. If the tip becomes too big (and DiI leaks), replace the needle.
When you have completed each open-book, use a transfer pipette to suck away and discard any excess, leaked DiI. Failure to do this will result in high background.
Leave the preparations for approximately three days at 4 °C to enable the DiI to spread along the axons.
3.5 Mounting for imaging
Use a syringe with an 18 G needle to spread a thin, uninterrupted border of vacuum grease (e.g. Dow Corning #976V) around the edges of a 24 mm x 24 mm glass coverslip. Add several drops of sterile PBS to the well. Note that mounting medium containing glycerol with n-propyl gallate may not be compatible with DiI11. Similarly, vacuum grease of low viscosity may mix with the PBS and result in high background.
Remove the pins from the open-book and transfer it into the PBS droplet. Immerse the open-book and position it in the middle of the well.
Gently place another 24 mm x 24 mm coverslip on top, making sure the open-book stays open. If necessary, the coverslip can be removed, and the open-book repositioned. Press gently around the edges of the preparation to create a complete seal of grease. Excess PBS will be squeezed out during this step. Avoid air bubbles.
Keep the preparations in the dark at 4 °C until ready for inspection and documentation by fluorescent microscopy. This should be done expeditiously (within one week), to ensure that the preparations do not dry out.
4. Representative Results
Electroporation and expression of plasmids
Under the conditions described above, fluorescent protein should be clearly detectable in the appropriate cell type without the need for additional amplification of the signal by antibody labeling. The fluorescent protein should only be detectable in the desired cell type/s. Representative examples of open-book preparations and cross sections of embryos electroporated with the different plasmids are shown in Figure 2.
Efficiency of artificial miRNAs
Artificial miRNAs against a novel gene of interest must first be screened for efficiency and specificity of their knockdown effects. We find that β-actin promoter-driven constructs, electroporated at 0.25 μg/μl, are appropriate for this3. Knockdown in vivo can be tested by immunohistochemistry or in situ hybridization.
DiI labeling
Appropriately targeted DiI injections into wildtype embryos should yield more than 80% of injection sites with ideal, archetypal trajectories3, as shown in Figure 3. Animal to animal variability should be low.
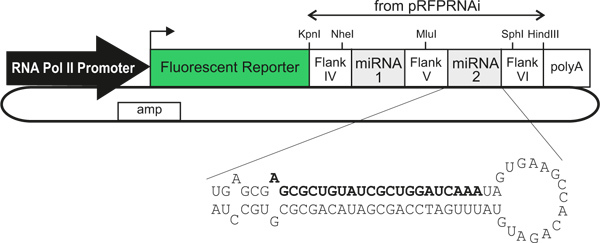 Figure 1. Generalized schematic of the miRNA-expressing plasmid vectors. The use of different RNA polymerase II promoters/enhancers enables cell type-specific expression. The transfected cells are identifiable by the expression of a fluorescent reporter that is directly linked (within a single transcript) to one or two artificial miRNAs, which knock down gene expression. Bold text indicates the sense strand of an artificial miRNA against LacZ, as described in the text.
Figure 1. Generalized schematic of the miRNA-expressing plasmid vectors. The use of different RNA polymerase II promoters/enhancers enables cell type-specific expression. The transfected cells are identifiable by the expression of a fluorescent reporter that is directly linked (within a single transcript) to one or two artificial miRNAs, which knock down gene expression. Bold text indicates the sense strand of an artificial miRNA against LacZ, as described in the text.
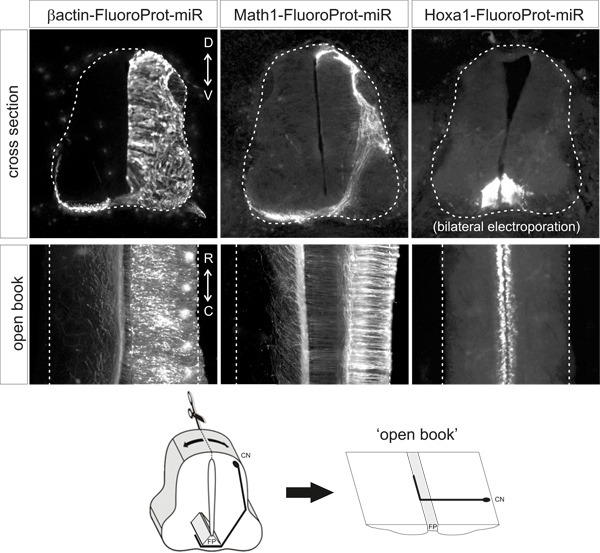 Figure 2. Representative examples of fluorescent protein expression patterns obtained following electroporation of the indicated plasmid vectors. Cross sections and open books are from HH25-26 chicken embryos that were electroporated at HH18. β-actin promoter drives ubiquitous expression, Math1 enhancer drives expression in dI1 neurons and Hoxa1 enhancer drives expression specifically in the floor plate. CN, commissural neuron; FP, floor plate.
Figure 2. Representative examples of fluorescent protein expression patterns obtained following electroporation of the indicated plasmid vectors. Cross sections and open books are from HH25-26 chicken embryos that were electroporated at HH18. β-actin promoter drives ubiquitous expression, Math1 enhancer drives expression in dI1 neurons and Hoxa1 enhancer drives expression specifically in the floor plate. CN, commissural neuron; FP, floor plate.
 Figure 3. Application and analysis of DiI injection sites in open book preparations. DiI should be injected in a punctate pattern, close to the lateral margin of the open book, on the electroporated side (identified by fluorescent protein expression). After 3 days of diffusion, commissural axon trajectories should be able to be visualized under fluorescent microscopy. Normal axon trajectories will grow towards the floor plate, cross the floor plate and then turn and grow rostrally. Abnormal phenotypes arising from gene knock down can be compared to this archetypal trajectory. In the example, some axons stall in the floor plate or make erroneous turning decisions on the contralateral side.
Figure 3. Application and analysis of DiI injection sites in open book preparations. DiI should be injected in a punctate pattern, close to the lateral margin of the open book, on the electroporated side (identified by fluorescent protein expression). After 3 days of diffusion, commissural axon trajectories should be able to be visualized under fluorescent microscopy. Normal axon trajectories will grow towards the floor plate, cross the floor plate and then turn and grow rostrally. Abnormal phenotypes arising from gene knock down can be compared to this archetypal trajectory. In the example, some axons stall in the floor plate or make erroneous turning decisions on the contralateral side.
Discussion
This simple, vector-based artificial miRNA expression strategy can be used to knockdown endogenous gene expression in the chicken neural tube. These functional tools offer multiple gene silencing, temporal control and cell-type specificity, to facilitate the elucidation of complex developmental pathways. In particular, we have demonstrated the utility of these plasmids in commissural axon guidance, since the plasmids can be used to knockdown distinct genes in commissural neurons or in their intermediate target, the floorplate3.
The intensity and location of fluorescent protein expression generated from the miRNA-expressing plasmids (and hence, the silenced cells) will depend on the following parameters:
- Enhancer used:
- β-actin promoter is ubiquitously active
- Math1 enhancer is active only in dI1 neurons12
- Hoxa1 enhancer III is active only in the floor plate13
- Concentration of plasmid. If plasmid concentration is too high, it may cause 'leakiness' of the enhancer activity and subsequent expression in undesired cells, while too little plasmid may reduce the expression of the fluorescent protein and prevent the electroporated cells from being traceable. High concentrations of RNAi plasmids should also be avoided to reduce off-target effects and minimize the chance of non-specific toxicity arising from saturation of the endogenous miRNA biogenesis pathway. We use:
- β-actin promoter: 0.25 μg/μl
- Math1 enhancer: 0.7 μg/μl
- Hoxa1 enhancer III: 0.5-1.0 μg/μl
Accuracy of electroporation.Handling chicken embryos in ovo requires manual skills that must be acquired through training. We and others have previously published troubleshooting tips for this procedure14,15.
Several miRNAs may need to be tested before an appropriate candidate is found. This process should incorporate the identification of several independent miRNAs to confirm an observed phenotype, negative controls (scrambled and/or unrelated miRNAs) and rescue constructs16.
Open-book preparations must be fixed optimally and injected with DiI into the appropriate location in order to visualize structural details of the axonal projections of commissural neurons. High background may be caused by prolonged fixation of the open-books or spillage of the DiI. Injection of DiI into cells located too dorsally will usually result in a lack of labeled axonal projections towards the floor plate. Cells located too ventrally will have many ipsilateral and contralateral projections at the midline, and should thus be avoided. For beginners, we recommend practicing on open-books taken from wildtype embryos to ensure accurate, reproducible positioning of the DiI.
Disclosures
No conflicts of interest declared.
Acknowledgments
Work in the lab of E.S. is supported by the Swiss National Science Foundation. We would like to thank Dr. Beat Kunz for assistance with filming.
References
- Chédotal A. Further tales of the midline. Current Opinion in Neurobiology. 2011;21:68–75. doi: 10.1016/j.conb.2010.07.008. [DOI] [PubMed] [Google Scholar]
- Avraham O, Hadas Y, Vald L, Zisman S, Schejter A, Visel A, Klar A. Transcriptional control of axonal guidance and sorting in dorsal interneurons by the Lim-HD proteins Lhx9 and Lhx1. Neural Development. 2009;4:21. doi: 10.1186/1749-8104-4-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson NH, Stoeckli ET. Cell type specific, traceable gene silencing for functional gene analysis during vertebrate neural development. Nucleic Acids Research. 2011;39:e133. doi: 10.1093/nar/gkr628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das RM, van Hateren NJ, Howell GR, Farrell ER, Bangs FK, Porteous VC. A robust system for RNA interference in the chicken using a modified microRNA operon. Developmental Biology. 2006;294:554–563. doi: 10.1016/j.ydbio.2006.02.020. [DOI] [PubMed] [Google Scholar]
- Perrin FE, Stoeckli ET. Use of lipophilic dyes in studies of axonal pathfinding in vivo. Microscopy Research and Technique. 2000;48:25–31. doi: 10.1002/(SICI)1097-0029(20000101)48:1<25::AID-JEMT4>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- Timmer J, Johnson J, Niswander L. The use of in ovo electroporation for the rapid analysis of neural-specific murine enhancers. Genesis. 2001;29:123–132. doi: 10.1002/gene.1015. [DOI] [PubMed] [Google Scholar]
- Kieleczawa J. Simple modifications of the standard DNA sequencing protocol allow for sequencing through siRNA hairpins and other repeats. Journal of Biomolecular Techniques. 2005;16:220–223. [PMC free article] [PubMed] [Google Scholar]
- Hamburger V, Hamilton HL. A series of normal stages in the development of the chick embryo. Journal of Morphology. 1951;88:49–92. doi: 10.1002/aja.1001950404. [DOI] [PubMed] [Google Scholar]
- Boulland J, Halasi G, Kasumacic N, Glover JC. Xenotransplantation of Human Stem Cells into the Chicken Embryo. J. Vis. Exp. 2010. p. e2071. [DOI] [PMC free article] [PubMed]
- Kim BG, Dai H-N, McAtee M, Vicini S, Bregman BS. Labeling of dendritic spines with the carbocyanine dye DiI for confocal microscopic imaging in lightly fixed cortical slices. Journal of Neuroscience Methods. 2007;162:237–243. doi: 10.1016/j.jneumeth.2007.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy MC, Fox EA. Anterograde tracing method using DiI to label vagal innervation of the embryonic and early postnatal mouse gastrointestinal tract. Journal of Neuroscience Methods. 2007;163:213–225. doi: 10.1016/j.jneumeth.2007.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helms AW, Abney AL, Ben-Arie N, Zoghbi HY, Johnson JE. Autoregulation and multiple enhancers control Math1 expression in the developing nervous system. Development. 2000;127:1185–1196. doi: 10.1242/dev.127.6.1185. [DOI] [PubMed] [Google Scholar]
- Li X, Lufkin T. Cre recombinase expression in the floorplate, notochord and gut epithelium in transgenic embryos driven by the Hoxa-1 enhancer III. Genesis. 2000;26:121–122. [PubMed] [Google Scholar]
- Mauti O, Baeriswyl T, Stoeckli ETGene. Silencing by Injection and Electroporation of dsRNA in Avian Embryos. Cold Spring Harbor Protocols. 2008. [DOI] [PubMed]
- Krull CE. A primer on using in ovo electroporation to analyze gene function. Developmental Dynamics. 2004;229:433–439. doi: 10.1002/dvdy.10473. [DOI] [PubMed] [Google Scholar]
- Cullen BR. Enhancing and confirming the specificity of RNAi experiments. Nature Methods. 2006;3:677–681. doi: 10.1038/nmeth913. [DOI] [PubMed] [Google Scholar]