

Abstract
Classical methods for studying neuronal circuits are fairly low throughput. Transsynaptic viruses, particularly the pseudorabies (PRV) and rabies virus (RABV), and more recently vesicular stomatitis virus (VSV), for studying circuitry, is becoming increasingly popular. These higher throughput methods use viruses that transmit between neurons in either the anterograde or retrograde direction.
Recently, a modified RABV for monosynaptic retrograde tracing was developed. (Figure 1A). In this method, the glycoprotein (G) gene is deleted from the viral genome, and resupplied only in targeted neurons. Infection specificity is achieved by substituting a chimeric G, composed of the extracellular domain of the ASLV-A glycoprotein and the cytoplasmic domain of the RABV-G (A/RG), for the normal RABV-G1. This chimeric G specifically infects cells expressing the TVA receptor1. The gene encoding TVA can been delivered by various methods2-8. Following RABV-G infection of a TVA-expressing neuron, the RABV can transmit to other, synaptically connected neurons in a retrograde direction by nature of its own G which was co-delivered with the TVA receptor. This technique labels a relatively large number of inputs (5-10%)2 onto a defined cell type, providing a sampling of all of the inputs onto a defined starter cell type.
We recently modified this technique to use VSV as a transsynaptic tracer9. VSV has several advantages, including the rapidity of gene expression. Here we detail a new viral tracing system using VSV useful for probing microcircuitry with increased resolution. While the original published strategies by Wickersham et al.4 and Beier et al.9 permit labeling of any neurons that project onto initially-infected TVA-expressing-cells, here VSV was engineered to transmit only to TVA-expressing cells (Figure 1B). The virus is first pseudotyped with RABV-G to permit infection of neurons downstream of TVA-expressing neurons. After infecting this first population of cells, the virus released can only infect TVA-expressing cells. Because the transsynaptic viral spread is limited to TVA-expressing cells, presence of absence of connectivity from defined cell types can be explored with high resolution. An experimental flow chart of these experiments is shown in Figure 2. Here we show a model circuit, that of direction-selectivity in the mouse retina. We examine the connectivity of starburst amacrine cells (SACs) to retinal ganglion cells (RGCs).
Keywords: Neuroscience, Issue 68, Genetics, Molecular Biology, Virology, Virus, VSV, transsynaptic tracing, TVA, retrograde, neuron, synapse
Protocol
1. Making Virus from cDNA: Recovery of VSV from cDNA using Vaccinia-T7 System10
One day prior to experiment, split BsrT7 cells into 60 mm dish containing DMEM + 10% FBS. Seed 2E6 cells per dish. BsrT7 cells are derived from BHK21, or baby hamster kidney, cells.
Warm PBS with 1 mM magnesium and 1 mM calcium to 37 °C.
Obtain a small aliquot vTF7-3, a vaccinia virus expressing T7 polymerase, and thaw to room temperature. vTF7-3 is an infectious vaccinia virus expressing the T7 polymerase11, and should be used only in Biosafety level 2 containment. This expression system is used to generate maximal levels of the desired transcripts from plasmids transfected in step 1.9. Vortex virus for 30 sec at max speed. Sonicate in a tabletop sonicator for 2 min in water bath at room temperature. Vortex again for 30 sec to break up viral clumps.
Remove media from the BSRT7 cells.
Add 11.7 μl vTF7-3 to 600 μl PBS with magnesium and calcium and add mixture onto the cells.
Move plates into a 34 °C incubator with 5% CO2. Rock plates gently once every 10 min.
After 45 min have elapsed, begin transfection. Add 20 μl lipofectamine 2,000 to 1 ml DMEM and mix. Wait 5 min.
Add 5.25 μg pN, 2.3 μg pP, 1.2 μg pl, 1 μg pCAG RABV-G, and 6 μg VSV genomic cDNA to 500 μl DMEM. pN, pP and pl are plasmids driving expression of VSV viral genes under the T7 promoter10, with the amounts determined empirically for optimum rescue of the virus.
Combine both tubes and mix. Wait 15 min at room temperature.
Remove PBS and wash the plates once with 1.5 ml DMEM.
Aspirate rinse media. Add transfection mix to plate. Move plates into 34 °C incubator.
Wait 5 hr, then remove media.
Change media to 4 ml DMEM + 2% FBS + 1x Penicillin-Streptomycin + 10 mM HEPES pH 7.4. The addition of Penicillin-Streptomycin, to prevent contamination, and HEPES, to maintain the pH of the media, are very important. The FBS content can be dropped to 2%, as the cells will be destroyed in a matter of days by the virus and thus do not need 10% FBS.
Place plates into 34 °C incubator for 48-72 hr.
Collect media at 3 and 6 days post-transfection, and immediately syringe filter through a 0.20 μm filter. This size filter removes the vaccinia virus, but not the VSV.
Since we want to supply RABV-G to the viral envelope, and the genome does not encode the RABV-G gene, it must be continually supplied in trans. To do this, make a separate plate of 293T TVA800 (TVA800) cells. These are cells that constitutively express the TVA receptor, permitting EnvA-mediated viral infection12. At 80% confluency, change media to DMEM only, then transfect the cells with 5 μg of plasmid encoding G protein (i.e. pCAG RABV-G) per plate. We use the PEI transfection reagent, a polycationic polymer which is much cheaper than comparable lipid-based methods. However, lipofectamine can also be used effectively for this purpose. The optimum PEI : DNA ratio needs to be determined empirically. Do this in a separate experiment with a fluorescent reporter, i.e. pCAG GFP. We use 10 μl PEI: 4 μg DNA.
Apply the supernatant from step 16 onto the transfected plate of TVA800 cells.
Observe this plate the following day for evidence of fluorescence of the virally expressed fluorophore. Viral fluorescence is first observed as a single cell, and gradually spreads over time (i.e.Figure 3). The virus should spread within a few hours.
2. Passage and Concentration of VSV
Split TVA800 cells into media containing DMEM + 10% FBS such that you will have four 10-cm plates at ~80% confluency the next day. 293T-based cells work well due to their high transfectability, and the use of TVA-expressing cells enhances the speed at which the cells in the dish become infected. Other highly transfectable cell lines may serve the purpose as well.
At 80% confluency, change media to DMEM only, then transfect the cells with 5 μg of plasmid encoding the G protein that you want to use (i.e. pCAG RABV-G) per plate. We use PEI, but FUGENE/Lipofectamine work as well.
Wait one day after the transfection for glycoprotein expression/surface accumulation. Change the media (5 ml/plate) to DMEM + 10% FBS, then infect with rVSV(A/RG) virus at an multiplicity of infection (MOI) of 0.01 < x < 0.1.
Check the amount of infection 24 hr later. You should see infected cells (identified by GFP fluorescence), some with surrounding patches of infected cells (i.e.Figure 3). Typically it's only worth collecting the supernatant if you see >50% of cells infected, otherwise the viral titers obtained from concentration will be suboptimal.
If > 50%, collect the 5 ml of media/plate, and replace with 5 ml of fresh media. If too few cells are infected, wait for another 12-24 hr, and check again. Freeze the collected supernatants at -80 °C.
Collect every 24 hr thereafter for 3 days, for a total of 4 days.
Altogether, if you started with 4 plates, you should have 20 ml of media from each day - so 80 ml total, or 2 ultracentrifuge tubes worth of supernatant. Thaw the supernatants at 37 °C and filter over 0.45 μm filters on ice. Aliquot 36 ml of virus into Beckman centrifuge tubes.
Concentrate the supernatant (21,000K in an SW28 rotor (=80,000 x g) for 90 min) at 4 °C. Decant the supernatant into a beaker containing 10% bleach, and while the tube is inverted, use an aspirator to remove as much media from the side of the tube as possible.
While covered. shake the viral tubes at 4 °C for 1 hr. A standard shaker at about 120 rpm is sufficient.
Gently triturate pellet in the remaining media by gently pipetting up and down 30 times. Be careful not to introduce air bubbles. The remaining media volume should be about 30 μl. Aliquot this volume into multiple tubes so as to prevent multiple freeze-thaw cycles for any particular stock, as this can result in a decrease in viral titer. These aliquots can be frozen at -80 °C.
Titer the virus. Titering at about 2 days post-infection is optimal. Initial infection can be observed in a matter of hours, but you get an underrepresentation of titer if you wait only 1 day. In order to obtain the viral titer, perform a limiting dilution experiment of the concentrated virus onto the appropriate cell line (293T cells work well) in a 24-well plate, diluting the virus 10-fold from 1 to 1 : 1,000,000. The titers we get are typically in the range of 108-1010 focus forming units (ffu)/ml.
3. Virus Injection
We use mice, typically between 6-10 weeks of age, for these experiments. Any age after which circuitry is established would suffice. We will choose the transmission from retinal ganglion cells (RGCs) to starburst amacrine cells (SACs) as an example. This experiment, as diagramed in Figure 2, requires a triple transgenic animal and only a single injection of virus. (All procedures shown were approved by the IACUC at Harvard Medical School, and were in accordance with institutional guidelines).
First, the proper animals need to be generated. The goal is to have TVA expressed in the cell type that one wishes to map into, by means of a virus (i.e. adeno-associated virus, AAV), or by a transgenic allele. The cell type specificity is usually obtained by Cre expression. Here, we also crossed in a conditional expression TVA allele7 to a Choline Acetyltransferase (ChAT)-Cre allele13, such that TVA is expressed in all cells with a Cre expression history (the SACs). We also crossed a conditional expression tdTomato allele (R26TdT) for visualization of TVA-expressing cells.
The coordinates for the injection location need to be identified. These coordinates of interest can be found in a brain atlas, i.e. the Franklin and Paxinos mouse brain atlas14. These coordinates need to be noted relative to a given landmark on the mouse skull. We use bregma, but any coordinate system is sufficient.
Prepare the stereotaxic apparatus. Turn on the microinjector controller, and move the plunger and electrode holder into place. Then back-fill the injection needle with mineral oil to provide an interface with the virus. Put the needle then onto the plunger, making sure that no air bubbles remain in the needle.
Thaw the virus, and place on ice to prevent a drop in viral titer. Set the microinjector to "withdraw" and move the tube of virus such that that is contacting the capillary on the microinjector. Withdraw the necessary amount of rVSV(A/RG) pseudotyped with RABV-G for the experiment.
Animals are given a pre-operative dose of buprenorphine (0.05-0.1 mg/kg) before the surgery begins. For anaesthetizing the animal, either isoflurane inhalation, or an intraperitoneal injection of a ketamine (40-80 mg/kg) and xylazine (5-10 mg/kg) mixture is sufficient. Choice of anesthetic is dependent on the user. A toe pinch is then performed to ensure that animals are fully anesthetized; if the animal does not respond, you can proceed with procedure. Be sure to obtain proper licenses before using these drugs.
Animals are placed on a heated pad, which is propped on a movable stand for positioning the mouse. The animal remains there throughout the duration of the surgical procedure. Apply eye ointment to prevent drying of the cornea while the animal is anesthetized.
The mouse's head then needs to be stabilized in the stereotaxic apparatus. Use the ear bars on the stereotaxic apparatus to ensure that the head is thoroughly secured, such that the head is parallel to the ground, and does not rotate laterally. Once this is completed, adjust the bite bar to help stabilize the head.
The animal's fur is shaved in the region of incision on top of the head. This area is then washed three times with ethanol, followed by three times with iodine. An incision is then made in the skin with a scalpel, revealing the skull.
Once the mouse and injection setup are prepared, bregma needs to be located. This is the area in the center of the head that the sagittal and two coronal sutures meet. Once this area is located, the dials on the stereotaxic apparatus can be used to adjust the microinjector to the proper coordinates. Here, we inject the lateral geniculate nucleus (LGN), using coordinates A/P -2.46 from bregma, L/M 2, D/V 2.75.
Once the coordinates have been dialed in, the drill must be assembled. Place the drill bit into the drill, then turn on the drill. A small hole should be drilled into the site identified as the injection site. Be very gentle with the drill in order to not cause damage to the brain parenchyma. When the bone is very thin, blood vessels on the dura become clear. At this point, carefully perforate the edges of the craniotomy with a small (30 gauge) needle and a fine (#5) forceps to remove the remaining bone.
Adjust the capillary to a position just dorsal of the brain surface, where the hole was just drilled. The needle should be sharp enough to penetrate the dura mater with ease. Then lower the needle to the desired depth, and begin injection. We inject virus at a rate of 100 nl/min.
The injection procedure to this point takes roughly ten minutes. Following injection, the needle remains in the brain for about seven minutes. This is crucial, as it permits diffusion of virus from the injection site. Rapidly removing the needle form the injection site results in a reduced infection efficiency at the site of injection, and infection along the needle tract.
The needle is very slowly raised from the injection site, over a span of about two minutes. This is again intended to reduce infection along the needle tract.
Once removed, the skin is then sutured. Animals are monitored continuously until they recover from anesthesia. Buprenorphine (0.05-0.1 mg/kg) is administered every 12 hr for 2 days.
4. Harvesting Tissue/Tissue Preparation
For harvesting the tissue of interest, the optimal time depends on the goals of the experiment. Generally, VSV transsynaptic spread can first be observed by less than 24 hr post-injection. However, waiting longer times will result in more spread.
To harvest the retina, animals are first euthanized using carbon dioxide inhalation and cervical dislocation is performed. The eyeball is then removed, and placed into a tube containing 4% formaldehyde in PBS for 1 hr at room temperature.
The eyeball is then placed into a Petri dish with PBS. The retina is then dissected away from other tissues.
For whole mount analysis, the retinae are cut so as to lay flat, placed onto a glass slide, then mounted with prolong gold mounting media and covered with a zero-thickness glass coverslip. Silicone spacers are placed between the slide and coverslip to minimize compression forces on the retina. For section analysis, the retina is placed in 30% sucrose in PBS until the retina submerges. It is then placed into a mixture of 50% OCT and 50% 30% sucrose solution for 3 hr. It is then flash frozen, and kept at -80 °C until ready to cut.
5. Representative Results
The virus that is rescued from cDNA should be able to infect TVA800 cells, but not 293T cells (Figure 3). Supplying the RABV-G permits the infection of multiple cell types, including 293T. However, since the A/RG genome is encoded in the genome, spread among cells occurs at a much higher rate in TVA-expressing cells.
Once the virus is concentrated, typical titers range between 108 ffu/ml and 1010 ffu/ml. These titers are adequate for use in vivo.
During LGN injection into the mouse, we do not distinguish dorsal LGN (dLGN) from ventral LGN (vLGN), as both typically become infected. Therefore, multiple RGC types become labeled (Figure 4), including those that project to the vLGN, such as melanopsin RGCs (Figure 4D) and ON-DSGCs (Figure 4E), and those that project to the dLGN, such as the small-arbor RGCs (Figure 4C) and ON-OFF-DSGCs (Figure 4F). Although more than one type of RGC transmitted virus to SACs, as reported elsewhere (Beier et al., in review), here we focus only on the well-studied connections of ON-OFF-DSGCs to SACs.
When we inject mice of the genotype cTVA(+)/ChAT-Cre(-), where no TVA should be expressed, only RGCs are labeled (i.e.Figure 4). These are due to initial uptake of the RABV-G pseudotyped virus by the RGCs, and no spread occurs. No RGCs are infected from an LGN infection of rVSV(A/RG) virus not pseudotyped with RABV-G, due to an inability of these virions to transverse the long axons of these RGCs. However, when injecting cTVA(+)/ChATCre(+) (and R26TdT(+)) mice into the LGN, viral spread does occur, only to red cells, which express the Cre reporter (i.e.Figure 5). These cells also co-label with the anti-ChAT antibody, and stratify only in the two ChAT laminae, confirming their identity as SACs (Figures 5B', C'). The number of virally labeled SACs per DSGC ranged from one to nine.
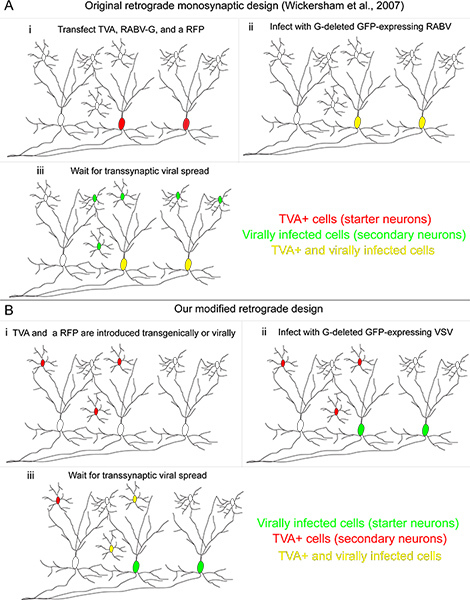 Figure 1. Our retrograde transsynaptic tracing system compared to the originally developed method. (A) (i) In the method developed by Wickersham and colleagues, "starter cells" are transfected with three genes: the TVA receptor, used to permit infection specifically of transfected cells; the RABV-G, used to complement the RABV, which itself had the G gene deleted; and a red fluorescent protein to identify transfected cells. (ii) A RABV pseudotyped with the A/RG protein infects TVA-expressing cells, making them yellow. (iii) Retrograde transsynaptic transmission occurs to upstream neurons. (B) (i) In our method, TVA-expressing cells are defined genetically, and are labeled with a conditional red fluorescent protein. (ii) The "starter cells" are those infected by a VSV encoding the A/RG gene in the viral genome. These starter cells do not express the TVA receptor. (iii) Retrograde transsynaptic transmission occurs to TVA-expressing neurons only if these cells provide synaptic input onto the starter neurons. Click here to view larger figure.
Figure 1. Our retrograde transsynaptic tracing system compared to the originally developed method. (A) (i) In the method developed by Wickersham and colleagues, "starter cells" are transfected with three genes: the TVA receptor, used to permit infection specifically of transfected cells; the RABV-G, used to complement the RABV, which itself had the G gene deleted; and a red fluorescent protein to identify transfected cells. (ii) A RABV pseudotyped with the A/RG protein infects TVA-expressing cells, making them yellow. (iii) Retrograde transsynaptic transmission occurs to upstream neurons. (B) (i) In our method, TVA-expressing cells are defined genetically, and are labeled with a conditional red fluorescent protein. (ii) The "starter cells" are those infected by a VSV encoding the A/RG gene in the viral genome. These starter cells do not express the TVA receptor. (iii) Retrograde transsynaptic transmission occurs to TVA-expressing neurons only if these cells provide synaptic input onto the starter neurons. Click here to view larger figure.
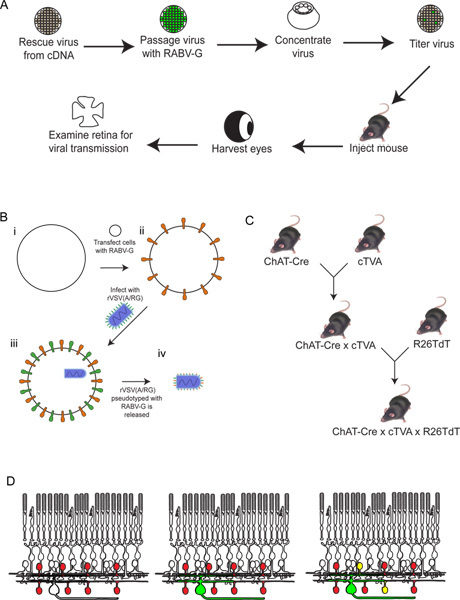 Figure 2. Schematic of the experimental procedure. (A) First, the virus is rescued from cDNA. It is then passaged and pseudotyped with RABV-G, and concentrated and titered. This virus is then injected into the brain, where it is allowed to incubate for the desired period of time. After this time, the eye is harvested, and the retina dissected and analyzed. (B) Schematic of pseudotyping the virus with RABV-G. This is necessary for infection of retinal ganglion cells from a brain injection. The RABV-G gene is first transfected into tissue culture cells expressing the TVA receptor. These cells are then infected with the rVSV(A/RG) virus. The virions released will have RABV-G in the viral envelope, but will not have the gene for RABV-G in the viral genome. (C) A schematic of the mouse crosses necessary for triple transgenic animals. Conditional TVA and tdTomato alleles are crossed to a Cre driver of choice, such that both TVA and tdTomato are expressed only in cells with a Cre expression history. In this example, we used ChAT-Cre. (D) A schematic of the retinal infection in order to trace DSGC-SAC circuits. The ChAT cells are made to express TVA and tdTomato, as shown in (C). RGCs are infected from an LGN infection of the virus produced in (B). As the virus expresses GFP, these RGCs will be green. The virus will then spread only to TVA-expressing ChAT amacrine cells if they are connected to the labeled RGC. These cells will be both red and green, or yellow. Click here to view larger figure.
Figure 2. Schematic of the experimental procedure. (A) First, the virus is rescued from cDNA. It is then passaged and pseudotyped with RABV-G, and concentrated and titered. This virus is then injected into the brain, where it is allowed to incubate for the desired period of time. After this time, the eye is harvested, and the retina dissected and analyzed. (B) Schematic of pseudotyping the virus with RABV-G. This is necessary for infection of retinal ganglion cells from a brain injection. The RABV-G gene is first transfected into tissue culture cells expressing the TVA receptor. These cells are then infected with the rVSV(A/RG) virus. The virions released will have RABV-G in the viral envelope, but will not have the gene for RABV-G in the viral genome. (C) A schematic of the mouse crosses necessary for triple transgenic animals. Conditional TVA and tdTomato alleles are crossed to a Cre driver of choice, such that both TVA and tdTomato are expressed only in cells with a Cre expression history. In this example, we used ChAT-Cre. (D) A schematic of the retinal infection in order to trace DSGC-SAC circuits. The ChAT cells are made to express TVA and tdTomato, as shown in (C). RGCs are infected from an LGN infection of the virus produced in (B). As the virus expresses GFP, these RGCs will be green. The virus will then spread only to TVA-expressing ChAT amacrine cells if they are connected to the labeled RGC. These cells will be both red and green, or yellow. Click here to view larger figure.
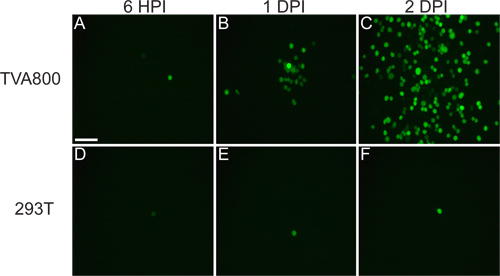 Figure 3. Behavior of rVSV(A/RG) virus on 293T and TVA800 cells. These cells were infected with a low titer rVSV(A/RG) pseudotyped with RABV-G. The virus spreads between TVA-expressing cells in a matter of hours, as shown at 6 hours post infection (HPI), 1 and 2 days post infection (DPI). However, the spread on 293T cells, which do not express the TVA receptor, though it does occur, happens much slower. Scale bar = 100 μm.
Figure 3. Behavior of rVSV(A/RG) virus on 293T and TVA800 cells. These cells were infected with a low titer rVSV(A/RG) pseudotyped with RABV-G. The virus spreads between TVA-expressing cells in a matter of hours, as shown at 6 hours post infection (HPI), 1 and 2 days post infection (DPI). However, the spread on 293T cells, which do not express the TVA receptor, though it does occur, happens much slower. Scale bar = 100 μm.
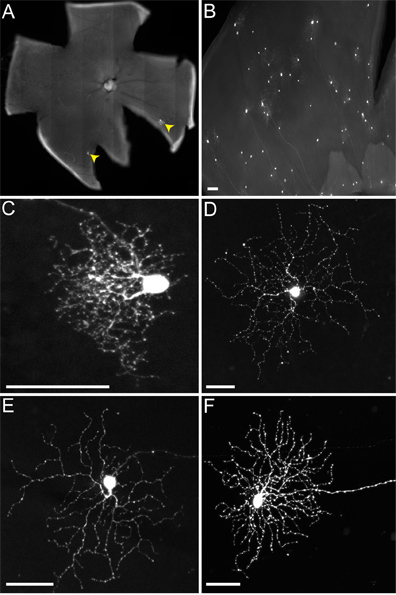 Figure 4. Representative infections of RGCs of non-TVA expressing mice. Animals were injected into the LGN with the rVSV(A/RG) virus pseudotyped with RABV-G, and tissue harvested 2 dpi. (A) Sparse (RGCs indicated by yellow arrowheads) or (B) dense infections can be obtained, depending on the titer of virus and quality of injection. Many different types of RGCs can be identified based on morphology, including (C) small arbor RGCs, (D) type 1 melanopsin RGCs, (E) ON-DSGCs, and (F) ON-OFF-DSGCs, among others. Scale bars = 50 μm.
Figure 4. Representative infections of RGCs of non-TVA expressing mice. Animals were injected into the LGN with the rVSV(A/RG) virus pseudotyped with RABV-G, and tissue harvested 2 dpi. (A) Sparse (RGCs indicated by yellow arrowheads) or (B) dense infections can be obtained, depending on the titer of virus and quality of injection. Many different types of RGCs can be identified based on morphology, including (C) small arbor RGCs, (D) type 1 melanopsin RGCs, (E) ON-DSGCs, and (F) ON-OFF-DSGCs, among others. Scale bars = 50 μm.
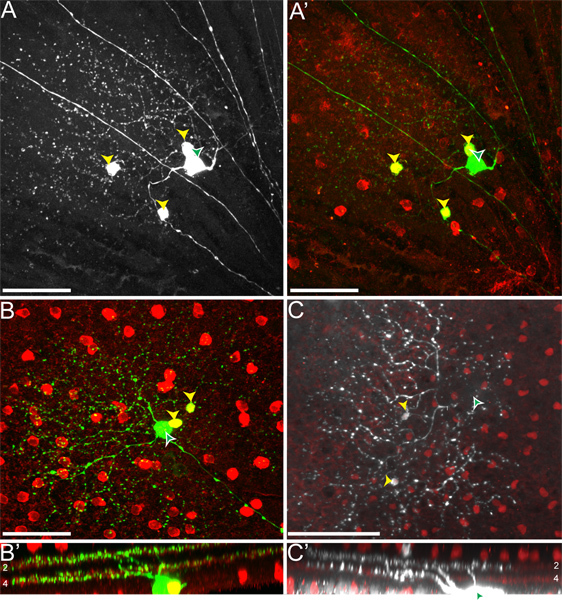 Figure 5. rVSV(A/RG) transmission from ON-OFF-DSGCs to SACs follows the expected pattern. (A) ON-OFF-DSGCs (white arrowhead with green fill) transmit virus to multiple SACs (yellow arrowheads). (A'). All green cells that are not the DSGC co-label with the Cre reporter, indicating that they are TVA-expressing SACs. (B-C) The DSGC (white arrowhead with green fill) and SACs (yellow arrowheads) stratify in the appropriate ChAT layers of the retinal inner plexiform layer (IPL). The cells to which the virus transmits include both ON and OFF-SACs. Numbers in panels B' and C' indicate the IPL laminae. The location of the DSGC soma is only indicated by the arrowhead but not shown in panel C to highlight the SACs. Scale bars = 50 μm.
Figure 5. rVSV(A/RG) transmission from ON-OFF-DSGCs to SACs follows the expected pattern. (A) ON-OFF-DSGCs (white arrowhead with green fill) transmit virus to multiple SACs (yellow arrowheads). (A'). All green cells that are not the DSGC co-label with the Cre reporter, indicating that they are TVA-expressing SACs. (B-C) The DSGC (white arrowhead with green fill) and SACs (yellow arrowheads) stratify in the appropriate ChAT layers of the retinal inner plexiform layer (IPL). The cells to which the virus transmits include both ON and OFF-SACs. Numbers in panels B' and C' indicate the IPL laminae. The location of the DSGC soma is only indicated by the arrowhead but not shown in panel C to highlight the SACs. Scale bars = 50 μm.
Discussion
Using viruses to study neural circuits is a relatively high throughput method of analyzing connected neurons. However, generating both VSV and RABV virions is not trivial. Although the protocol listed above for rescuing virus from cDNA is provided, it is still a low-probability event. The levels of each of the N, P, and L plasmids need to be finely adjusted, and many trials and replicates need to be done to ensure viral rescue. The formation of the ribonucleotide particle incorporating a full VSV genome RNA is a low-probability event, and therefore may take several attempts. This technique may require time for optimization, however, once it is optimized, rescuing viruses is much easier.
Analysis of viral transmission from individual RGCs is best done when RGC dendritic arbors do not overlap (i.e.Figure 4A). The amount of virus injected and injection coordinates can be adjusted for optimal infection. Also, the location of injection will affect the numbers and types of RGCs labeled (i.e. superior colliculus or superchiasmatic nucleus will label different numbers and types of RGCs than an LGN injection).
VSV has a number of advantages relative to PRV and RABV. The expression level of VSV is very rapid, being visible within hours9,15. This permits shorter incubation times. In this experiment, the A/RG glycoprotein was encoded within the viral genome, making the virus replication-competent. While making replication-competent rabies virus makes a BL3 containment level necessary, for VSV, it is only BL2. The glycoprotein could have been provided in trans in the RGCs, i.e. by prior infection of an AAV encoding the A/RG fusion protein. However, this strategy relies on double infection, and the levels of glycoprotein expression may not be optimal. When expressed from the viral genome, the glycoprotein levels are finely tuned as in the case of wild-type VSV. Without the need for co-infection, all RGCs labeled have the potential for viral transmission. The major drawback of VSV is its relatively rapid cell toxicity. We have examined VSV-infected cells by physiology, and noted that they were physiologically normal by 18 hr post-infection, but were too sick to obtain physiological measurements by 48 hr post-infection. Generally, neurons look morphologically normal for 2-3 days post-infection, but begin to show signs of sickness (blebbing) shortly thereafter. This is after the time in which transsynaptic transmission can be observed (24 hr). However, all transsynaptic viruses, including RABV and PRV, are toxic to the cells. Therefore, VSV is at present best used for an anatomical labeling tool, though we are in the process of making it better suited for physiological manipulations.
VSV is a particularly powerful transsynaptic neuronal tracer. By nature of its ability to accept different glycoproteins, it can trace neurons in the anterograde or retrograde directions9. Here, we show that it can also be used to probe a different question - whether or not a genetically-defined cell type inputs onto a starter cell population. Here we demonstrate its utility in the retina, however we also have used it to label genetically defined interneuronal populations the brain, with equal success. It can also be modified to contain a different glycoprotein, to permit transsynaptic anterograde transmission to TVA-expressing cells. The possibility of further manipulations offers the potential to further help analyze microcircuits and decode the complex computations of neural connections.
Disclosures
No conflicts of interest declared.
Acknowledgments
We would like to acknowledge Sean Whelan for assistance with rescuing recombinant VSV variants, and Didem Goz and Ryan Chrenek for technical assistance. This work was supported by HHMI (CLC), and #NS068012-01 (KTB).
References
- Wickersham IR. Monosynaptic restriction of transsynaptic tracing from single, genetically targeted neurons. Neuron. 2007;53:639–647. doi: 10.1016/j.neuron.2007.01.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshel JH, Mori T, Nielsen KJ, Callaway EM. Targeting single neuronal networks for gene expression and cell labeling in vivo. Neuron. 2010;67:562–574. doi: 10.1016/j.neuron.2010.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wall NR, Wickersham IR, Cetin A, De La Parra M, Callaway EM. Monosynaptic circuit tracing in vivo through Cre-dependent targeting and complementation of modified rabies virus. Proc. Natl. Acad. Sci. U.S.A. 2010;107:21848–21853. doi: 10.1073/pnas.1011756107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickersham IR. Monosynaptic restriction of transsynaptic tracing from single, genetically targeted neurons. Neuron. 2007;53:639–647. doi: 10.1016/j.neuron.2007.01.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yonehara K. Spatially asymmetric reorganization of inhibition establishes a motion-sensitive circuit. Nature. 2011;469:407–410. doi: 10.1038/nature09711. [DOI] [PubMed] [Google Scholar]
- Stepien AE, Tripodi M, Arber S. Monosynaptic rabies virus reveals premotor network organization and synaptic specificity of cholinergic partition cells. Neuron. 2010;68:456–472. doi: 10.1016/j.neuron.2010.10.019. [DOI] [PubMed] [Google Scholar]
- Beier KT, Samson MES, Matsuda T, Cepko CL. Conditional expression of the TVA receptor allows clonal analysis of descendents from Cre-expressing progenitor cells. Dev. Biol. 2011;353:309–320. doi: 10.1016/j.ydbio.2011.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seidler B. A Cre-loxP-based mouse model for conditional somatic gene expression and knockdown in vivo by using avian retroviral vectors. Proc. Natl. Acad. Sci. U.S.A. 2008;105:10137–10142. doi: 10.1073/pnas.0800487105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beier KT. Anterograde or retrograde transsynaptic labeling of CNS neurons with vesicular stomatitis virus vectors. Proc. Natl. Acad. Sci. U.S.A. 2011;108:15414–15419. doi: 10.1073/pnas.1110854108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whelan SP, Ball LA, Barr JN, Wertz GT. Efficient recovery of infectious vesicular stomatitis virus entirely from cDNA clones. Proc. Natl. Acad. Sci. U.S.A. 1995;92:8388–8392. doi: 10.1073/pnas.92.18.8388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuerst TR, Niles EG, Studier FW, Moss B. Eukaryotic Transient-Expression System Based on Recombinant Vaccinia Virus That Synthesizes Bacteriophage T7 RNA Polymerase. PNAS. 1986;83:8122–8126. doi: 10.1073/pnas.83.21.8122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young JA, Bates P, Varmus HE. Isolation of a chicken gene that confers susceptibility to infection by subgroup A avian leukosis and sarcoma viruses. J. Virol. 1993;67:1811–1816. doi: 10.1128/jvi.67.4.1811-1816.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madisen L. A robust and high-throughput Cre reporting and characterization system for the whole mouse brain. Nat Neurosci. 2010;13:133–140. doi: 10.1038/nn.2467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franklin K, Paxinos G. The Mouse Brain in Stereotaxic Coordinates. San Diego, CA: Academic Press; 1997. [Google Scholar]
- van den Pol AN. Viral strategies for studying the brain, including a replication-restricted self-amplifying delta-G vesicular stomatis virus that rapidly expresses transgenes in brain and can generate a multicolor golgi-like expression. J. Comp. Neurol. 2009;516:456–481. doi: 10.1002/cne.22131. [DOI] [PMC free article] [PubMed] [Google Scholar]