

Abstract
Programmed cell death (PCD) occurs in adults to maintain normal tissue homeostasis and during embryological development to shape tissues and organs1,2,6,7. During development, toxic chemicals or genetic alterations can cause an increase in PCD or change PCD patterns resulting in developmental abnormalities and birth defects3-5. To understand the etiology of these defects, the study of embryos can be complemented with in vitro assays that use differentiating embryonic stem (ES) cells.
Apoptosis is a well-studied form of PCD that involves both intrinsic and extrinsic signaling to activate the caspase enzyme cascade. Characteristic cell changes include membrane blebbing, nuclear shrinking, and DNA fragmentation. Other forms of PCD do not involve caspase activation and may be the end-result of prolonged autophagy. Regardless of the PCD pathway, dying cells need to be removed. In adults, the immune cells perform this function, while in embryos, where the immune system has not yet developed, removal occurs by an alternative mechanism. This mechanism involves neighboring cells (called "non-professional phagocytes") taking on a phagocytic role-they recognize the 'eat me' signal on the surface of the dying cell and engulf it8-10. After engulfment, the debris is brought to the lysosome for degradation. Thus regardless of PCD mechanism, an increase in lysosomal activity can be correlated with increased cell death.
To study PCD, a simple assay to visualize lysosomes in thick tissues and multilayer differentiating cultures can be useful. LysoTracker dye is a highly soluble small molecule that is retained in acidic subcellular compartments such as the lysosome11-13. The dye is taken up by diffusion and through the circulation. Since penetration is not a hindrance, visualization of PCD in thick tissues and multi-layer cultures is possible12,13. In contrast, TUNEL (Terminal deoxynucleotidyl transferase dUTP nick end labeling) analysis14, is limited to small samples, histological sections, and monolayer cultures because the procedure requires the entry/permeability of a terminal transferase.
In contrast to Aniline blue, which diffuses and is dissolved by solvents, LysoTracker Red DND-99 is fixable, bright, and stable. Staining can be visualized with standard fluorescent or confocal microscopy in whole-mount or section using aqueous or solvent-based mounting media12,13. Here we describe protocols using this dye to look at PCD in normal and sonichedgehog null mouse embryos. In addition, we demonstrate analysis of PCD in differentiating ES cell cultures and present a simple quantification method. In summary, LysoTracker staining can be a great complement to other methods of detecting PCD.
Keywords: Developmental Biology, Issue 68, Molecular Biology, Stem Cell Biology, Cellular Biology, mouse embryo, embryonic stem cells, lysosome, programmed cell death, imaging, sonic hedgehog
Protocol
1. LysoTracker Staining of Mouse Embryos
Generate mouse embryos by placing young (5-6 week old) females into male stud cages. Monitor on following mornings for the appearance of a vaginal plug indicating that mating has occurred. If the female is pregnant, noon on that day is called 0.5 dpc (days post coitus). The procedure presented here is ideal for embryos 7-13 dpc.
On the embryonic day of interest, euthanize the female according to approved protocols and remove the uterus. Remove the embryos from the decidua in a 10 cm Petri dish with Hanks BSS (without phenol red). Remove the extra-embryonic membranes and do at least one rinse with Hanks BSS to remove extraneous tissues and blood.
Prepare LysoTracker Embryo Staining Solution (5 mM LysoTracker in Hanks BSS). A convenient volume for 1-2 litters is 5 ml. Gently mix to disburse the dye evenly.
Add embryos to the LysoTracker Embryo Staining Solution in a microfuge tube or vial and incubate at 37 °C for 45 min.
Rinse very gently in Hanks BSS four times, 5 min each wash.
Fix in 4% Paraformaldehyde overnight.
Rinse once in Hanks BSS, 10 min, to remove the fix.
Dehydrate through a methanol series (50%, 75%, 80%, 100%, 5 min each step) to eliminate background staining. This is important for achieving good signal-to-noise during imaging.
At this point you can store the embryo samples indefinitely in methanol at -20 °C, protected from light.
Note: A tissue sample can be taken at any step for DNA genotyping, however, it can be most convenient to take a sample just before storage so that up to this step all the samples can be batch-processed. Collect a sample of tissue from the tail, limb, or head from each embryo and put all into separate tubes. Rehydrate the tissue sample and prepare for genotyping.
2. LysoTracker Staining of Differentiating ES Cultures
Prepare embryoid bodies via the Hanging Drop Method15 with a starting density of 500 cells per 20 μl drop.
After 3 days, transfer the embryoid bodies to suspension culture on 10 cm non-adherent Petri dishes (i.e. those used for bacteriological work). Culture for 5 days, changing the media every 2 days.
On day 7, transfer the embryoid bodies to gelatinized 8-well chamber slides (add 0.1% Gelatin to the chambers, wait 5 min, remove, and add media). Transfer 1-2 embryoid bodies per chamber.
Culture for 10 days, changing media every other day. Be careful to place the pipette at the corner of the chamber slide so as to not perturb the embryoid body attaching to the glass surface. Also when adding fresh media also place the pipette at the corner of the chamber and release the media slowly.
After 10 days of culture, to induce apoptosis as a positive control, treat some of the wells with 0.1 mM and 1.0 mM H2O2 (add the H2O2 to fresh media, mix, then add to the cells) for 60 min. Rinse twice with D-PBS and culture overnight in regular media.
Gently aspirate existing media and add LysoTracker Cell staining solution (500 nM LysoTracker (final concentration) in media, 300 μl per chamber).
Incubate at 37 °C for 15 min.
Rinse twice with D-PBS 2 times, 5 min each.
Fix in 4% paraformaldehyde for 15 min at RT.
Rinse twice with D-PBS, 5 min.
3. Viewing LysoTracker Stained Embryos or ES Cultures
Prepare the cell samples for microscopy by aspirating the D-PBS; removing the chamber off the slide; air drying for about 5 min; and then adding an aqueous mounting medium (Vectashield with DAPI or similar). Place a foil barrier while drying to decrease bleaching of the fluorophore. Mount embryos in a deep-depression glass dish or small Petri dish in Vectashield or similar.
Visualize with a dissecting or compound microscope outfitted with a rhodamine or Texas Red filter (LysoTracker RED DND-99, Excitation/Emission: 577/590 nm). The staining is typically quite bright so long exposures are usually not necessary.
4. Quantification of Results Using Imaging Techniques
Take digital photos of your samples under the same exposure conditions in manual mode.
Open the images in Adobe Photoshop or a similar imaging program. To determine the amount of LysoTracker staining, select the red channel and threshold the image (converts the image to black-and-white) such that the red staining is now represented by white pixels. Select the white pixels and use the histogram function to obtain the total count.
Use the blue channel to count the number of nuclei in the field. It can be useful to keep a record of the count by annotating the image.
Record the values and then calculate the average staining level per cell. Repeat this analysis with the same threshold setting for all images you wish to sample.
5. Representative Results
There is a normal pattern of PCD during embryonic development that can change when a growth factor gene, such as Sonic hedgehog (Shh), is removed16,17 . Since LysoTracker is a small molecule dye that is highly diffusible, the whole embryo can be visualized to assess areas of PCD even deep within the mesenchyme12,13. Figure 1 shows the LysoTracker staining pattern for normal and Shh-/- mutant mouse embryos at 10.5dpc. At this stage, PCD is greatly increased in Shh-/- mutant embryos, particularly in the limb and somite regions. Portions of the embryo can be imaged in whole-mount at higher magnification (1B) to assess the overall pattern. Dissected portions can also be sectioned with a vibrating microtome (1C) or a cryostat (D-F) to reveal PCD regions in greater detail.
TUNEL analysis is best done on thin sections where penetration of the terminal transferase is less problematic. LysoTracker staining and a TUNEL assay can be done in the same tissue (Figure 1D-G). These results show that although there is not a strict 1:1 correlation between LysoTracker and TUNEL staining, the general regions of PCD overlap considerably. At a higher magnification, it is possible to see puncta that stain positive with the TUNEL assay but not with the LysoTracker dye, puncta that stain only with the LysoTracker dye, and puncta that have overlapping staining.
When an EB is plated on a gelatin-coated surface, differentiating cells will migrate outward. After 10 days of culture, cells will have differentiated into a variety of cell types including neurons, muscle cells, and cardiomyocytes. The cultures shown in Figure 2 contain mixed populations of these cell types, however one could adapt our protocol to study PCD of a particular cell-type of interest. For example, the EB could be treated with Retinoic Acid to enhance the differentiation of neurons19 , and then the LysoTracker assay could be used to assess neuronal cell death under different conditions. In the normal culture of differentiated ES cells, there are a few cells that undergo PCD and these can be detected with LysoTracker and/or TUNEL assays (Figure 2A). PCD can be induced by a number of different cytotoxic agents such as hydrogen peroxide (H2O2) which induces oxidative damage and initiates the apoptotic pathway20,21 . A low dose of H2O2 increases both LysoTracker and TUNEL staining compared to no treatment, while a high dose results in cell shrinkage and widespread LysoTracker and TUNEL positivity. Similar to the results in the embryo, it is possible to see puncta that stain positive with the TUNEL assay but not with the LysoTracker dye, puncta that stain only with the LysoTracker dye, and puncta that have overlapping staining. The consequence of the application of a cytotoxic agent that induces apopotosis is compared to serum starvation conditions which induce autophagy (Figure 2D). Cells that have undergone a period of serum starvation (1 hour) exhibit a different pattern where LysoTracker and TUNEL staining do not highly correlate. LysoTracker staining is much increased compared to untreated cultures, however TUNEL positivity is at normal levels.
Employing standard tools available in Adobe Photoshop, LysoTracker staining can be quantified with a simple thresholding method that allows one to calculate the average staining level for a population of cells. Figure 3A-B shows how a sample image looks during the quantification process. Starting with a baseline image with LysoTracker staining in the red channel and the DAPI-stained nuclei in the blue channel (panel 3A), the red channel of this image was thresholded to convert the image to black-and-white and the white pixels were then selected and counted (Figure 3A'). A thresholding level was chosen that represents the LysoTracker staining pattern. As with any quantification method that uses images, it is important to avoid overexposing your photos as this could potentially result in over-emphasizing differences. The number of the nuclei in the field can then be counted by looking at just the blue channel (Figure 3B) and marking the nuclei using a drawing tool such as a paintbrush while counting (Figure 3B'). One can then express the level of staining by calculating the average level of staining per cell (number of pixels/nuclei count). If one wishes to use this algorithm for high through-put screening, most screening packages can measure brightness in a particular channel and also have an object identifier function that will count nuclei in a field, however for most typical applications the manual method is quick and simple. To give an example, a sample region from Figures 2A-C was quantified and the results confirm that increasing concentrations of H2O2 results in a higher level of LysoTracker staining (Figure 3C).
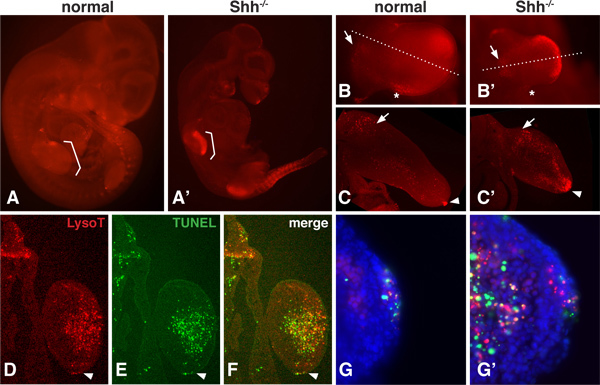 Figure 1. LysoTracker staining in normal and Shh-/- embryos. A,A'. Normal and Shh-/-mouse embryos at 10.5 dpc, stained with LysoTracker Red, mounted in Vectashield and photographed under an epiflourescence dissecting microscope. Brackets indicate the location of the forelimbs. B,B'. Forelimb of normal and Shh-/- embryos imaged at higher magnification. Note the normal domains of cell death including the 'Posterior Necrotic Zone' (marked with an asterisk) and a region in the proximal mesenchyme of the limb bud (marked with an arrow). Shh null limbs exhibit stronger staining in the medial and distal portion. C,C'. Medial sections through the forelimb bud of both normal and null limb buds. Note the presence of staining in the proximal mesenchyme (arrow) and AER (arrowhead) of the normal limb bud and the strong staining within the dorsal and ventral and distal mesenchyme within the null limb bud. D-F. The same section (12 micron, frozen section) was imaged for LysoTracker and TUNEL staining (Roche 1684795). The section passes through the distal portion of the Shh null mesenchyme. The arrowhead points to the AER. Note how the overall pattern is very similar using both methods, however the staining does not overlap completely. G,G'. At a higher magnification of the AER and mesenchyme, one can see both the presence of puncta that are LysoTracker or TUNEL-positive only as well as puncta that have overlapping staining.
Figure 1. LysoTracker staining in normal and Shh-/- embryos. A,A'. Normal and Shh-/-mouse embryos at 10.5 dpc, stained with LysoTracker Red, mounted in Vectashield and photographed under an epiflourescence dissecting microscope. Brackets indicate the location of the forelimbs. B,B'. Forelimb of normal and Shh-/- embryos imaged at higher magnification. Note the normal domains of cell death including the 'Posterior Necrotic Zone' (marked with an asterisk) and a region in the proximal mesenchyme of the limb bud (marked with an arrow). Shh null limbs exhibit stronger staining in the medial and distal portion. C,C'. Medial sections through the forelimb bud of both normal and null limb buds. Note the presence of staining in the proximal mesenchyme (arrow) and AER (arrowhead) of the normal limb bud and the strong staining within the dorsal and ventral and distal mesenchyme within the null limb bud. D-F. The same section (12 micron, frozen section) was imaged for LysoTracker and TUNEL staining (Roche 1684795). The section passes through the distal portion of the Shh null mesenchyme. The arrowhead points to the AER. Note how the overall pattern is very similar using both methods, however the staining does not overlap completely. G,G'. At a higher magnification of the AER and mesenchyme, one can see both the presence of puncta that are LysoTracker or TUNEL-positive only as well as puncta that have overlapping staining.
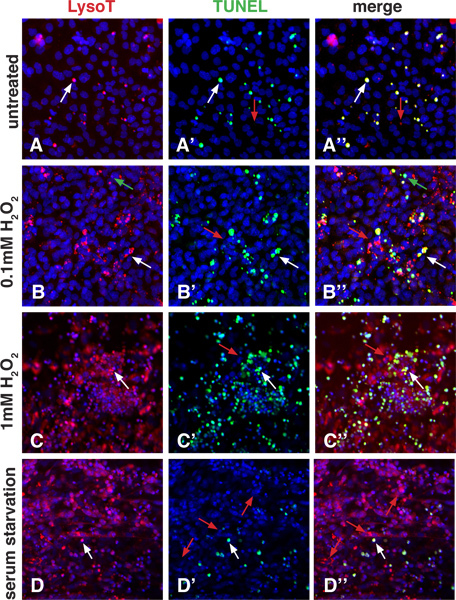 Figure 2. LysoTracker staining in differentiated ES cells. Differentiating cells were plated on 8-chamber slides in the absence of LIF. To show how LysoTracker and TUNEL staining compare, the apoptosis pathway was induced with 2 different doses of H2O2, while the autophagy pathway was induced using 1 hour of serum starvation. These images were taken with a Zeiss LSM5 Confocal Microscope. A-A'': Untreated cultures show some normal apoptosis as indicated by LysoTracker and TUNEL staining. The pattern using both techniques mostly overlaps (white arrows), however, one can see a few puncta that are LysoTracker positive only (red arrows). B-B'', C-C": Treatment with increasing amounts of H2O2 stimulates apoptosis. The lower dose shows more LysoTracker and TUNEL positive puncta when compared to untreated cells. The higher dose induces in even more LysoTracker and TUNEL positive puncta. White arrows point to overlapping staining, while red arrows point to puncta that are positive for LysoTracker only. It is also possible to see puncta that are only positive for TUNEL staining (green arrows). D-D": Serum starvation can induce autophagy, which can stimulate an accumulation of material in lysosomes and autophagolysosomes. Note how many more LysoTracker puncta are visible compared to untreated cultures. TUNEL positive puncta are present (most of which have overlapping LysoTracker staining, white arrows), however, the number is not greatly increased compared to untreated conditions. In addition, under serum starvation conditions, LysoTracker-only positive puncta are highly prevalent (red arrows).
Figure 2. LysoTracker staining in differentiated ES cells. Differentiating cells were plated on 8-chamber slides in the absence of LIF. To show how LysoTracker and TUNEL staining compare, the apoptosis pathway was induced with 2 different doses of H2O2, while the autophagy pathway was induced using 1 hour of serum starvation. These images were taken with a Zeiss LSM5 Confocal Microscope. A-A'': Untreated cultures show some normal apoptosis as indicated by LysoTracker and TUNEL staining. The pattern using both techniques mostly overlaps (white arrows), however, one can see a few puncta that are LysoTracker positive only (red arrows). B-B'', C-C": Treatment with increasing amounts of H2O2 stimulates apoptosis. The lower dose shows more LysoTracker and TUNEL positive puncta when compared to untreated cells. The higher dose induces in even more LysoTracker and TUNEL positive puncta. White arrows point to overlapping staining, while red arrows point to puncta that are positive for LysoTracker only. It is also possible to see puncta that are only positive for TUNEL staining (green arrows). D-D": Serum starvation can induce autophagy, which can stimulate an accumulation of material in lysosomes and autophagolysosomes. Note how many more LysoTracker puncta are visible compared to untreated cultures. TUNEL positive puncta are present (most of which have overlapping LysoTracker staining, white arrows), however, the number is not greatly increased compared to untreated conditions. In addition, under serum starvation conditions, LysoTracker-only positive puncta are highly prevalent (red arrows).
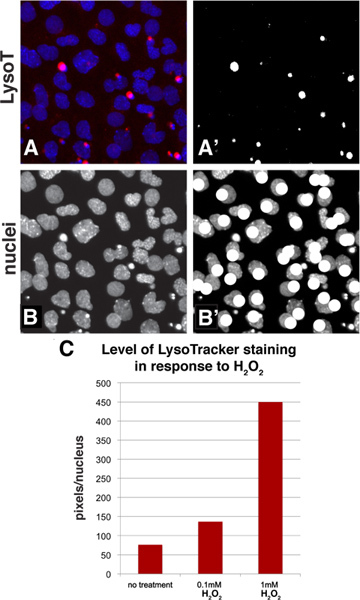 Figure 3. Quantification method to assess the level of staining in a population of cells using Adobe Photoshop. A: A portion of Figure 2A was chosen and the LysoTracker (red channel) and DAPI staining (nuclei, blue channel) is shown in this panel. A': The red channel of Figure 3A was thresholded to convert it to a black-and-white image. The white pixels can then be selected and counted using the histogram function. B-B': The blue channel shows the nuclei and while counting them, the image can be annotated with a drawing function such as a paintbrush. C: This is an example quantification of a sample from panels 2A-C. The data is represented as pixels per nuclei. Click here to view larger figure.
Figure 3. Quantification method to assess the level of staining in a population of cells using Adobe Photoshop. A: A portion of Figure 2A was chosen and the LysoTracker (red channel) and DAPI staining (nuclei, blue channel) is shown in this panel. A': The red channel of Figure 3A was thresholded to convert it to a black-and-white image. The white pixels can then be selected and counted using the histogram function. B-B': The blue channel shows the nuclei and while counting them, the image can be annotated with a drawing function such as a paintbrush. C: This is an example quantification of a sample from panels 2A-C. The data is represented as pixels per nuclei. Click here to view larger figure.
Discussion
Important hallmarks of apoptosis include the detection of DNA damage with the TUNEL assay, along with the detection of caspase activity (detected with mAbs or a binding molecule such as ZVAD-fmk), the observation of cellular changes, and the presentation of phosphatidylserine (PS) on the membrane surface (detected by Annexin V binding). There are some disadvantages with each of these assays. For example, the terminal transferase used in the TUNEL assay does not penetrate beyond a few cell layers. Also Annexin V can mark cells that are not undergoing apoptosis such as those that have membrane damage (Annexin V gets inside the cell and marks the PS on the inner leaflet). In addition caspase assays can detect caspase activity that is not associated with apoptosis. LysoTracker is not a substitute for these assays, however it can be a useful complement assay depending on the context.
LysoTracker staining has several advantages over other methods. Because it is a small molecule dye as opposed to a protein such as terminal transferase (used in the TUNEL assay) or Annexin V, it readily diffuses through thick tissues. This means that LysoTracker staining is particularly useful to look for changes in the overall pattern of apoptosis. Thus, a particularly useful application of LysoTracker staining is as a first-pass assay to determine patterns of PCD in embryos or cells of different genotypes or to look at the overall effect of the application of certain toxins or drug treatments. In addition, the high signal-to-noise ratio of the dye, its ease of use (cells or tissues can simply be immersed), its ability to be fixed and the endurance of the fluorophore make the use of LysoTracker ideal for high-volume screening of cells or tissues that may exhibit PCD.
As with any assay for PCD, however, the context of the experiment is important to consider. During early mouse embryonic development, in the absence of an immune system, dying cells can be phagocytosed by neighboring cells22. It is not known how quickly this occurs. However, since it is possible to find TUNEL positive puncta that are not LysoTracker positive (Figure 1G), cells may be undergoing the later stages of apoptosis (DNA fragmentation) before being removed by phagocytosis. When TUNEL and LysoTracker staining overlap, this may indicate that a cell or cell fragment containing fragmented DNA is being phagocytosed. Finally, LysoTracker-positive-only puncta could indicate cellular debris that does not contain DNA but is being cleared by neighboring cells. Thus, during early mouse development, regardless of the assay (TUNEL or LysoTracker), the staining pattern can indicate both cells that are dying and neighboring cells containing debris.
Our results (Figure 2A-C) show that LysoTracker can be very useful for assessing PCD in cultured differentiating ES cells and therefore is likely to be useful for a variety of cultured cell types. Interestingly, when cells are stimulated with H2O2 to undergo apoptosis, most TUNEL positive puncta are also LysoTracker positive (Figure 2B", 2C"). This suggests that in monolayer culture, neighboring cells may still be able to phagocytose nearby dying cells. Indeed with a considerably higher dose of H2O2 (10mM), the vast majority of cells contain TUNEL positive puncta and there is relatively little LysoTracker staining, suggesting that when so many cells are dying, none are healthy enough to act as non-professional phagocytes (data not shown). An important point that is not well appreciated is that regardless of assay, it may be difficult to determine if one is looking at a dying cell that has high lysosomal activity or the corpse of a dying cell being engulfed by a neighbor cell, especially when cells are in close proximity.
LysoTracker has also been a useful tool for assessing autophagy23-26 and in response to serum starvation, differentiated ES cells display a marked increase in the number of LysoTracker staining puncta (compare Figure 2D with 2A). This likely indicates an increase in the formation of autophagolysosomes and lysosomes in response to decreased nutrient availability (as has been shown in other contexts using LysoTracker24,25). In contrast, TUNEL staining, while still largely overlapping with LysoTracker staining, is not greatly increased, indicating that although the cells experienced stress from loss of serum, after 24 hours, this stress was not sufficient to induce apoptosis.
Thus one caveat to consider (or potential advantage, depending on what one wants to assay) is that while LysoTracker can mark regions of apoptosis (dying cells as well as the neighboring non-professional phagocytes), it can also mark cells that are undergoing autophagy. To determine the precise mechanism of PCD, it is best to do several assays and if possible, use high magnification optical or electron microscopy techniques to distinguish between the various possibilities. In summary, these examples show that LysoTracker is a great tool for assessing PCD but that the context of the assay should be considered carefully since LysoTracker positive puncta could represent an increase in lysosome activity due to non-professional phagocytosis or due to autophagy27,28.
Disclosures
No conflicts of interest declared.
Acknowledgments
We thank members of the Mariani lab for help editing the protocol. This work was funded by a CIRM Postdoctoral Training Grant (JLF), a CIRM BRIDGES internship (TZTT), the Robert E. and May R. Wright Foundation (FVM), and the University of Southern California (FVM).
References
- Baehrecke EH. How death shapes life during development. Nat. Rev. Mol. Cell Biol. 2002;3:779–787. doi: 10.1038/nrm931. [DOI] [PubMed] [Google Scholar]
- Meier P, Finch A, Evan G. Apoptosis in development. Nature. 2000;407:796–801. doi: 10.1038/35037734. [DOI] [PubMed] [Google Scholar]
- Ikonomidou C. Ethanol-induced apoptotic neurodegeneration and fetal alcohol syndrome. Science. 2000;287:1056–1060. doi: 10.1126/science.287.5455.1056. [DOI] [PubMed] [Google Scholar]
- Dunty WC, Chen SY, Zucker RM, Dehart DB, Sulik KK. Selective vulnerability of embryonic cell populations to ethanol-induced apoptosis: implications for alcohol-related birth defects and neurodevelopmental disorder. Alcohol Clin. Exp. Res. 2001;25:1523–1535. [PubMed] [Google Scholar]
- Price OT, Lau C, Zucker RM. Quantitative fluorescence of 5-FU-treated fetal rat limbs using confocal laser scanning microscopy and Lysotracker. Cytometry A. 2003;53:9–21. doi: 10.1002/cyto.a.10036. [DOI] [PubMed] [Google Scholar]
- Jacobson MD, Weil M, Raff MC. Programmed cell death in animal development. Cell. 1997;88:347–354. doi: 10.1016/s0092-8674(00)81873-5. [DOI] [PubMed] [Google Scholar]
- Fuchs Y, Steller H. Programmed cell death in animal development and disease. Cell. 2011;147:742–758. doi: 10.1016/j.cell.2011.10.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qu X. Autophagy gene-dependent clearance of apoptotic cells during embryonic development. Cell. 2007;128:931–946. doi: 10.1016/j.cell.2006.12.044. [DOI] [PubMed] [Google Scholar]
- Grimsley C, Ravichandran KS. Cues for apoptotic cell engulfment: eat-me, don't eat-me and come-get-me signals. Trends Cell Biol. 2003;13:648–656. doi: 10.1016/j.tcb.2003.10.004. [DOI] [PubMed] [Google Scholar]
- Lauber K, Blumenthal SG, Waibel M, Wesselborg S. Clearance of apoptotic cells: getting rid of the corpses. Mol. Cell. 2004;14:277–287. doi: 10.1016/s1097-2765(04)00237-0. [DOI] [PubMed] [Google Scholar]
- Haller T, Dietl P, Deetjen P, Volkl H. The lysosomal compartment as intracellular calcium store in MDCK cells: a possible involvement in InsP3-mediated Ca2+ release. Cell Calcium. 1996;19:157–165. doi: 10.1016/s0143-4160(96)90084-6. [DOI] [PubMed] [Google Scholar]
- Zucker RM, Hunter S, Rogers JM. Confocal laser scanning microscopy of apoptosis in organogenesis-stage mouse embryos. Cytometry. 1998;33:348–354. doi: 10.1002/(sici)1097-0320(19981101)33:3<348::aid-cyto9>3.0.co;2-c. [DOI] [PubMed] [Google Scholar]
- Zucker RM, Hunter ES, 3rd, Rogers JM. Apoptosis and morphology in mouse embryos by confocal laser scanning microscopy. Methods. 1999;18:473–480. doi: 10.1006/meth.1999.0815. [DOI] [PubMed] [Google Scholar]
- Gavrieli Y, Sherman Y, Ben-Sasson SA. Identification of programmed cell death in situ via specific labeling of nuclear DNA fragmentation. J. Cell Biol. 1992;119:493–501. doi: 10.1083/jcb.119.3.493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foty R. A Simple Hanging Drop Cell Culture Protocol for Generation of 3D Spheroids. J. Vis. Exp. 2011. p. e2720. [DOI] [PMC free article] [PubMed]
- Chiang C. Manifestation of the limb prepattern: limb development in the absence of sonic hedgehog function. Dev. Biol. 2001;236:421–435. doi: 10.1006/dbio.2001.0346. [DOI] [PubMed] [Google Scholar]
- Ishibashi M, McMahon AP. A sonic hedgehog-dependent signaling relay regulates growth of diencephalic and mesencephalic primordia in the early mouse embryo. Development. 2002;129:4807–4819. doi: 10.1242/dev.129.20.4807. [DOI] [PubMed] [Google Scholar]
- Schuldiner M. Induced neuronal differentiation of human embryonic stem cells. Brain Res. 2001;913:201–205. doi: 10.1016/s0006-8993(01)02776-7. [DOI] [PubMed] [Google Scholar]
- Bain G, Kitchens D, Yao M, Huettner JE, Gottlieb DI. Embryonic stem cells express neuronal properties in vitro. Dev. Biol. 1995;168:342–357. doi: 10.1006/dbio.1995.1085. [DOI] [PubMed] [Google Scholar]
- Dumont A. Hydrogen peroxide-induced apoptosis is CD95-independent, requires the release of mitochondria-derived reactive oxygen species and the activation of NF-kappaB. Oncogene. 1999;18:747–757. doi: 10.1038/sj.onc.1202325. [DOI] [PubMed] [Google Scholar]
- Hampton MB, Orrenius S. Dual regulation of caspase activity by hydrogen peroxide: implications for apoptosis. FEBS Lett. 1997;414:552–556. doi: 10.1016/s0014-5793(97)01068-5. [DOI] [PubMed] [Google Scholar]
- Wood W. Mesenchymal cells engulf and clear apoptotic footplate cells in macrophageless PU.1 null mouse embryos. Development. 2000;127:5245–5252. doi: 10.1242/dev.127.24.5245. [DOI] [PubMed] [Google Scholar]
- Scott RC, Juhasz G, Neufeld TP. Direct induction of autophagy by Atg1 inhibits cell growth and induces apoptotic cell death. Curr. Biol. 2007;17:1–11. doi: 10.1016/j.cub.2006.10.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez-Enriquez S, Kim I, Currin RT, Lemasters JJ. Tracker dyes to probe mitochondrial autophagy (mitophagy) in rat hepatocytes. Autophagy. 2006;2:39–46. doi: 10.4161/auto.2229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bampton ET, Goemans CG, Niranjan D, Mizushima N, Tolkovsky AM. The dynamics of autophagy visualized in live cells: from autophagosome formation to fusion with endo/lysosomes. Autophagy. 2005;1:23–36. doi: 10.4161/auto.1.1.1495. [DOI] [PubMed] [Google Scholar]
- Boya P, Mellen MA, de la Rosa EJ. How autophagy is related to programmed cell death during the development of the nervous system. Biochem. Soc. Trans. 2008;36:813–817. doi: 10.1042/BST0360813. [DOI] [PubMed] [Google Scholar]
- Galluzzi L. Guidelines for the use and interpretation of assays for monitoring cell death in higher eukaryotes. Cell Death Differ. 2009;16:1093–1107. doi: 10.1038/cdd.2009.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klionsky DJ. Guidelines for the use and interpretation of assays for monitoring autophagy in higher eukaryotes. Autophagy. 2008;4:151–175. doi: 10.4161/auto.5338. [DOI] [PMC free article] [PubMed] [Google Scholar]