

Abstract
Resistance to platinum-based chemotherapy frequently poses a serious problem in the treatment of head and neck squamous cell carcinoma. In this study, we isolated cisplatin-resistant cells from a head and neck squamous cell carcinoma cell line. The mismatch repair (MMR) system is known as one of the cisplatin-resistant mechanisms. When the expression levels of hMLH1 and hMSH2, a mismatch repair gene and its gene product, were analyzed, the hMLH1 mRNA and protein expression levels were significantly decreased in the cisplatin-resistant cell lines compared with a cisplatin-sensitive cell line. In addition, the microsatellite instability (MSI) phenotype was examined for the absence of MMR. Our data support the hypothesis that hMLH1 mRNA and protein expression levels are predictors of cisplatin sensitivity, but MSI was not involved in cisplatin resistance. The status of hMLH1 predicts the sensitivity of head and neck squamous cell carcinoma to platinum-based chemotherapy.
Keywords: head and neck cancer, cisplatin, Mismatch Repair System, hMLH1, microsatellite instability
Introduction
Cisplatin is a commonly used drug in head and neck cancer chemotherapy in combination with 5-FU or docetaxel. However, one of the major limitations to its use in the treatment of head and neck cancer is natural or acquired resistance to cisplatin (1). The mechanism of resistance to cisplatin is unclear, but several hypotheses have been suggested in previous reports. Resistance to cisplatin is generally multi-factorial and has been shown to be the result of reduced drug accumulation, inactivation by thiol-containing species, increased repair/tolerance of platinum-DNA adducts, and alterations in proteins involved in apoptosis (2,3).
It is generally accepted that the futile attempt to repair cisplatin-induced DNA damage may finally result in the triggering of apoptosis (4). The mismatch repair (MMR) system, one of the signal transduction pathways, is involved in inducing apoptosis. Many studies have demonstrated that the loss of MMR in cisplatin resistance is associated with microsatellite instability (MSI) and reduced apoptosis (5). Cells in which the MMR system has been inactivated display an MSI phenotype identified in tumors of several different origins, both heredity and sporadic (6). Microsatellite sequences are tandem repeat sequences of 1–4 nucleotide units, and more than tens of thousands of different microsatellite sequences are distributed throughout human chromosomes. MSI characterizes the mutator phenotype and is the hallmark of MMR deficiency (7).
In this study, cisplatin-resistant UM-SCC 23 C/R and UM-SCC 81B cells were isolated from the head and neck squamous cell carcinoma UM-SCC 23 cell line. In cisplatin-resistant cells, hMLH1 gene and protein expression levels were decreased in the cisplatin-resistant cell lines. The MSI phenotype was absent in all the cell lines. Our data support the hypothesis that hMLH1 is an important predictor of cisplatin sensitivity, while MSI was not involved in cisplatin sensitivity.
Materials and methods
Cells and cell culture
The UM-SCC 23 and UM-SCC 81B cells (head and neck squamous cell carcinoma cell lines) were kindly donated by Dr Thomas E. Carey, Laboratory of Head and Neck Cancer Biology at the University of Michigan. The cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM; Sigma, MO, USA) supplemented with 10% fetal bovine serum (FBS; Invitrogen, CA, USA) in a humidified atmosphere of 5% CO2 at 37°C.
Isolation of cisplatin-resistant cells
UM-SCC 23 cells (10×106) were inoculated into a 10-cm dish and cultured for 24 h in DMEM with 10% FBS. Cells were then treated with cisplatin (Nihonkayaku, Tokyo, Japan) at a concentration of 0.5 mg/ml for 24 h, then cultured in DMEM without cisplatin until returning to stable growth. The concentration of cisplatin treatment was stepwisely increased from 1.0, 2.0, 3.0, 4.0 to 5.0 mg/ml.
Colony formation assay for cisplatin sensitivity
The appropriate number of cells were inoculated in a 6-cm dish, and treated with each concentration of cisplatin for 24 h. The cells were washed twice with PBS, and the culture medium was exchanged for a fresh one. Seven to fourteen days after inoculation, colonies were stained with 0.05% crystal violet. Colonies of ≥50 cells were scored as originating from a single clonogenic cell.
Analysis of hMLH1 and hMSH2 mRNA expression
Expression of hMLH1 and hMSH2 mRNA in each cell line was determined by real-time RT-PCR. Total RNA was extracted with TRIzol reagent (Invitrogen) from the cell lines, and first cDNA strand synthesis, performed with ThermoScript™ (Invitrogen) for the detection of hMLH1 and hMSH2 mRNA, was amplified under the following conditions: 10 min at 95°C and 40 cycles of 5 sec at 95°C, 20 sec at 60°C and 40 sec at 72°C. The LightCycler System (Roche Diagnostics, Sandhoferstrase, Mannheim, Germany) with SYBR Green PCR Core Reagents (PE Biosystems, Werrinton, UK) was used. Expression levels of hMLH1 and hMSH6 mRNA for each sample were determined by standardizing with the expression level of β-actin.
Western blot analysis
To observe the expression of hMLH1 and hMSH2, proteins were extracted with RIPA solution (1% NP-40, sodium deoxycholate and 0.05% SDS in PBS). Total protein (10 μg) was loaded onto a 10% SDS gel and blotted onto a nitrocellulose membrane after electrophoresis. The primary mouse polyclonal anti-hMLH1 (Pharmingen, CA, USA), polyclonal anti-hMSH2 (Serotec Ltd., Oxford, UK) and polyclonal anti-β-actin (Abcam, Cambridge, UK) were used in 1:200, 1:200 and 1:10,000 dilutions, respectively.
The secondary antibodies were peroxidase-conjugated anti-mouse IgG used in a 1:10,000 dilution. Immunoreactive proteins were detected using enhanced chemiluminescence (ECL; Amersham Pharmacia Biotech Inc., NJ, USA).
DNA preparation and microsatellite analysis
Approximately 50–100 cells were seeded onto a 10-cm dish and cultured at 37°C in 5% CO2 for 7 days, and the colony was prepared. Using small cloning cylinders, each single clone was isolated, inoculated into a well of a 48-well plate, and grown to confluence. DNA samples for PCR amplification were prepared by treating the cells with cytolytic solutions (10 mM Tris-HCl of 100 μl, 1 mM EDTA, 5 μg/ml Proteinase K) for 2 h at 65°C and 15 min at 95°C. Extracted DNA was amplified on microsatellite loci D9S171 and D13S175 by PCR using microsatellite primer (ABI PRISM® Linkage Mapping Sets, version 2.5; Applied Biosystems, CA, USA). The reaction was conducted under the following conditions: 12 min at 95°C, 10 cycles of 15 sec at 95°C, 15 sec at 55°C, 15 sec at 72°C and 20 cycles of 15 sec at 89°C, 15 sec at 55°C, 15 sec at 72°C. Microsatellite analysis was performed with the ABI PRISM 310 Genetic Analyzer (Applied Biosystems).
Results
Cisplatin sensitivity of each cell line
The cisplatin sensitivity of the UM-SCC 23 cells, and of the UM-SCC 81B and UM-SCC 23 C/R cells isolated from the UM-SCC 23 cell line by colony formation assay, was analyzed. The results are shown in Fig. 1. UM-SCC 81B cells, the intrinisic cisplatin-resistant cell line for cisplatin, were ∼2-fold more resistant than UM-SCC 23 cells. UM-SCC 23 C/R cells were ∼3.5-fold more resistant to cisplatin than UM-SCC 23 cells.
Figure 1.
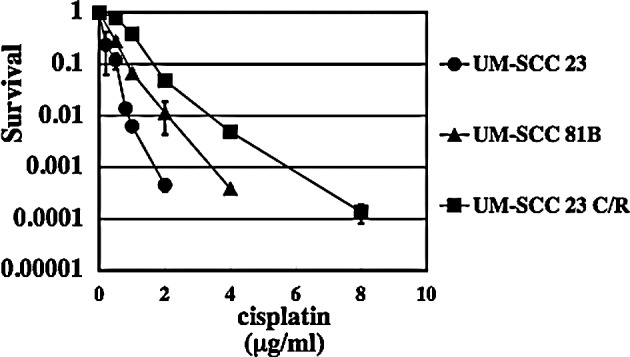
Sensitivities of head and neck squamous cell carcinoma and resistant cell lines to cisplatin. UM-SCC 23, UM-SCC 81B and UM-SCC 23 C/R cells were treated with cisplatin for 24 h, and the cytotoxicity was analyzed using a colony-formation assay. Each data point is the mean of three independent experiments. Vertical bars represent standard deviations.
hMLH1 and hMSH2 mRNA expression levels
Expression of hMLH1 and hMSH2 mRNA in UM-SCC 23 and UM-SCC 23 C/R cells was analyzed by real-time RT-PCR. Expression levels of hMLH1 mRNA in UM-SCC 81B and UM-SCC 23 C/R cells were decreased ∼60% as compared with UM-SCC 23 cells. A difference in hMSH2 mRNA expression level was not found among the three cell lines (Fig. 2).
Figure 2.
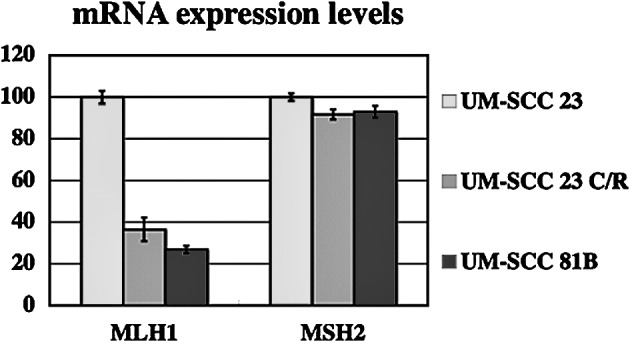
mRNA expression levels of hMLH1 and hMSH2. UM-SCC 23, UM-SCC 81B and UM-SCC 23 C/R cells were analyzed using real-time RT-PCR. Each data point is the mean of three independent experiments. Vertical bars represent standard deviations.
hMLH1 and hMSH2 protein expression level
hMLH1 and hMSH2 mismatch repair proteins were analyzed by Western blot analysis. The hMLH1 protein expression level was decreased to a greater extent in the UM-SCC 23 C/R than in the UM-SCC 23 cells. The hMLH1 expression level was further decreased in the UM-SCC 81B cells. hMSH2 was examined using the same method, but no change was found among the three cell lines (Fig. 3).
Figure 3.
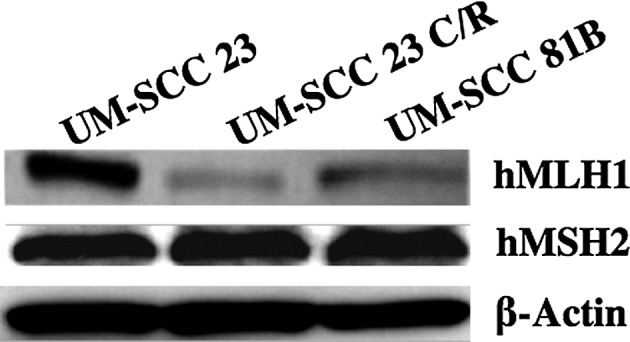
Expression levels of hMLH1 and hMSH2 mismatch repair proteins. UM-SCC 23, UM-SCC 81B and UM-SCC 23 C/R cells were examined by Western blot analysis.
Microsatellite instability
Microsatellite instability was analyzed with Gene Scan. A change in the microsatellite was found in 1 of 55 samples in D9S171 for the UM-SCC 23 cells. Sixty-six samples of UM-SCC 23 C/R cells were analyzed, but changes in the microsatellite were not observed. The micro-satellite changes were found in 2 samples each in D9S171 and D13S175 among 61 samples for UM-SCC 81B cells (Fig. 4; Table I).
Figure 4.
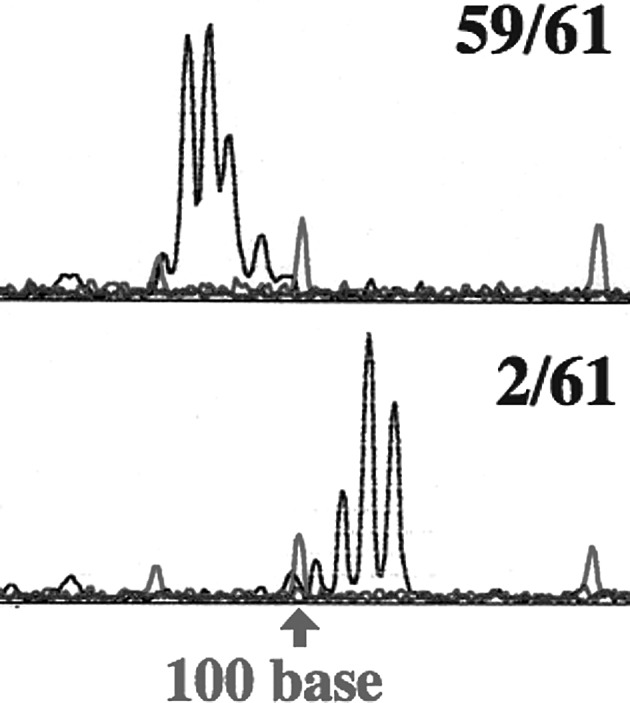
Changes in the microsatellite sequence. Microsatellite sequence changes were detected with Gene Scan. Results in D9S171 for UM-SCC 81B are shown. The upper column shows results noted in 59 of 61 samples, the lower column shows microsatellite sequence changes in 2 of 61 samples. The red lines indicate base size marker and the black lines indicate the samples. The arrow shows 100 bases.
Table I.
The ratio of the microsatellite change in each cell line.
| Change
|
||
|---|---|---|
| D9S171 | D13S175 | |
| UM-SCC 23 | 1/55 (1.81%) | 0/55 (0.00%) |
| UM-SCC 23 C/R | 0/66 (0.00%) | 0/66 (0.00%) |
| UM-SCC 81B | 2/61 (3.03%) | 2/61 (3.03%) |
Discussion
The MMR system plays an important role in the control of genomic instability in cells. In order to ensure genomic stability, it is necessary that the repair of DNA occurs prior to DNA replication (8). Before repair is initiated, the damage to DNA must be recognized by specific proteins. Indeed, a number of DNA damage recognition proteins have been identified, but studies to define their involvement in cisplatin-resistant tumor cells have largely been confined to the MMR complex (9). MMR serves a critical purpose in maintaining the integrity of the genome through the repair of DNA mismatch lesions, but does not actually repair cisplatin adducts. One proposed theory is that MMR attempts to repair the lesion, but in failing to do so activates the apoptotic signal (10).
In this study, no difference was observed in hMSH2 mRNA and protein expression levels among the three cell lines. However, hMLH1 mRNA and protein expression levels were significantly decreased in the cisplatin-resistant cells. The MMR system involves at least five proteins (hMLH1, hMSH2, hMSH3, hMSH6 and hPMS2) and functions as an ATP-dependent repair process that corrects misincorporated nucleotides. hMSH2/hMSH6 heterodimers directly bind, as the first mismatch recognizing complex, to GpG intrastrand adducts of cisplatin, and hMLH1/hPMS2 heterodimers are subsequently recruited to play an important role as the hMSH2/hMSH6/hMLH1/hPMS2 complex, and induce the stabilization and pro-apoptotic activation of p73 (11). This process requires both the MMR system and the c-Abl kinase. Previous studies have shown a decreased hMLH1 expression level or a defect caused by methylation of the promoter domain of hMLH1 in cisplatin-resistant cells (12). It is thought that the expression level of hMLH1 involved in apoptotic induction is decreased through recognition of the cisplatin adduct in the process in which cells acquire cisplatin resistance. Therefore, the cisplatin-DNA-adduct is recognized by hMSH2; however, the decrease of the apoptosis signal pathway mediated by the mismatch repair system decreases hMLH1 expression levels, resulting in the development of cisplatin resistance (13). In light of this observation regarding cisplatin sensitivity, hMLH1 mRNA and gene product expression levels may be predictors of natural and acquired cisplatin resistance.
Analysis of the microsatellite sequence in UM-SCC 23, UM-SCC 81B and UM-SCC 23 C/R cells indicated changes in the microsatellite in 2 samples each of D9S171 and D13S175 among 61 samples in UM-SCC 81B cells, a natural cisplatin-resistant cell line. The frequency of microsatellite changes was much lower than in other reports of MSI. In addition, a microsatellite change was not found in UM-SCC 23 C/R cells established as cisplatin-resistant cells. MSI was hardly evident in the three cell lines. The absence of MSI in sporadic colon cancer can be a predictive marker of sensitivity for the first post-operative adjuvant chemotherapy (14), and sensitivity of cisplatin-based chemotherapy is not associated with MSI in cervical cancer (15,16).
In conclusion, since hMLH1 mRNA and protein expression levels were decreased in the UM-SCC 81B and UM-SCC 23 C/R natural and acquired cisplatin-resistant cell lines, cisplatin adduct recognition was deduced to be involved in the acquisition of cisplatin resistance. Therefore, hMLH1 gene and gene product expression levels are effective predictors of the sensitivity to cisplatin in head and neck cancer chemotherapy. In addition, MSI was not present in conjunction with decreased hMLH1 expression levels; therefore, cisplatin-based chemotherapy is not associated with the frequency of MSI.
References
- 1.Choong N, Vokes E. Expanding role of the medical oncologist in the management of head and neck cancer. CA Cancer J Clin. 2008;58:32–53. doi: 10.3322/CA.2007.0004. [DOI] [PubMed] [Google Scholar]
- 2.Wozniak K, Blasiak J. Recognition and repair of DNA-cisplatin adducts. Acta Biochim Pol. 2002;49:583–596. [PubMed] [Google Scholar]
- 3.Siddik ZH. Cisplatin: mode of cytotoxic action and molecular basis of resistance. Oncogene. 2003;22:7265–7279. doi: 10.1038/sj.onc.1206933. [DOI] [PubMed] [Google Scholar]
- 4.Yang Z, Faustino PJ, Andrews PA, Monastra R, Rasmussen AA, Ellison CD, Cullen KJ. Decreased cisplatin/DNA adduct formation is associated with cisplatin resistance in human head and neck cancer cell lines. Cancer Chemother Pharmacol. 2000;46:255–262. doi: 10.1007/s002800000167. [DOI] [PubMed] [Google Scholar]
- 5.Papouli E, Cejka P, Jiricny J. Dependence of the cytotoxicity of DNA-damaging agents on the mismatch repair status of human cells. Cancer Res. 2004;64:3391–3394. doi: 10.1158/0008-5472.CAN-04-0513. [DOI] [PubMed] [Google Scholar]
- 6.Colella G, Marchini S, D’Incalci M, Brown R, Broggini M. Mismatch repair deficiency is associated with resistance to DNA minor groove alkylating agents. Br J Cancer. 1999;80:338–343. doi: 10.1038/sj.bjc.6690360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Geisler JP, Goodheart MJ, Sood AK, Holmes RJ, Hatterman-Zogg MA, Buller RE. Mismatch repair gene expression defects contribute to microsatellite instability in ovarian carcinoma. Cancer. 2003;98:2199–2206. doi: 10.1002/cncr.11770. [DOI] [PubMed] [Google Scholar]
- 8.Ishizaki K, Nishizawa K, Kato T, Kitao H, Han ZB, Hirayama J, Suzuki F, Cannon TF, Kamigaichi S, Tawarayama Y, Masukawa M, Shimazu T, Ikenaga M. Genetic changes induced in human cells in Space Shuttle experiment (STS-95) Aviat Space Environ Med. 2001;72:794–798. [PubMed] [Google Scholar]
- 9.Manic S, Gatti L, Carenini N, Fumagalli G, Zunino F, Perego P. Mechanisms controlling sensitivity to platinum complexes: role of p53 and DNA mismatch repair. Curr Cancer Drug Targets. 2003;3:21–29. doi: 10.2174/1568009033333727. [DOI] [PubMed] [Google Scholar]
- 10.Pani E, Stojic L, El-Shemerly M, Jiricny J, Ferrari S. Mismatch repair status and the response of human cells to cisplatin. Cell Cycle. 2007;15:1796–1802. doi: 10.4161/cc.6.14.4472. [DOI] [PubMed] [Google Scholar]
- 11.Shimodaira H, Yoshioka-Yamashita A, Kolodner RD, Wang JY. Interaction of mismatch repair protein PMS2 and the p53-related transcription factor p73 in apoptosis response to cisplatin. Proc Natl Acad Sci USA. 2003;100:2420–2425. doi: 10.1073/pnas.0438031100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cejka P, Stojic L, Mojas N, Russell AM, Heinimann K, Cannavó E, di Pietro M, Marra G, Jiricny J. Methylation-induced G(2)/M arrest requires a full complement of the mismatch repair protein hMLH1. EMBO J. 2003;22:2245–2254. doi: 10.1093/emboj/cdg216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stojic L, Brun R, Jiricny J. Mismatch repair and DNA damage signaling. DNA Repair. 2004;3:1091–1101. doi: 10.1016/j.dnarep.2004.06.006. [DOI] [PubMed] [Google Scholar]
- 14.Ribic CM, Sargent DJ, Moore MJ, Thibodeau SN, French AJ, Goldberg RM, Hamilton SR, Laurent-Puig P, Gryfe R, Shepherd LE, Tu D, Redston M, Gallinger S. Tumor microsatellite-instability status as a predictor of benefit from fluorouracil-based adjuvant chemotherapy for colon cancer. N Engl J Med. 2003;349:247–257. doi: 10.1056/NEJMoa022289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Helleman J, van Staveren IL, Dinjens WN, van Kuijk PF, Ritstier K, Ewing PC, van der Burg ME, Stoter G, Berns EM. Mismatch repair and treatment resistance in ovarian cancer. BMC Cancer. 2006;31:201. doi: 10.1186/1471-2407-6-201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Magnowska M, Surowiak P, Nowak-Markwitz E, Michalak M, Magnowski P, Rokita W, Kedzia H, Zabel M, Spaczyński M. Analysis of hMLH1 and hMSH2 expression in cisplatin-treated ovarian cancer patients. Ginekol Pol. 2008;79:826–834. [PubMed] [Google Scholar]