

Abstract
We recently reviewed the status of peptide and nonpeptide agonists and antagonists for the V1a, V1b and V2 receptors for arginine vasopressin (AVP) and the oxytocin receptor for oxytocin (OT). In the present review, we update the status of peptides and nonpeptides as: (i) research tools and (ii) therapeutic agents. We also present our recent findings on the design of fluorescent ligands for V1b receptor localisation and for OT receptor dimerisation. We note the exciting discoveries regarding two novel naturally occurring analogues of OT. Recent reports of a selective VP V1a agonist and a selective OT agonist point to the continued therapeutic potential of peptides in this field. To date, only two nonpeptides, the V2/V1a antagonist, conivaptan and the V2 antagonist tolvaptan have received Food and Drug Administration approval for clinical use. The development of nonpeptide AVP V1a, V1b and V2 antagonists and OT agonists and antagonists has recently been abandoned by Merck, Sanofi and Pfizer. A promising OT antagonist, Retosiban, developed at Glaxo SmithKline is currently in a Phase II clinical trial for the prevention of premature labour. A number of the nonpeptide ligands that were not successful in clinical trials are proving to be valuable as research tools. Peptide agonists and antagonists continue to be very widely used as research tools in this field. In this regard, we present receptor data on some of the most widely used peptide and nonpeptide ligands, as a guide for their use, especially with regard to receptor selectivity and species differences.
Keywords: vasopressin, oxytocin, agonists, antagonists, peptide, non-peptide
Introduction
Subsequent to the pioneering original synthesis of oxytocin (OT) (2) and arginine vasopressin (AVP) (3) by Vincent du Vigneaud and his associates, thousands of analogues of both of these neurohypophysial peptides have been synthesised in many laboratories throughout the world. The Merrifield solid phase method (4) has been of inestimable importance in facilitating the rapid and efficient synthesis of agonists and antagonists for the AVP V1a, V1b and V2 receptors and for the OT uterine receptor (5). Many of these ligands have found widespread use as pharmacological tools for studies on the peripheral and central effects of OT and AVP. These design and synthetic studies have been the subject of numerous reviews (1, 6–20).
Structure activity and design studies carried out in other laboratories over the past five decades have laid the foundation for the design studies that we present here. These pivotal contributions by others have been fully documented (11, 14, 18, 20).
Oxytocin and AVP mediate their biological effects by acting on specific receptors (21–23). OT and AVP receptors belong to a G-protein coupled receptor family, characterised by seven putative transmembrane helices. Reviews on AVP and OT receptors are available elsewhere (12, 22, 23). OT receptors are expressed in the uterus, the mammary gland, the ovary, the brain, the kidney, the heart, bone and in endothelial cells (23). In the uterus, OT receptors mediate the uterine contracting (oxytocic) effect of OT (23). The central effects of OT continue to be the focus of intense investigative scrutiny in animals (24–28) and in humans (29–35), as a possible therapeutic agent for the treatment of autism and other anxiety disorders.
Arginine vasopressin mediates its actions through three known receptors: V1a, V1b and V2. V1a receptors are expressed in the liver, vascular smooth muscle cells, brain and in many other tissues (12, 21, 22). In the vasculature, V1a receptors mediate the pressor actions of AVP by a phospholipase C-mediated pathway. In the brain, V1a receptors mediate the anxiety producing responses to AVP (27, 36). V1b receptors, discovered long after the V1a and V2 receptors, present in the anterior pituitary, mediate the adrenocorticotrophic hormone-releasing effects of AVP, also by a phospholipase C-mediated pathway (22) Evidence for the presence of V1b receptors in extra-pituitary tissues such as brain, the kidney and the adrenal medulla has also been reported (37). Recently, the V1b receptor has been shown to mediate anxiety and stress in rats and in humans (45). V2 receptors, present in the collecting duct of the kidney, mediate the antidiuretic action of AVP by an adenylate cyclase-mediated pathway (12, 21, 22).
Scope of the present review
We have previously reviewed the status of developments in the design and synthesis of peptide and nonpeptide AVP and OT agonists and antagonists (1). Here, we focus on the properties of the most widely used peptides requested from the Manning laboratory or purchased from suppliers, together with some recently reported potential clinically useful peptides from the Ferring Laboratory (38, 39). Space considerations preclude our being able to present or to discuss recent synthetic studies carried out in other laboratories (40–43). We also update the current status of the pre-clinical and clinical development of nonpeptide AVP and OT antagonists and of the pre-clinical development of nonpeptide OT agonists (44). The excellent reviews on nonpeptide AVP antagonists (45) and on nonpeptide OT antagonists (46) should be consulted for more in-depth presentations of their chemistry and pharmacology. We also review the merits of peptide versus nonpeptide AVP and OT agonists and antagonists as: (i) research tools and (ii) therapeutic agents. We present human and rat receptor data for a number of selective peptide agonists and for both peptide and nonpeptide antagonists. We illustrate the need to be aware of: (i) species differences, (ii) selectivity differences and (iii) in vitro–in vivo differences when using a specific ligand for receptor characterisation. Finally, we present the highlights of our recent studies aimed at: (i) the development of selective fluorescent ligands for the rat and human V1b receptors (47) and (ii) the development of fluorescence based strategies that have been used to prove the existence of OT receptor dimers in native tissue (48).
Peptide synthesis
All the OT and AVP agonists, antagonists, radiolabelled and fluorescent ligands from our laboratories were synthesised using the Merrifield solid-phase method (4, 49). The synthetic strategy relies very heavily on methodology developed in the du Vigneaud laboratory for the original syntheses of OT and AVP (2, 3). The procedures used are described in the original publications cited here. For other references, see Manning et al. (1).
Bioassays
All of the published peptides from our laboratories, presented in Tables 1, 3–8, were assayed for agonistic and antagonistic activities in in vitro and in vivo rat oxytocic assays, in the rat vasopressor assay and in the rat antidiuretic assay in the laboratories of our collaborators Drs Wilbur H. Sawyer, W. Y. Chan and Hazel Szeto. For agonists, the four-point assay design (50) was used and for antagonists, Schild’s pA2 method (51) was employed. The pA2 is the negative logarithm of the molar concentration of the antagonist that requires a two-fold increase in agonist concentration to achieve the same effect as that found in the absence of antagonist. In practice, this concentration is estimated by finding concentrations above and below the pA2 dose and interpolating on a logarithmic scale.
Table 1.
Potent and Selective Agonists for the Uterine Oxytocin Receptor in the Rat.
| Ratios | |||||||
|---|---|---|---|---|---|---|---|
| Number | Peptide | OT receptor oxytocic (O) (units/mg) | V1a receptor vasopressor (P) (units/mg) | V2 receptor antidiuretic (A) (units/mg) | O/A | O/P | Reference |
| OT | 520 | 4 | 4 | 130 | 130 | 75 | |
| 1 | [Thr4]OT | 923 | 0.4 | 0.9 | 1025 | 2307 | 75 |
| 2 | HO[Thr4]OT | 4179 | 4.92 | 5.3 | 790 | 850 | 75 |
| 3 | [Thr4, Gly7]OT | 166 | < 0.01 | ∼0.002 | 83 000 | >16 600 | 75 |
| 4 | HO[Thr4, Gly7]OT | 218 | < 0.01 | 0.004 | 54 500 | >21 800 | 75 |
| 5 | Carba-1-[4-FBzlGly7]dOT(FE 202767)* | ND | ND | ND | 38 | ||
OT, oxytocin; HO, 1-hydroxy (hydroxyl group replaces α-amino group); FBzl, fluorobenzyl. *In vitro potency EC50a (nm) hOT 0.08, hV2 330, hV1a>10000 selectivity versus receptor hV2 4100, hV1b > 120000. aEC50 is the concentration of agonist leading to half-maximal activity. ND, Not determined.
Table 3.
Potent and Selective Agonists for the Vasopressin V2 Receptor in the Rata.
| Ratios | |||||||
|---|---|---|---|---|---|---|---|
| Number | Peptide | OT receptor oxytocic (O) (units/mg) | V1a receptor vasopressor (P) (units/mg) | V2 receptor antidiuretic (A) (units/mg) | A/P | A/O | Reference |
| AVP | 14 | 373 | 320 | 0.9 | 22.8 | 79 | |
| 1 | dDAVP (desmopressin)* | 1.5 | 0.39 | 1200 | 3000 | 800 | 79, 80 |
| 2 | VDAVP | 0.60 | 0.037 | 653 | 17 650 | 1 090 | 79 |
| 3 | dVDAVP | 8 | Antagonist (pA2 = 7.03) | 1230 | Infinite | 79 | |
OT, oxytocin; AVP, arginine vasopressin; d, 1-deamino; DAVP, D-Arg8VP; V, Val4. *Desmopressin is the drug of choice for the treatment of diabetes insipidus. aThe data given are obtained from Sawyer et al. (79).
Table 8.
Some Nonselective and Selective Oxytocin Antagonists in the Rat.
| Antioxytocic (anti-OT) | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| In vitro pA2a | In vivo | Antivasopressor (anti-V1a) | Antidiuretic activity (V2) | ||||||
| Number | Peptide | No Mg2+ | EDa | pA2c | EDb | pA2c | Units/mg | ED ratiod | Ref |
| 1 | d[D-Tyr(Et)2,Thr4]OVT (atosiban) | 8.29 | Antagonist | Agonist 0.02 (IU/μmol) | Agonist 0.04 (IU/μmol) | 106 | |||
| 7.71 | 5.95 | 7.05 | 48.5 | 6.14 | Antagonist (pA2 ≍ 5.9) | 8 | 104 | ||
| 2 | d(CH2)5[Tyr(Me)2]OVT | 8.52 | 4.2 | 7.37 | 0.80 | 7.96 | ≍ 0.01 | 0.2 | 103 |
| 3 | desGly-NH2,d(CH2)5[Tyr(Me)2, Thr4]OVT | 7.89 | 1.3 | 7.69 | 23 | 6.48 | Antagonist (pA2 ≍ 5.5) | 17.7 | 104 |
| 4 | d(CH2)5[Tyr(Me)2, Thr4, Tyr-NH29 ]OVT | 7.63 | 1.0 | 7.83 | 6.6 | 7.02 | ≍ 0.015 | 6.6 | 104, 105 |
| 5 | desGly-NH2,d(CH2)5[D-Tyr2, Thr4]OVT | 7.77 | 2.85 | 7.37 | 272 | 5.39 | Antagonist (pA2 < 5.5) | 95 | 104 |
In vitro pA2 values represents the negative logarithm to the base 10 of the average molar concentration [m] of antagonist which reduces the response to 2 × units of agonist to the response with × units of agonist. bThe effective dose (ED) is defined as the dose (in nm/kg) of antagonist that reduces the response to 2 × units of agonist to the response with × units of agonist administered in the absence of antagonist. cEstimated in vivo pA2 values represent the negative logarithms of the ‘effective dose’ divided by the estimated volume of distribution (67 ml/kg) (52). dED ratio = anti-vasopressor ED/antioxytocic ED.
In the rat in vivo assays, the pA2 (effective dose) is divided by an arbitrarily assumed volume of distribution of 67 ml/kg (52) in an attempt to derive the approximate molar concentration [M] of the pA2 dose in the vicinity of the receptors. Thus, in vivo pA2 values are very approximate estimates. The USP Posterior Pituitary Reference Standard or synthetic OT and AVP, which had been standardised in oxytocic and vasopressor units against this standard, were used as agonists for working standards in all bioassays. In vitro oxytocic assays were performed on isolated uteri from diethylstilbestrol-primed rats in a Mg2+-free van Dyke Hasting’s solution (53). In vivo anti-OT potencies were determined in urethane-anaesthetised diethylstilbestrol-primed rats as described previously (54, 55). Vasopressor assays were performed on urethane-anaesthetised and phenoxybenzamine-treated rats as described by Dekanski (55), and antidiuretic assays on water-loaded rats under ethanol anesthesia as described by Sawyer (56).
Receptor binding and functional assays
Membranes and/or cell lines that express the rat and human AVP V1a, V1b and V2 receptors (57–64) and the human OT receptor (65) were used for binding and functional assays: inositol phosphate accumulation (66) for V1a, V1b and OT receptors and cyclic AMP accumulation (67) for V2 receptors, as described previously (68–74). These receptor studies were carried out in Montpellier and Milan.
Selective oxytocin agonists (Table 1)
The pharmacological properties in rat bioassays for OT and the four analogues (peptides 1–4), which are more potent and/or more selective than OT (75), are given in Table 1. [Thr4,Gly7]OT (peptide 3), also referred to as TGOT, has been widely used as a selective OT agonist. Its human and rat receptor affinities are given in Table 11. The recently reported (38) highly selective OT analogue FE 202767 (peptide 5) has not been evaluated in standard rat bioassays. It exhibits high affinity and selectivity for the human OT receptor. It thus offers promise as a potential new OT therapeutic (38).
Table 11.
Common Agonists to Oxytocin (OT)/Arginine Vasopressin (AVP) Receptorsa.
 |
The two new OT-related analogues given in Table 2 (1, 2) have not yet been evaluated in standard rat bioassays. The discovery of [Pro8]OT in new world monkeys by Lee et al. (76), independently confirmed by Jeffrey French and colleague (E. B. Harrison and J. A. French, personal communication), is an exciting new development in this field. The novel C-terminal extended analogue of OT; oxytocin-Gly-Lys-Arg, reported by Gutkowska et al. (77) and Danalache et al. (78), opens up new possibilities for the design of potential new cardiomyogenic therapies.
Table 2.
New Oxytocin (OT)-Related Peptides.
| Number | Peptide | Reference |
|---|---|---|
| 1 | Oxytocin-Gly-Lys-Arg | 77, 78 |
| 2 | [Pro8]OT | 76 |
Selective vasopressin V2 receptor agonists (Table 3)
AVP is equipotent as an antidiuretic agonist and as a vasopressor agonist (79) (Table 3). Thus, it is totally nonselective. It is also not selective with respect to its oxytocic activity. The three analogues of AVP, peptides 1–3 in Table 3 namely; dDAVP, VDAVP and dVDAVP, exhibit striking gains in antidiuretic/vasopressor selectivity. All three peptides have been widely used as selective V2 agonists. dDAVP, first synthesised by the Zaoral et al. (80) in Prague and later licensed to Ferring, has long been the drug of choice for the treatment of diabetes insipidus. It has been marketed under the trademark Desmopressin (Minirin). The human receptor affinities for dDAVP and dVDAVP given in Table 11 shows clearly that dVDAVP has a ten-fold higher affinity for the human VP V2 receptor than dDAVP. However, both peptides also exhibit high affinities for the human V1b receptor and to somewhat lesser extent for the human OT receptor (Table 11). So clearly they are not selective V2 agonists in humans with respect to both hV1b or hV1a receptors. The search for a V2 agonist that is selective with respect to the V1a and V1b receptors in humans is still a challenging goal in this field. Yet, in the rat, dDAVP could be considered as a relatively good selective V2 agonist (Table 11).
Selective vasopressin V1a receptor agonists (Table 4)
Table 4.
Potent and Selective Agonists for the Vasopressin V1a Receptor in the Rat.
| Ratios | |||||||
|---|---|---|---|---|---|---|---|
| Number | Peptide | OT receptor oxytocic (O) (units/mg) | V1a receptor vasopressor (P) (units/mg) | V2 receptor antidiuretic (A) (units/mg) | P/A | P/O | Reference |
| AVP | 373 | 320 | 14 | 1.2 | 26.6 | 79, 84 | |
| LVP ([Lys8]VP) | 270 | 284 | 10 | 0.95 | 27 | 85 | |
| 1 | [Phe2]LVP (felypressin, octapressin) | 57 | 21 | 0.3 | 2.7 | 190 | 17 |
| 2 | [Phe2]OVT, [Phe2,Orn8]vasotocin | 124 | 0.55 | 1 | 225 | 124 | 81 |
| 3 | F-180 | 164 | 0.19 | 863 | 82 | ||
| 4 | FE 202158* | ND | ND | ND | 39, 83 | ||
OT, oxytocin; F180, Hmp-Phe-Ile-Hgn-Asn-Cys-Pro-Dab(Abu)-Gly-NH2; where Hmp, 2-hydroxy-3-mercaptopropionic acid; Hgn, homoglutamine; Dab, 2,4-diaminobutyric acid; Abu, 2-aminobutyric acid; *FE 202158, [Phe2,Ile3,Hgn4,Orn(iPr)8]AVP, where Hgn is homoglutamine and iPr is isopropyl.
In rat bioassays, [Phe2]OVT (peptide 2; Table 4) is a fairly potent vasopressor agonist (81). Its vasopressor (P) activity is 124 units/mg. In antidiuretic (A) assays, it exhibits only 0.55 units/mg. Its P/A ratio is 225 (81). Thus, for many years, it has been considered to be a selective V1a agonist and has been widely used as a selective V1a agonist. However, based on its rat V1a receptor affinity data in Table 11, it is not selective for the rat V1a receptor in this assay. In this regard, the selective V1a agonist F-180 (82), which is a highly selective V1a agonist in rat bioassays (peptide 3; Table 4), is even more puzzling. In rat receptor assays (Table 11), it is clearly nonselective for V1a receptors. By contrast, F-180 exhibits high affinity and selectivity for the human V1a receptor. The exciting new V1a agonist (peptide 4; Table 5), FE202158, recently reported by Ferring (39, 83) is currently undergoing clinical trials for the treatment of vasodilating hypotension. Compared to F180, this peptide exhibits better selectivity for the human V1a receptor (Table 11). In the rat, this agonist is very specific for the V1a receptor compared to the V2 receptor (selectivity higher than 800), yet it has not been tested for the OT and V1b receptors. This intriguing new V1a agonist is not yet available to other scientists for use as a pharmacological research tool.
Table 5.
Lys8 Analogues of d[Cha4]AVP (1) and d[Leu4]AVP (3) exhibit High Affinities and Selectivities for both Rat and Human V1b Receptors.
| Affinity (Ki) (nm) | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Number | Peptide | Rat antidiuretic activity (U/mg) | rV1b-R | hV1b-R | rV2-R | hV2-R | rV1a-R | hV1a-R | rOT-R | hOT-R | Reference |
| AVP | 323 | 0.29 | 0.68 | 0.45 | 1.2 | 2.6 | 1.1 | 1.7 | 1.7 | 74, 84 | |
| dAVP | 1745 | 0.20 | 0.37 | 0.76 | 5 | 10.8 | 3.8 | 0.97 | – | 74, 84 | |
| 1 | d[Cha4]AVP | 133.6 | 1.40 | 1.2 | 12.7 | 750 | 2297 | 151 | 1430 | 240 | 69, 72, 74 |
| 2 | d[Cha4, Lys8]VP | 0.82 | 1.9 | 2.2 | 596 | 11 484 | 9093 | 283 | 586 | 141 | 85, 86 |
| 3 | d[Leu4]AVP | 378 | 0.04 | 0.23 | 3.1 | 245 | 1252 | 44.1 | 481 | 211 | 72, 74 |
| 4 | d[Leu4, Lys8]VP | 10.5 | 0.16 | 0.51 | 101 | 6713 | 3786 | 69.3 | 64 | 29 | 74, 86, 87 |
V1b receptor agonists (Table 5)
AVP was synthesised in 1954 (3) (Table 5).The first ‘selective’ V1a agonist [Phe2]OVT was synthesised 10 years later in 1964 (81). The first selective V2 agonist dDAVP was reported in 1967 (80). Yet, it was not until 2002, almost 50 years after the synthesis of AVP, that the first selective agonist for the human V1b receptor, d[Cha4]AVP was synthesised (69). The reasons why this discovery took so long have been documented by Manning et al. (1).
Table 5 lists four analogues of dAVP (peptides 1–4) that exhibit high affinities for both the rat and human V1b receptors. d[Cha4]AVP (peptide 1) was the first V1b agonist that was shown to be selective for the human V1b receptor (69). d[Leu4]AVP (peptide 3) has later been shown to be a selective agonist for the human V1b receptor (72). Both (peptides 1 and 3) exhibit high affinities for the rat V1b receptor. However, they also possess high in vivo antidiuretic activity. Thus, neither is a selective V1b agonist in the rat.
Replacement of the Arg8 residue in (peptides 1 and 3) by a Lys8 residue to give d[Cha4,Lys8]VP (peptide 2) and d[Leu4,Lys8]VP (peptide 4), respectively, resulted in the first peptides that are selective V1b agonists in the rat (73, 74). It was subsequently shown that both peptides 1 and 2 are also highly selective for human V1b receptors (86). It bears noting that d[Leu4,Lys8]VP had been reported to be a weak antidiuretic V2 agonist/weak vasopressor agonist in the rat (87), 30 years before the V1b receptor was first predicted and/or cloned (88). The selective V1b agonists d[Cha4]AVP and d[Leu4,Lys8]VP have been used as a research tool in a number of studies (1, 20, 62, 73, 89, 90). Furthermore, d[Leu4,Lys8]VP has been utilised in the design of a series of fluorescent ligands for the V1b receptor (47).
Selective V1a antagonists (Table 6)
Table 6.
Design of Highly Selective V1a Antagonists.
| Anti-OT (in vitro) | Anti-OT (in vivo) | Anti-V1a (in vivo) | ||||
|---|---|---|---|---|---|---|
| Number | Peptide | pA2a | pA2b | pA2b | Anti-V1a/anti-OT selectivity | Reference |
| 1 | d(CH2)5[Tyr(Me)2]AVP (Manning compound) | 8.13 | 6.62 | 8.62 | 100 | 91 |
| 2 | d(CH2)5[Tyr(Me)2, Dap5]AVP | 5.83 | NDc | 7.49 | Infinite | 92 |
| 3 | d(CH2)5[Tyr(Me)2, Dab5]AVP | NDc | NDc | 6.71 | Infinite | 92 |
OT, oxytocin; d(CH)5 = β-mercapto-β,β-cyclopentamethylenepropionyl. aIn vitro pA2 values represent the negative logarithm to the base 10 of the average molar concentration [m] of the antagonist that reduces the response to 2 × units of agonist to the equal the response seen with 1 × units of agonist administered in the absence of the antagonist. bIn vivo pA2 values are estimated because the molar concentration for the in vivo pA2 is estimated by dividing the effective dose (ED) by the estimated volume of distribution of (67 ml/kg) (52). ED is defined as the dose (nmol/kg intravenously) of the antagonist that reduces the response to 2 × units of agonist to the response with 1 × units of agonist administered in the absence of the antagonist. cND, not detectable (weak agonist, < 0.03 U/mg).
d(CH2)5[Tyr(Me)2]AVP, also known as Manning compound (peptide 1; Table 6) is a potent VP V1a antagonist/weak VP V2 agonist (91). It is thus highly selective for V1a receptors versus V2 receptors. It is, however, a potent in vitro OT antagonist and a fairly potent OT antagonist in vivo (91). It has found widespread use as a selective V1a antagonist in a variety of studies on the peripheral and central effects of AVP. Indeed, it has become the most widely used V1a antagonist reported to date. Isosteric modifications of d(CH2)5[Tyr(Me)2]AVP at position 5 with diaminopropionic acid (Dap) and diaminobutyric acid (Dab) led to d(CH2)5[Tyr(Me)2,Dap5]AVP (peptide 2; Table 6) and d(CH2)5[Tyr(Me)2,Dab5]AVP (peptide 3; Table 6), respectively. Both peptides are devoid of anti OT activity in vivo (92). Although both peptides are much less potent than d(CH2)5[Tyr(Me)2]AVP as V1a antagonists, because they lack anti OT potency in vivo, they are highly selective for V1a receptors in the rat. Their use is recommended for in vivo studies that require discrimination between V1a and OT receptors in the rat.
Nonselective and selective cyclic and linear V2/V1a antagonists for rat receptors
It was not until 1981, almost 30 years after the first laboratory synthesis of OT, that the first cyclic AVP V2/V1a antagonists were reported (8) (Table 7). Six years later, the unexpected discovery of the first linear V2/V1a antagonists was reported (93). The early cyclic and linear V2/V1a antagonists were nonselective for V2 receptors. Further modifications of the early cyclic V2/V1a antagonists led to the discovery of selective cyclic V2 antagonists (94). Some of the most commonly used nonselective cyclic and linear V2/V1 antagonists (peptides 1, 2, 7) and selective cyclic V2 antagonists (peptides 3–6) are given in Table 7. Peptide 6 (101) has been very useful for the design of a lanthanide cryplate-labelled ligand as a fluorescent probe for measuring receptor dimerisation (48) Peptide 8 (HO-LVA) (95), a potent linear V1a antagonist, has served as a precursor for the radioiodinated V1a ligand [125I]HO-LVA (97). This radioligand has found widespread use as a selective probe for V1a receptors. Its affinities for rat receptors are given in Table 12. A number of these V2/V1a antagonists (peptides 1–3; Table 7) exhibit oxytocic antagonism in vivo. The remaining peptides 4–8 have not been evaluated in anti-OT assays. Caution should be exercised in using any of the peptides in Table 7 as selective V2 ligands. The affinities of peptides 1 and 5 for the human and rat V2 receptors are given in Table 12.
Table 7.
Nonselective and Selective Cyclic and Linear V2/V1a Antagonists for Rat Receptors.
| Antiantidiuretic (A) (anti-V2) | Antivasopressor (P) (anti-V1a) | Antioxytocic In vivo | ||||||
|---|---|---|---|---|---|---|---|---|
| Number | Peptide | EDa | pA2b | EDa | pA2b | pA2b | ED ratio A/P | Reference |
| 1 | d(CH2)5[D-Tyr(Et)2]VAVP | 1.1 | 7.81 | 8.22 | 7.47* | 0.4 | 98 | |
| 2 | desGly,d(CH2)5[D-Tyr(Et)2, Val4]AVP | 1.5 | 7.69 | 0.45 | 8.17 | 6.98* | 0.3 | 99 |
| 3 | d(CH2)5[D-Ile2, Ile4]AVP | 0.67 | 8.04 | 0.45 | 6.42 | 6.90* | 39 | 94 |
| 4 | desGly-NH2,d(CH2)5[D-Ile2, Ile4]AVP | 0.90 | 7.88 | 26 | ∼5.2 | ∼440 | 100 | |
| 5 | d(CH2)5[D-Ile2, Ile4, Ala-NH29 ]AVP | 0.46 | 8.16 | ∼400 | 6.25 | 83 | 100 | |
| 6 | d(CH2)5[D-Tyr(Et)2, Ile4, Eda9]AVP | 0.77 | 8.00 | 38 | 8.33 | 101 | ||
| 7 | Aaa-D-Tyr(Et)-Phe-Val-Asn-Abu-Pro-Arg-Arg-NH2 | 0.53 | 8.11 | 0.32 | 7.75 | 2.3 | 48, 95 | |
| 8 | 4-HO-Phaa-D-Tyr(Me)-Phe-Gln-Asn-Arg-Pro-Arg-NH2(HO-LVA) | Agonist | 0.056 U/mg | 1.2 | 8.47 | 12, 96, 97 | ||
Aaa, adamantaneacetyl; Eda, ethylenediamine; 4-HO-Phaa, 4-hydroxyphenylacetyl. *In vivo anti-oxytocin (OT) potencies were reported previously (10), aThe effective dose (ED) is defined as the dose (in nmol/kg) that reduces the response to 2 × units of agonist to equal the response to 1 × unit. b Estimated in vivo pA2 values represent the negative logarithms of the EDs divided by the estimated volume of distribution (67ml/kg) (52).
Table 12.
Common Antagonists to Oxytocin (OT)/Arginine Vasopressin (AVP) Receptorsa.
 |
Some nonselective and selective oxytocin antagonists (Table 8)
The OT antagonists listed in Table 8 have all found widespread use as pharmacological tools. Under the tradename Tractocile, atosiban (peptide 1) has been approved for clinical use in Europe for the prevention of premature labour (102). All of these OT antagonists exhibit varying degrees of anti-V1a potency in the rat. Thus, they are far from being selective. Indeed, d(CH2)5[Tyr(Me)2]OVT, one of our original OT antagonists (103), is five-fold more potent as a vasopressor antagonist than as an OT antagonist (103). Peptide 5, desGly-NH2, d(CH2)5 [D-Tyr2,Thr4]OVT (104) is the most selective OT antagonist in Table 8. It has been used in a variety of studies. These are listed under ‘Research Uses’.
This OT receptor antagonist also exhibits a high affinity and selectivity for the human OT receptor (Table 12). Peptide 3, desGly-NH2,d(CH2)5[Tyr(Me)2Thr4]OVT, although much less selective than OTA No. 5, has proved to be very useful as a research tool in the Neumann laboratory for a wide variety of studies on the central effects of OT. Some examples of these studies are listed under ‘Research Uses’.The 125I derivative of peptide 4, d(CH2)5[Tyr(Me)2,Thr4,Tyr-NH29]OVT (105) has found widespread use as a probe of OT receptors.
Nonpeptide vasopressin antagonists as pharmacological tools and therapeutic agents (Table 9)
Table 9.
Nonpeptide Vasopressin Antagonists as Pharmacological Tools and Therapeutic Agents.
| Number | Receptor Type | Company | Code | Name | Supplier | Status | Reference: synthesis | Reference: pharmacological use | Reference: clinical use |
|---|---|---|---|---|---|---|---|---|---|
| 1 | V1a | Sanofi | SR49059 | Relcovaptan | Tocris | Phase II (terminated) | 45, 116 | 117 | |
| 2 | V1a | Pfizer | PF-00738245 | No name | Pfizer | New compound | 118 | ||
| 3 | V1a | Otsuka | OPC-21268 | No name | Tocris Sigma-Aldrich | Phase II Japan stopped US/Europe | 107 | ||
| 4 | V1b | Sanofi | SSR149415 | Nelivaptan | Axon Medchem | Preclinical (terminated) | 45, 119, 164 | 120, 147, 148, 150 | |
| 5 | V2 | Otsuka | OPC-41061 | Tolvaptan | Shanghai DND Pharm-Technology Co.,Inc. | Approved by US Food and Drug Administration oral use (Samsca) | 108 | 115 | 111–115, 121–123 |
| 6 | V2 | Sanofi | SR121463(B) | Satavaptan | None | Phase III (terminated) | 45 | 124–126 | |
| 7 | V2 | Otsuka | OPC-31260 | Mozavaptan | Otsuka; Anhui Pharmaceutical Co., LTD | Phase II | 127 | ||
| 8 | V2/V1a | Astellas | YM-087 | Conivaptan | LGM Pharma, Beijing HuameiHuli Biochem Ltd | Approved by US Food and Drug Administration i.v. use (Vaprisol) | 127 | 152 | 111, 112, 128, 129 |
The search for nonpeptide antagonists for the AVP, V1a, V1b and V2 receptors has been pursued with vigour by many pharmaceutical companies, most notably Otsuka, Sanofi, Azevan, Astellas, Wyeth-Ayerst, Johnson & Johnson, Yamanouchi and Pfizer (Table 9). In 1991, Otsuka reported the first nonpeptide V1a antagonist OPC-21268 (No. 3; Table 9) (107). During the subsequent 20 years, a number of promising nonpeptide V1, V1b, and V2 antagonists have been reported (1, 45). Table 9 lists a number of these, together with references to: (i) their original synthesis (45, 107, 108, 110, 116–119, 127, 164); (ii) some research uses (115, 117, 120, 124, 126, 147, 148, 150, 152); and (iii) their clinical uses (109, 111–115, 121-123, 128, 129). Most notable, are the Otsuka V2 antagonist (Tolvaptan, No. 5; Table 9) first reported in 1998 by Yamamura et al., (108) and shown to be effective in the treatment of hyponatraemia (109) and the Astellas V2/V1a antagonist (Conivaptan, No. 8; Table 9) first reported in 1997 by Tahara et al. (110). Both conivaptan and tolvaptan have been approved for clinical use by the Food and Drug Administration (111–115). The development of nonpeptide V1a, V1b and V2 antagonists at other companies has recently been abandoned as a resut of failures in clinic trials. Thus, Sanofi, which had reported the V1a antagonist SR-49059, Relcovaptan (No. 1; Table 9), the V1b antagonist SSR-149415, Nelivaptan (No. 4; Table 9) and the V2 antagonist SR-121463 (B) Satavaptan (No. 6; Table 9), all highly promising candidates for therapeutic development (45), has recently abandoned its entire AVP nonpeptide antagonist programme. It should be noted that these three Sanofi nonpeptide antagonists are highly useful pharmacological tools. The commercial availability of some of these nonpeptide VP antagonists is shown in Table 9. Human and rat receptor affinities for all three Sanofi V1a, V1b and V2 nonpeptides are given in Table 12.
Nonpeptide oxytocin antagonists and a nonpeptide oxytocin agonist (Table 10)
Table 10.
Non peptide Oxytocin Antagonists and a Nonpeptide Agonist.
| Compound | Number | Code (name) | Company | Supplier | Status | Reference: original synthesis, structure and pharmacologocal properties) | Reference: use |
|---|---|---|---|---|---|---|---|
| OT antagonist | 1 | L-368,899 | Merck | Tocris | Phase II Discontinued | 46, 130 | 134–136 |
| 2 | L-371,257 | Merck | Tocris | Phase II Discontinued | 46 | 141 | |
| 3 | WAY-162720 | Wyeth-Ayerst (now Pfizer) | No | Failed in preclinical | 131 | 141 | |
| 4 | GSK2211149A (Retosiban) | Glaxo SmithKline | Simagchem; Manus Aktteva Biopharma LLP | Phase II completed | 132, 133 | ||
| OT agonist | 1 | WAY-267464 | Wyeth-Ayerst (now Pfizer) | Tocris | Failed in preclinical | 138, 140 | 137, 141 |
A number of companies have been active in this area. In the mid-1990s, Merck reported a number of promising nonpeptide OT antagonists (46, 130) (Table 10). Most notable were L-368,899 (No. 1; Table 10) and L-371, 257 (No. 2; Table 10). Both of these failed in clinical development for the treatment of premature labour. Merck subsequently abandoned its nonpeptide OT antagonist programme. Similarly, Wyeth-Ayerst (now Pfizer) reported that its nonpeptide OT antagonist WAY-1627720 (131) (No. 3; Table 10) failed in preclinical development. Glaxo SmithKline is now the only company pursuing the clinical development of a nonpeptide OT antagonist. Its promising nonpeptide OT antagonist GSK 2211149A (Retosiban) (132, 133) is currently in a Phase II clinical trial. Its human and rat OT receptor affinities are given in Table 12. Its OT selectivities for human OT/VP receptors are excellent. Although the Merck and Wyeth-Ayerst nonpeptide OT antagonists have failed in clinical development, they are proving to be very useful as research tools (134–138) The Merck compounds are now available from TOCRIS. The nonpeptide OT agonist WAY-267464 reported by Pfizer (131) appeared to have promise as a therapeutic agent for the treatment of anxiety disorders such as autism spectrum disorders (139). However, its failure in preclinical development led to the abandonment of the nonpeptide OT agonist programme at Pfizer (44). WAY 267464 is now available from TOCRIS. Presently, there are no other companies pursuing the development of nonpeptide OT agonists.
The use of radiolabelled molecules, agonists and antagonists for characterising receptor affinities for OT and VP (Tables 11 and 12)
Some history
Initially in the 1960, the affinity, selectivity and potency of analogues for the different VP/OT receptors were deduced by in vivo bioassays such as oxytocic, antidiuretic and pressor tests (see above), which reflected their activity through the OT receptor, V2 and V1a receptors, respectively. The characterisations performed at that time did not take into account the V1b receptor, which was discovered only in the 1980s (142). As noted above, this led to a long delay in the discovery of selective ligands for the V1b receptor. Nevertheless, the use of bioassays allowed the identification of key structure/function relationships of a large number of analogues and still represents a milestone in our understanding of OT/AVP selectivity (1, 7–18). Furthermore, because the data obtained using these in vivo tests integrated several ADME (Absorption, Distribution, Metabolism and Elimination) parameters, the values obtained reflect the in vivo physiological activity of the peptide being studied. These values sometimes differed from those obtained by in vitro pharmacological tests.
In the 1970s, the development of radiolabelled VP/OT analogues (143) and the discovery of second messenger cascades such as cAMP (67), calcium and inositol phosphate (144) made possible the determination of more reliable pharmacological parameters reflecting more precisely the interaction between analogues and their specific receptors. Binding assays with radiolabelled ligands conducted on plasma membrane preparations allowed the determination of the affinity (Kd) of a given molecule for a given VP/OT receptor subtype, a parameter which intrinsiquely characterises the analogue/receptor association (145). Second messenger measurements allowed the characterisation of its functional activity in order to measure precise functional effects. Comparison of the affinities of one analogue for the all receptors of the VP/OT family allowed the determination of its selectivity towards a given receptor isoform. Moreover, the ability of a given analogue to activate, inhibit or leave unaffected second messenger production in cell cultures, provided important insights into its pharmacological status (agonist, partial agonist, pure antagonist, inverse agonist). Such classical pharmacological assays have efficiently served the scientific community for the last three decades and allowed the characterisation of the numerous VP/OT analogues designed and synthesised during this period (1).
Conundrums posed by pharmacological data
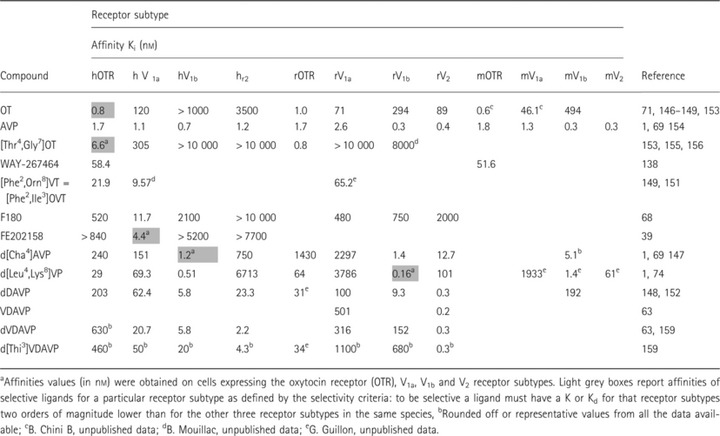
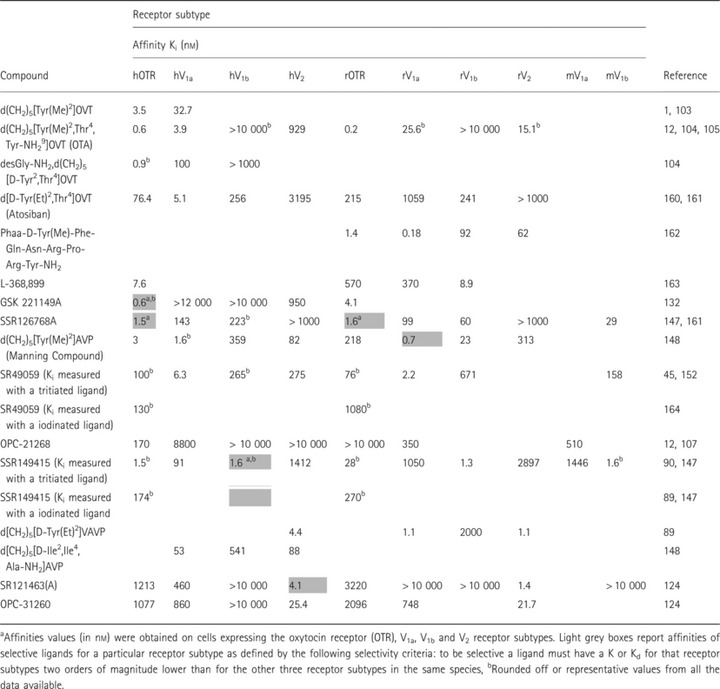
From all the in vitro and in vivo pharmacological studies carried out on OT and VP agonists and antagonists, three intriguing features have emerged; namely: (i) lack of receptor selectivity; (ii) species differences; and (iii) in vitro in vivo difference. In Tables 11 and 12, we have listed the most commonly used agonists and antagonists available for each VP/OT receptor isoform in three mammalian species: human, rat and mouse.
The affinities (Ki) of these isoforms have been measured using classical pharmacological tests and their agonist or antagonists properties determined using classical second messenger assays. As proposed in previous reviews (156, 157), receptor subtype selectivity can be defined, within a single species, on the basis of the ability of a compound to bind to a single VP/OT receptor isoform with a nanomolar affinity, at the same time displaying, for the three other receptor isoforms, an affinity at least two orders of magnitude lower. The compounds that fullfill these requirements are highlighted with light grey in Tables 11 and 12. It immediately appears from this criterion that only a very few analogues are selective. A major problem is also that selectivity is not conserved among species as a result of subtle but nevertheless crucial differences in receptor pharmacology. Despite these limitations, the use of selective compounds still represents the best experimental strategy to unambiguously characterise VP/OT receptors in a given biological sample, keeping in mind that receptor selectivity for any given compound is: (i) strictly dependent upon the receptor species considered; (ii) usually lost if high doses (100-fold the Ki) of a selective compound are used; and (iii) dependent upon the biological models tested. Experiments performed on membrane preparations or on cell cultures generally need lower concentrations of selective analogue compared to experiments performed on organ slices, where drugs need to diffuse within the tissues and may be rapidly degraded.
It should also be noted that the pharmacological profile of any given compound determined by classical tests on membranes or cell models cannot be directly translated in vivo without adequate controls. Adsorption, distribution in different biological compartments, and metabolism greatly interfere with the biological activity of drugs, sometimes completely altering their pharmacological properties.
For example, [Phe2Orn8]VT (also known as [Phe2]OVT), which does not display any V1a selectivity in classical binding experiments (Table 11), has been characterised as a selective V1a agonist in vivo in rats (Table 4) (81).
Concerning selective agonists, it should also be noted that a major difference exists between the two natural hormones, OT and VP. Although OT is selective for the human OT receptor, VP is not, because it binds with similar affinities to V1a, V1b V2 and OT receptors. This may explain why VP may trigger physiological functions in vivo via OT receptors, as described previously (158). However, fully characterised selective agonists for human V1a receptor (F 180, FE202158), human V1b receptor (d[Cha4]AVP); rat V1b receptor (d[Leu4,Lys8]AVP), rat OT receptor [Thr4,Gly7]OT and rat V2 receptor (dDAVP) are now available (Table 11). For the rat, V2 receptor d[Thi3]VDAVP (15, 159) appears to be the best selective agonist as a result of its good V2 versus V1b selectivity.
Among the several antagonists reported and currently employed, only a few have been fully characterised and have been demonstrated to be selective whithin a species (Table 12).
Among the OT receptor antagonists, SSR126768A has been shown to be a very selective antagonist for both human and rat OTR and GSK 221149A for human OT receptor. Manning compound is relatively selective for the rat V1a receptor (but not for the human V1a receptor) for which SSR49059 should be preferred. Finally, SSR149415 (147) is selective for both the human and the rat V1b receptor isoforms, whereas SSR121463(A) is highly selective for the human V2 receptor. Concerning the SSR149415 and SSR49059, it should be noted that different laboratories have obtained different values for their affinities, probably depending on the binding assay employed (i.e. competition against a radioactive agonist or antagonist) (89). In our opinion, the values obtained in competition experiments using radiolabelled agonists will better correlate with biological antagonistic activity in vitro and in vivo and should be preferred.
Until now, other analogues commonly employed as ‘selective’ have not been fully characterised and, when they are used at high doses, could lead to ambiguous results in species in which their pharmacological properties have not been assessed. It should be noted that the pharmacology of OT/VP analogues on mouse receptors is still very limited, representing a gap that needs to be filled; in particular, for the relevance that genetically-modified mouse models have acquired in translational medicine.
The lack of selective analogues for some rat, mouse and human receptor isoforms makes the design and synthesis of new molecules very necessary. The restriction of radioactivity approaches in laboratory practice and the need to easily test a large number of molecules led to the development of new assays using the gene reporters. Such tests using the measurement of reporter gene activities allows an easy screening of a large number of molecules and rapid identification of ‘lead molecules’ (83).
Yet, these ‘in vitro methodologies’ also have some limitations. First, they can be used only in transfected cells and not in native models. Moreover, according to the second messenger cascade associated with the receptor being considered (cAMP for the V2, InsP3 for the V1a, V1b and OT receptor isoforms), such assays require the use of different reporter genes. This may introduce a bias in the determination of receptor selectively. It is also well known that assays using luciferase gene expression and luciferase activity measurements involve a strong amplification of the initial receptor-mediated second messenger accumulation. This prevents the good determination of the agonist or partial agonist properties of the analogue tested. One needs to be aware of the limitations of these recent ‘in vitro methodologies’ and to verify, using classical pharmacological tests, the selectivity, affinity and functional potencies of the lead compounds characterised by this approach. Obviously, to move to clinical development, the best approach would be to test the compounds of interest by in vivo technologies similar to those used to evaluate virtually all of the peptides in Tables 1, 3–8.
New technologies for screening more selective VP/OT analogues
Recently, new physical techniques involving label-free biosensors have been proposed for pharmacological screening of muscarinic and corticotrophic analogues (165). These methods are based on the measurement of cell shape changes induced by ligand-receptor interactions. Such techniques have the advantage of being performed on native cells and do not require the use of radioactive molecules. Their efficiency for testing new VP/OT molecules may represent another alternative for screening new analogues.
Finally, a new bioluminescence or fluorescence resonance energy transfer (BRET or FRET) approach in which analogues could be screened for their capability to promote receptor coupling and activation of single G-protein isoforms has been recently applied to the human OT receptor (166). This technique allows the precise characterisation of which G protein is associated with which receptor isoform. Thus, for example, d(CH2)5[D-2-Nal2,Thr4,Tyr-NH29]OVT (OTA) and atosiban (160) (Table 12) were found to be entirely biased respectively toward Gi1 or Gi3 activation (166). However, this technique cannot be used on native tissues or primary cultures.
The recent development of fluorescent ligands for a better knowledge of central and peripheral VP/OT receptors
Design and use of classical fluorophores
Receptors of the AVP and OT family are important in the regulation of the stress processes (167). Centrally, the V1a, V1b and OT receptors have been involved in stress and especially in learning and memory processes. Important data have been obtained by the use of knockout animals but, after a period of cloning and pharmacological characterisation in the last decade, it became necessary to elucidate the distribution of these receptors to better understand their central functions in vivo.
Although several publications describe the AVP V1a, V2 and OT receptor distribution by using autoradiography (105, 168, 169) or immunodetection (170), the lack of selective V1b radio-labelled VP analogues or of receptor antibodies has hindered progress in the detection of receptor distribution in native tissues. Results obtained by molecular approaches such as reverse transcriptase-polymerase chain reaction (62, 88) or mRNA detection by in situ hybridisation (171–173), although more accurate, did not provide clear information regarding the brain regions detected by immunostaining. Thus, developing fluorescent ligands to decipher AVP receptor distribution in the brain and at the periphery, and to study molecular interactions such as receptor dimerisation, appeared as an absolute necessity.
Various fluorescent analogues of AVP and OT have been synthesised for several receptors of the VP/OT family (174). Thus, good fluorescent V1a and OT ligands have been produced, although no good fluorescent specific ligand was available to selectively detect central and peripheral V1b receptors.
In our previous work (69, 73, 74), by replacing the glutamine4 of the natural AVP with a cyclohexylalanine or a leucine, the arginine8 by a lysine and by removing the NH2 of the cysteine1 to increase stability towards aminopeptidases, we produced analogues showing an increased selectivity for the V1b receptors (Table 5). d[Leu4,Lys8]VP (Peptide 4; Table 5) was found to be selective for the rat V1b receptors and, to a lesser extent, for the human hV1b receptors, conserving a nanomolar affinity for these receptor isoforms (73, 74). We have taken advantage of the Lys8 residue in d[Leu4,Lys8]VP with its epsilon NH2 group to introduce fluorophores on its side chain. This allowed us to create fluorescent tools that would conserve the pharmacology of the d[Leu4,Lys8]VP, to resist degradation and to selectively decorate the plasma membrane of Chinese hamster ovary cells expressing V1b and/or OT receptors with an excellent resolution (47).
Different fluorophores were attached to the d[Leu4,Lys8]VP: First, the antraniloyl group (Atn), a small fluorescent molecule of 97 Da highly sensitive to microenvironmental changes (175) may also be a good donor in FRET experiments to identify V1b receptor homodimers in vivo. We have also selected the Alexas (Molecular Probes) for their brightness and their resistance to photobleaching (176). We have used Alexa 488 and Alexa 647, with the latter being one of the brightest fluorescent molecules reported so far (177).
The pharmacological properties (binding, coupling to phospholipase C) of fluorescent analogues of d[Leu4,Lys8]VP indicate that they conserved a very good selectivity for V1b versus V1a and V2 receptors, and remained full agonists. These properties allow receptor labelling and measurement of biological activity at the cellular level. Thus, these new fluorescent analogues are promising tools for the detection of functional V1b or OT receptors in human (47) and in rat native tissues.
Use of long life fluorophores
However, it should be noted that, except for very recent ones, all the ligands previously reported were designed with classical fluorophores, exhibiting short-lived fluorescence properties (fluorescence half-time life in the 10 ns range). Most of them were essentially used to follow internalisation in cell lines (146, 178) or to label receptors in a native context (179). Interestingly, a first nonpeptide antagonist with a nanomolar affinity for the human V2 receptor has been developed (180). This ligand will find application in fluorescence polarisation-based binding assays aiming to screen for small organic molecule libraries.
Recently, fluorescence-based strategies have been extensively used to study molecular interactions. Thus, the FRET approach was used to demonstrate G protein-coupled receptors oligomerisation (181, 182). Regarding AVP and OT receptors, various experimental approaches based on chimeric receptor expression in cell lines have been developed to analyse receptor oligomers. The studies have used receptors fused either to small tags recognised by fluorescent antibodies (183, 184), or to bioluminescent or fluorescent proteins (185, 186), or to suicide enzymes (48). However, these strategies were not relevant for proving the existence of such receptor complexes in native tissues. Therefore, a FRET strategy based on the indirect labelling of receptors with fluorescent donor and acceptor ligands has been recently developed (48). Unfortunately, because of the overlap between excitation and emission spectra of the donor and acceptor fluorophores on one hand, and of the high autofluorescence of the biological preparation on the other hand, specific FRET could hardly be detected. To improve the signal-to-noise ratio, lanthanide cryptate-labelled ligands were designed and characterised. Despite the size of the cage, these ligands still display very good affinities for the V1a and OT receptors (48). Lanthanide cryptates display interesting fluorescent properties because they have a fluorescent half-time life of approximately 1 ms (i.e. 100 000-fold greater than classical fluorophores), allowing time-resolved FRET experiments to be set up (187). Experiments using these new probes have been performed not only on AVP V1a and V2 receptors and on OT receptors expressed in cell lines, but also on OT receptors naturally expressed in lactating rat mammary gland. The sensitivity is such that it has been possible to prove the existence of OT receptor dimers in this latter native tissue (48).
These newly-synthesised ligands and those that exhibit high quantum yield have also been used to develop original binding assays. These assays, based on time-resolved FRET between compatible fluorophores carried by tagged receptors and ligands, display very good sensitivities and are safer than radioactive-based assays (188–190).
Therapeutics uses of peptide and nonpeptide oxytocin and vasopressin agonists and antagonists (Table 13)
13.
Food and Drug Administration Approved Peptide and Nonpeptide Drugs from the Oxytocin (OT)/Arginine Vasopressin (AVP) Field.
| OT/AVP field | |
|---|---|
| Peptidesa | Nonpeptidesb |
| Carbetocin (Duratocin, Depotocin, Sofla, Pabal) | Conivaptan hydrochloride (Vaprisol) |
| Desmopressin (Minirin, DDAVP) | Tolvaptan |
| Ornithine vasopressin (Ornipressin, POR-8) | |
| Lypressin (Diapid, LVP) | |
| Oxytocin | |
| Terlipressin (glypressin) | |
| Vasopressin (Pitressin) | |
| Atosiban (in Europe) |
See Reichert (194). bSee Ferguson-Myrthil (115).
Table 13 lists the seven peptides and two nonpeptide drugs in the OT and AVP field that have been approved for therapeutic use. Numerous recent studies point to the use of OT as a potential new therapy for the treatment of a broad range of psychiatric disorders (24, 33, 34). Also, a very recent report suggests the exciting prospect that OT may have potential for the treatment of human obesity and type 2 diabetes (191).
To date, only two nonpeptides, the VP V2/V1a antagonist conivaptan and the VP V2 antagonist tolvaptan have been approved for clinical use (112–115, 123, 192). The nonpeptide OT antagonist retosiban (132, 133) is currently in a Phase II clinical trial. Clinical trials with other nonpeptide VP, V1a and V1b antagonists shown in Table 9 and with the other OT antagonists and the OT agonist shown in Table 10 have been terminated (44). No new nonpeptides in this field are currently in clinical trial. Thus, the early promise of nonpeptides as therapeutic agents in this field (45, 46) has clearly not been realised. This should be a cautionary tale for those in pharmaceutical companies (138), in granting agencies (139) and on study sections (44) who have strongly promoted the development of nonpeptides over peptides as therapeutic agents. This philosophy led to the abandonment of peptides as potential drugs by Big Pharma almost two decades ago. Clearly, it is now time for Big Pharma to reassess the value of peptides as therapeutic drugs (44). In this regard, recent progress in the development of peptide drugs (193, 194) provides very compelling support for the thesis that peptides are clearly superior to nonpeptides as therapeutic agents, thus bolstering the case for continued support for the design and synthesis of peptides as potential therapies. In this regard, in the OT field, there is an urgent need for functionally selective OT ligands (166) and for a long lasting OT agonist as a potential therapy for the treatment of autism and other anxiety disorders (139).
Research uses of peptides
During the period 1980–2012, over 3000 samples of OT and AVP agonists and antagonists from the Manning laboratory have been and continue to be donated as research tools to over 700 investigators (some multiple times) in the USA and worldwide for their own independent studies. Studies carried out with these donated peptides and with those purchased from commercial suppliers, such as Sigma, Bachem and Peninsula, have resulted in more than 2000 publications by these and other investigators during this period. Examples of studies carried out since 2008 with some of these peptides are available (90, 117, 195–246).
Research uses of nonpeptides
With the exception of the Sanofi nonpeptide V2 antagonist satavaptan, all of the nonpeptide AVP antagonists listed in Table 9 are now available from Tocris or other suppliers. To date, a small number of studies that have utilised the Sanofi V1a antagonist relcovaptan, the Sanofi V1b antagonist nelivaptan and the Sanofi V2 antagonist satavaptan have been reported (117–120, 125, 126) (Table 9).
The two Merck nonpeptide OT antagonists L-368,899 and L-371,257 and the Pfizer nonpeptide OT agonist Way-267464 shown in Table 10 are all available from Tocris. The Glaxo SmithKline nonpeptide OT antagonist GSK-2211149A (Retosiban) is also available from a number of suppliers. The Pfizer nonpeptide OT antagonist WAY-162720 is not yet commercially available. A number of studies that have utilised some of these nonpeptide ligands as research tools have been reported (138–141, 145) (Table 10).
Conclusions
In our 2008 review (1), we examined the status (as of 2007) of both peptide and nonpeptide agonists and antagonists of the OT receptor and of the VP V1a, V1b and V2 receptors as: (i) research tools and (ii) therapeutic agents. Although the research uses of both peptide and nonpeptide ligands have continued to grow during the intervening 4 years, by contrast, the therapeutic development of nonpeptide AVP and OT antagonists has been drastically curtailed. Merck, Pfizer and Sanofi have all abandoned their nonpeptide programmes. The nonpeptide V2/V1a the antagonist, conivaptan and the nonpeptide V2 antagonist tolvaptan, which have been approved by the Food and Drug Administration, have not as yet found widespread acceptance in the clinic (113, 192). Pfizer has also abandoned its nonpeptide OT agonist programme (44). It remains to be seen how the Glaxo SmithKline nonpeptide OT antagonist retosiban will fare in its current Phase II clinical trial. All in all, since our 2008 review (1), interest in the development of nonpeptides as therapeutics has greatly diminished. On the other hand, as noted above, Ferring has a promising V1a agonist (Table 4) and a promising OT agonist (Table 1), awaiting clinical development. The design and synthesis of: (i) functionally selective OT peptides and (ii) of a long lasting OT analog as a potential therapy for autism spectrum disorders and other anxiety disorders remain as pressing needs in this field. Both OT and VP peptides and nonpeptides are continuing to be very valuable research tools. In this regard, we have addressed here the issues of the: (i) lack of receptor selectivity, (ii) species differences and (iii) in vitro–in vivo differences, all of which need to be taken into account when using a given peptide or nonpeptide ligand as a research tool. Finally, the development of new fluorescent ligands as powerful new tools for localising and characterising OT and VP receptors, which we have presented here, points to the continued usefulness of OT and AVP peptide ligands (agonists, antagonists and fluorescent derivatives) as incisive molecular pharmacological probes.
Acknowledgments
Work from the authors’ laboratories was supported in part by research grants from the National Institute of General Medical Sciences (No. GM-25280: Maurice Manning); the MIUR (FIRB grant RBPRO5JH2P ITALNANONET) and the Carpilo Foundation (Grant 200610882) (Bice Chini); and INSERM, Institute de la Santé et de la Recherche Médicale and CNRS (Centre National de la Recherche Scientifique) (Thierry Durroux; Bernard Mouillac; Maithe Corbani; Gilles Guillon). We also thank, Dr Rao Makineni, Robert and Suzanne Tyner, and Frederick Paulsen for generous research support (to M.M.). We are greatly indebted to our former collaborators: Dr Wilbur H. Sawyer, Dr W. Y. Chan, Dr Hazel Szeto, Ms Becky Wo, Dr Serge Jard, Dr Claude Barberis, Dr Jean Jacque Drefuss and Dr Eliene Tribollet, as well as the co-workers who have participated in these synthetic studies over many years. Dr William Morris Baxter, Dr Ting-Chi Wuu, Ms Karen Morton, Dr Victor Smart Abbey, Ms Esther J. Coy, Dr John Lowbridge, Ms Jacqueline Judd, Dr Lajos Balaspiri, Dr Andras Turan, Dr Marian Kruszynski, Dr Bernard Lammek, Dr Zbigniew Grzonka, Dr Aleksander Kolodziejczyk, Dr Aleksandra Kolodziejczyk, Dr Wieslaw A. Klis, Dr Eleonora Nawrocka, Dr Joseph Przybylski, Dr Svetlana Pancheva, Dr Krasimira Miteva and Dr Ling Ling Cheng. We also wish to thank Ms Ann Chlebowski for her expert assistance in the preparation of this manuscript.
References
- 1.Manning M, Stoev S, Chini B, Durroux T, Mouillac B, Guillon G. In: Peptide and Nonpeptide Agonists and Antagonists for the Vasopressin and Oxytocin V1a, V1b, V2 and OT Receptors: Research Tools and Potential Therapeutic Agents. Landgraf R, Neumann I, editors. Prog Brain ResAmsterdam: Elsevier; 2008. pp. 473–512. [DOI] [PubMed] [Google Scholar]
- 2.du Vigneaud V, Ressler C, Swan JM, Roberts CW, Katsoyannis PG. The synthesis of oxytocin. J Am Chem Soc. 1954;76:3115–3121. [Google Scholar]
- 3.du Vigneaud V, Gish DT, Katsoyannis PG. A synthetic preparation possessing biological properties associated with arginine-vasopressin. J Am Chem Soc. 1954;76:4751–4752. [Google Scholar]
- 4.Merrifield RB. Solid phase peptide synthesis. I. The synthesis of a tetrapeptide. J Am Chem Soc. 1963;85:2149–2154. [Google Scholar]
- 5.Manning M. Impact of the Merrifield solid phase method on the design and synthesis of selective agonists and antagonists of oxytocin and vasopressin: a historical perspective. Biopolymers. 2008;90:203–212. doi: 10.1002/bip.20802. [DOI] [PubMed] [Google Scholar]
- 6.Sawyer WH, Manning M. Synthetic analogues of oxytocin and vasopressin. Annual Rev Pharmacol. 1973;13:5–17. doi: 10.1146/annurev.pa.13.040173.000253. [DOI] [PubMed] [Google Scholar]
- 7.Manning M, Grzonka Z, Sawyer WH. Synthesis of posterior pituitary hormones and hormone analogues. In: Beardwell C, Robertson G, editors. The Pituitary. Kent: Butterworth; 1981. pp. 265–296. [Google Scholar]
- 8.Sawyer WH, Grzonka Z, Manning M. Neurohypophyseal peptides: design of tissue-specific agonists and antagonists. Mol Cell Endocrinol. 1981;22:117–134. doi: 10.1016/0303-7207(81)90086-1. [DOI] [PubMed] [Google Scholar]
- 9.Manning M, Sawyer WH. In: Development of Selective Agonists and Antagonists of Vasopressin and Oxytocin, Vasopressin. Schrier R, editor. New York, NY: Raven Press; 1985. pp. 131–144. [Google Scholar]
- 10.Manning M, Bankowski K, Sawyer WH. Selective agonists and antagonists of vasopressin. In: Gash DM, Boer GJ, editors. Vasopressin: Principles and Properties. New York, NY: Plenum Press; 1987. pp. 335–368. [Google Scholar]
- 11.Manning M, Sawyer WH. Discovery, development, and some uses of vasopressin and oxytocin antagonists. J Lab Clin Med. 1989;114:617–632. [Published erratum appears in J Lab Clin Med 1990; 115: 530] [PubMed] [Google Scholar]
- 12.Barberis C, Morin D, Durroux T, Mouillac B, Guillon G, Hibert M, Tribollet E, Manning M. Molecular pharmacology of vasopressin and oxytocin receptors and therapeutic potential. Manning. Drug News Perspect. 1999;12:279–292. [Google Scholar]
- 13.Manning M, Sawyer WH. Design, synthesis and some uses of receptor specific agonists and antagonists of vasopressin and oxytocin. J Receptor Res. 1993;13:195–214. doi: 10.3109/10799899309073655. [DOI] [PubMed] [Google Scholar]
- 14.Manning M, Sawyer WH. Antagonists of vasopressin and oxytocin: current status and future perspectives. In: Jard S, Jamison R, editors. Vasopressin. London: Coll Inserm/John Libbey Eurotext; 1991. pp. 297–309. [Google Scholar]
- 15.Chan WY, Wo NC, Stoev S, Cheng LL, Manning M. Discovery and design of novel and selective vasopressin and oxytocin agonists and antagonists: the role of bioassays. Exp Physiol. 2000;85(Suppl):7S–18S. doi: 10.1111/j.1469-445x.2000.tb00003.x. [DOI] [PubMed] [Google Scholar]
- 16.Manning M. Neurohypophyseal peptide agonists and antagonists: a five decade trail from du Vigneaud to 2004. In: Flegel M, Fritkin M, Gilon C, Slaninova S, editors. Peptides 2004. Geneva: Kenes International; 2005. pp. 101–106. [Google Scholar]
- 17.Berde B, Boissonnas RA. Basic pharmacological properties of synthetic analogues and homologues of the neurohypophysial hormones. In: Berde B, editor. Neurohypophysial Hormones and Similar Polypeptides, Handbook of Experimental Pharmacology 23. Berlin: Springer Verlag; 1968. pp. 802–870. [Google Scholar]
- 18.Lebl M. Analogs with inhibitory properties. In: Jost K, Lebl M, Brtnik F, editors. Handbook of Neurohypophyseal Hormone Analogs. Vol. 2. Boca Raton, FL: CRC Press; 1988. pp. 17–72. Part 1, Part 2. 127–267. [Google Scholar]
- 19.Pierzynski P. Oxytocin and vasopressin V1a receptors as new therapeutic targets in assisted reproduction. Reprod Biomed Online. 2011;22:9–16. doi: 10.1016/j.rbmo.2010.09.015. [DOI] [PubMed] [Google Scholar]
- 20.Hruby VJ, Smith CW. Structure-activity relationships of neurohypophyseal peptides. In: Udenfriend S, Meienhofer J, editors. The Peptides: Analysis, Synthesis, Biology. New York, NY: Academic Press; 1987. pp. 77–207. Vol. 8. In: Chemistry, Biology, and Medicine of Neurohypophyseal Hormones and Their Analogs, vol. ed. Smith CW. [Google Scholar]
- 21.Frank E, Landgraf R. The vasopressin system – from antidiuresis to psychopathology. Eur J Pharmacol. 2008;583:226–242. doi: 10.1016/j.ejphar.2007.11.063. [DOI] [PubMed] [Google Scholar]
- 22.Jard S. Vasopressin receptors. A historical survey. In: Zingg HH, Bourque CW, Bichet DG, editors. Adv Exp Med Biol. New York, NY: Plenum Press; 1998. pp. 1–13. [PubMed] [Google Scholar]
- 23.Gimpl G, Fahrenholz F. The oxytocin receptor system: structure function and regulation. Physiol Rev. 2001;81:629–683. doi: 10.1152/physrev.2001.81.2.629. [DOI] [PubMed] [Google Scholar]
- 24.Insel TR, Young LJ. The neurobiology of attachment. Nat Rev Neurosci. 2001;2:129–136. doi: 10.1038/35053579. [DOI] [PubMed] [Google Scholar]
- 25.Bosch OJ, Meddle SL, Beiderbeck DI, Douglas AJ, Neumann ID. Brain oxytocin correlates with maternal aggression: link to anxiety. J Neurosci. 2005;25:6807–6815. doi: 10.1523/JNEUROSCI.1342-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Huber D, Veinante P, Stoop R. Vasopressin and oxytocin excite distinct neuronal populations in the central amygdala. Science. 2005;308:245–248. doi: 10.1126/science.1105636. [DOI] [PubMed] [Google Scholar]
- 27.Ma S, Shipston MJ, Morilak D, Russell JA. Reduced hypothalamic vasopressin secretion underlies attenuated adrenocorticotropin stress responses in pregnant rats. Endocrinology. 2005;146:1626–1637. doi: 10.1210/en.2004-1368. [DOI] [PubMed] [Google Scholar]
- 28.Parker KJ, Buckmaster CL, Schatzberg AF, Lyons DM. Intranasal oxytocin administration attenuates the ACTH stress response in monkeys. Psychoneuroendocrinology. 2005;30:924–929. doi: 10.1016/j.psyneuen.2005.04.002. [DOI] [PubMed] [Google Scholar]
- 29.Kirsch P, Esslinger C, Chen Q, Mier D, Lis S, Siddhanti S, Gruppe H, Mattay VS, Gallhofer B, Meyer-Lindenberg A. Oxytocin modulates neural circuitry for social cognition and fear in humans. J Neurosci. 2005;25:11489–11493. doi: 10.1523/JNEUROSCI.3984-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hollander E, Bartz J, Chaplin W, Phillips A, Sumner J, Soorya L, Anagnostou E, Wasserman S. Oxytocin increases retention of social cognition in autism. Biol Psychiatry. 2007;61:498–503. doi: 10.1016/j.biopsych.2006.05.030. [DOI] [PubMed] [Google Scholar]
- 31.Hollander E, Novotny S, Hanratty M, Yaffe R, DeCaria CM, Aronowitz BR, Mosovich S. Oxytocin infusion reduces repetitive behaviors in adults with autistic and Asperger’s disorders. Neuropsychopharmacology. 2003;28:193–198. doi: 10.1038/sj.npp.1300021. [DOI] [PubMed] [Google Scholar]
- 32.Harony H, Wagner S. The contribution of oxytocin and vasopressin to mammalian social behavior: potential role in autism spectrum disorder. Neurosignals. 2010;18:82–97. doi: 10.1159/000321035. [DOI] [PubMed] [Google Scholar]
- 33.Meyer-Lindenberg A, Domes G, Kirsch P, Heinrich M. Oxytocin and vasopressin in the human brain: social neuropeptides for translational medicine. Nat Rev Neurosci. 2011;12:524–538. doi: 10.1038/nrn3044. [DOI] [PubMed] [Google Scholar]
- 34.Striepens N, Kendrick KM, Maier W, Hurlemann R. Prosocial effects of oxytocin and clinical evidence for its therapeutic potential. Front Neuroendocrinol. 2011;32:426–450. doi: 10.1016/j.yfrne.2011.07.001. [DOI] [PubMed] [Google Scholar]
- 35.Ellenbogen MA, Linnen AM, Grumet R, Cardoso C, Joober R. The acute effects of intranasal oxytocin on automatic and effortful attentional shifting to emotional faces. Psychophysiology. 2012;49:128–137. doi: 10.1111/j.1469-8986.2011.01278.x. [DOI] [PubMed] [Google Scholar]
- 36.Ring RH. The central vasopressinergic system: examining the opportunities for psychiatric drug development. Curr Pharm Des. 2005;11:205–225. doi: 10.2174/1381612053382241. [DOI] [PubMed] [Google Scholar]
- 37.Grazzini E, Lodboerer AM, Perrez-Martin A, Joubert D, Guillon G. Molecular and functional characterization of V1b vasopressin receptor in rat adrenal medulla. Endocrinology. 1996;137:3906–3914. doi: 10.1210/endo.137.9.8756565. [DOI] [PubMed] [Google Scholar]
- 38.Wisniewski K, Galyean R, Schteingart CD, Tariga H, Croston G, Alagarsamy S, Riviere PJM. Synthesis and in vitro characterization of new potent and selective oxytocin receptor agonist. In: Lebl M, Meldal M, Knud J, Jensen KJ, Høeg-Jensen T, editors. Peptides 2010. 2010. pp. 306–307. Proc of the 31st European Peptide Symposium, The European Peptide Society. [Google Scholar]
- 39.Laporte R, Kohan A, Heitzmann J, Wisniewska H, Toy J, La E, Tariga H, Alagarsamy S, Ly B, Dykert J, Qi S, Wisniewski K, Galyean R, Croston G, Schteingart CD, Rivière PJ. Pharmacological characterization of FE 202158, a novel, potent, selective, and short-acting peptidic vasopressin V1a receptor full agonist for the treatment of vasodilatory hypotension. J Pharmacol Exp Ther. 2011;337:786–796. doi: 10.1124/jpet.111.178848. [DOI] [PubMed] [Google Scholar]
- 40.Kwiatkowska A, Sobolewski D, Prahl A, Borovicková L, Slaninová J, Lammek B. Arginine vasopressin and its analogues-the influence of position 2 modification with 3,3-diphenylalanine enantiomers. Highly potent V2 agonists. Eur J Med Chem. 2009;44:2862–2867. doi: 10.1016/j.ejmech.2008.12.010. [DOI] [PubMed] [Google Scholar]
- 41.Magafa V, Borovicková L, Slaninová J, Cordopatis P. Synthesis and biological activity of oxytocin analogues containing unnatural amino acids in position 9: structure activity study. Amino Acids. 2010;38:1549–1559. doi: 10.1007/s00726-009-0372-2. [DOI] [PubMed] [Google Scholar]
- 42.Muttenthaler M, Andersson A, de Araujo AD, Dekan Z, Lewis RJ, Alewood PF. Modulating oxytocin activity and plasma stability by disulfide bond engineering. J Med Chem. 2010;53:8585–8596. doi: 10.1021/jm100989w. [DOI] [PubMed] [Google Scholar]
- 43.Slusarz R, Slusarz MJ. An influence of the aromatic side chains conformations in positions 2 and 3 of vasopressin analogs on interactions with vasopressin and oxytocin receptors. QSAR Comb Sci. 2009;28:1166–1175. [Google Scholar]
- 44.Manning M, Stoev S, Bankowski K. Peptides versus nonpeptides as therapeutics: an exiting challenge for Big Pharma. In: Lebl M, Meldal M, Jensen KJ, Hoeg-Jensen T, editors. Peptides 2010. Prompt Scientific Publishing: European Peptide Society; 2010. pp. 366–367. [Google Scholar]
- 45.Serradeil-Le Gal C, Wagnon J, Valette G, Garcia G, Pascal M, Maffrand JP, Le Fur G. Nonpeptide vasopressin receptor antagonists: development of selective and orally active V1a, V2 and V1b receptor ligands. Prog Brain Res. 2002;139:197–210. doi: 10.1016/s0079-6123(02)39017-4. [DOI] [PubMed] [Google Scholar]
- 46.Freidinger RM, Pettibone DJ. Small molecule ligands for oxytocin and vasopressin receptors. Med Res Rev. 1997;17:1–16. doi: 10.1002/(sici)1098-1128(199701)17:1<1::aid-med1>3.0.co;2-5. [DOI] [PubMed] [Google Scholar]
- 47.Corbani M, Trueba M, Stoev S, Murat B, Mion J, Boulay V, Manning M, Guillon G. Design, synthesis and pharmacological characterization of fluorescent peptides for imaging human V1b vasopressin or oxytocin receptors. J Med Chem. 2011;54:2864–2877. doi: 10.1021/jm1016208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Albizu L, Cottet M, Kralikova M, Stoev S, Seyer R, Brabet I, Roux T, Bazin H, Bourrier E, Lamarque L, Breton C, Rives ML, Newman A, Javitch J, Trinquet E, Manning M, Pin JP, Mouillac B, Durroux T. Time-resolved FRET between GPCR ligands reveals oligomers in native tissues. Nat Chem Biol. 2010;6:587–594. doi: 10.1038/nchembio.396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Stewart JM, Young JD. Solid Phase Synthesis. Rockford, IL: Pierce Chemical Company; 1984. [Google Scholar]
- 50.Holton PA. A modification of the method of Dale and Laidlaw for standardization of posterior pituitary extract. Br J Pharmacol. 1948;3:328–334. doi: 10.1111/j.1476-5381.1948.tb00396.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Schild HO. pA, a new scale of the measurement of drug antagonism. Br J Pharmacol Chemother. 1947;2:189–206. doi: 10.1111/j.1476-5381.1947.tb00336.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Dyckes DF, Nestor JJ, Jr, Ferger MF, du Vigneaud V. [1-β-mercapto-β,β-diethylpropionic acid]-8-lysine vasopressin, a potent inhibitor of 8-lysine-vasopressin and of oxytocin. J Med Chem. 1974;17:250–252. doi: 10.1021/jm00248a028. [DOI] [PubMed] [Google Scholar]
- 53.Munsick RA. Effect of magnesium ion on the response of the rat uterus to neurohypophysial hormones and analogues. Endocrinology. 1960;66:451–457. doi: 10.1210/endo-66-3-451. [DOI] [PubMed] [Google Scholar]
- 54.Chan WY, Kelly N. A pharmacological analysis on the significance of the chemical functional groups of oxytocin to its oxytocic activity and on the effect of magnesium on the in vitro and in vivo oxytocic activity of neurohypophysial hormones. J Pharmacol Exp Ther. 1967;156:150–158. [PubMed] [Google Scholar]
- 55.Dekanski J. The quantitative assay of vasopressin. Br J Pharmacol. 1952;7:567–572. doi: 10.1111/j.1476-5381.1952.tb00723.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sawyer WH. Biologic assays for oxytocin and vasopressin. Methods Med Res. 1961;9:210–219. [PubMed] [Google Scholar]
- 57.Morel A, O’Carroll AM, Brownstein MJ, Lolait SJ. Molecular cloning and expression of a rat V1a vasopressin receptor. Nature. 1992;356:523–526. doi: 10.1038/356523a0. [DOI] [PubMed] [Google Scholar]
- 58.Thibonnier M, Auzan C, Madhun Z, Wilkins P, Berti-Mattera L, Clauser E. Molecular cloning, sequencing, and functional expression of a cDNA encoding the human V1a vasopressin receptor. J Biol Chem. 1994;269:3304–3310. [PubMed] [Google Scholar]
- 59.Sugimoto T, Saito M, Mochizuki S, Watanabe Y, Hashimoto S, Kawashima H. Molecular cloning and functional expression of a cDNA encoding the human V1b vasopressin receptor. J Biol Chem. 1994;269:27088–27092. [PubMed] [Google Scholar]
- 60.De Keyzer Y, Auzan C, Lenne F, Beldjord C, Thibonnier M, Bertagna X, Clauser E. Cloning and characterization of the human V3 pituitary vasopressin receptor. FEBS Lett. 1994;356:215–220. doi: 10.1016/0014-5793(94)01268-7. [DOI] [PubMed] [Google Scholar]
- 61.Birnbaumer M, Seibold A, Gilbert S, Ishido M, Barberis C, Antaramian A, Brabet P, Rosenthal W. Molecular cloning of the receptor for human antidiuretic hormone. Nature. 1992;357:333–335. doi: 10.1038/357333a0. [DOI] [PubMed] [Google Scholar]
- 62.Lolait SJ, O’Carroll AM, McBride OW, Konig M, Morel A, Brownstein MJ. Cloning and characterization of a vasopressin V2 receptor and possible link to nephrogenic diabetes insipidus. Nature. 1992;357:336–339. doi: 10.1038/357336a0. [DOI] [PubMed] [Google Scholar]
- 63.Butlen D, Guillon G, Rajerison RM, Jard S, Sawyer WH, Manning M. Structural requirements for activation of vasopressin-sensitive adenylate cyclase, hormone binding, and antidiuretic actions: effects of highly potent analogues and competitive inhibitors. Mol Pharmacol. 1978;14:1006–1017. [PubMed] [Google Scholar]
- 64.Kirk CJ, Guillon G, Balestre MN, Jard S. Stimulation, by vasopressin and other agonists, of inositol-lipid breakdown and inositol phosphate accumulation in WRK1 cells. Biochem J. 1986;240:197–204. doi: 10.1042/bj2400197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kimura T, Tanizawa O, Mori K, Brownstein M, Okayama H. Structure and expression of a human oxytocin receptor. Nature. 1992;356:526–529. doi: 10.1038/356526a0. [DOI] [PubMed] [Google Scholar]
- 66.Bone EA, Fretten P, Palmer S, Kirk CJ, Michell RH. Rapid accumulation of inositol phosphates in isolated rat superior cervical sympathetic ganglia exposed to V1-vasopressin and muscarinic cholinergic stimuli. Biochem J. 1984;221:803–811. doi: 10.1042/bj2210803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Salomon Y, Londos C, Rodbell M. A highly sensitive adenylate cyclase assay. Anal Biochem. 1974;58:541–548. doi: 10.1016/0003-2697(74)90222-x. [DOI] [PubMed] [Google Scholar]
- 68.Andrés M, Trueba M, Guillon G. Pharmacological characterization of F-180: a selective human V1a vasopressin receptor agonist of high affinity. Br J Pharmacol. 2002;135:1828–1836. doi: 10.1038/sj.bjp.0704634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Derick S, Cheng LL, Voirol MJ, Stoev S, Giacomini M, Wo NC, Szeto HH, Ben Mimoun M, Andres M, Gaillard RC, Guillon G, Manning M. [1-deamino-4-cyclohexylalanine] arginine vasopressin: a potent and specific agonist for vasopressin V1b receptors. Endocrinology. 2002;143:4655–4664. doi: 10.1210/en.2002-220363. [DOI] [PubMed] [Google Scholar]
- 70.Guillon G, Pena A, Murat B, Derick S, Trueba M, Ventura MA, Szeto HH, Wo N, Stoev S, Cheng LL, Manning M. Position 4 analogues of [deamino-Cys1]arginine vasopressin exhibit striking species differences for human and rat V(2)/V(1b) receptor selectivity. J Pept Sci. 2006;12:190–198. doi: 10.1002/psc.710. [DOI] [PubMed] [Google Scholar]
- 71.Cantau B, Keppens S, De Wulf H, Jard S. 3H)-vasopressin binding to isolated rat hepatocytes and liver membranes: regulation by GTP and relation to glycogen phosphorylase activation. J Recept Res. 1980;1:137–168. doi: 10.3109/10799898009044096. [DOI] [PubMed] [Google Scholar]
- 72.Cheng LL, Stoev S, Manning M, Derick S, Pena A, Ben Mimoun M, Guillon G. Design of potent and selective agonists for the human vasopressin V1b receptor based on modifications of deamino-[Cys1]arginine vasopressin at position 4. J Med Chem. 2004;47:2375–2388. doi: 10.1021/jm030611c. [DOI] [PubMed] [Google Scholar]
- 73.Pena A, Murat B, Trueba M, Ventura MA, Bertrand G, Cheng LL, Stoev S, Szeto HH, Wo NC, Brossard G, Serradeil-Le Gal C, Manning M, Guillon G. Pharmacological and physiological characterization of d[Leu4,Lys8]Vasopressin, the first V1b selective agonist for rat vasopressin/oxytocin receptors. Endocrinology. 2007;148:4136–4146. doi: 10.1210/en.2006-1633. [DOI] [PubMed] [Google Scholar]
- 74.Pena A, Murat B, Trueba M, Ventura MA, Wo NC, Szeto HH, Cheng LL, Stoev S, Guillon G, Manning M. Design and synthesis of the first selective agonists for the rat vasopressin V1b receptor: based on modifications of deamino-[Cys1]arginine vasopressin at positions 4 and 8. J Med Chem. 2007;50:835–847. doi: 10.1021/jm060928n. [DOI] [PubMed] [Google Scholar]
- 75.Lowbridge J, Manning M, Haldar J, Sawyer WH. Synthesis and some pharmacological properties of [4-threonine, 7-glycine]oxytocin, [1-(L-2-hydroxy-3-mercaptopropanoic acid), 4-threonine, 7-glycine]oxytocin (hydroxyl[Thr4,Gly7]oxytocin), and [7-glycine]oxytocin, peptides with high oxytocic-antidiuretic selectivity. J Med Chem. 1977;20:120–123. doi: 10.1021/jm00211a025. [DOI] [PubMed] [Google Scholar]
- 76.Lee AG, Cool DR, Grunwald WC, Jr, Neal DE, Buckmaster CL, Cheng MY, Hyde SA, Lyons DM, Parker KJ. A novel form of oxytocin in New World monkeys. Biol Lett. 2011;7:584–587. doi: 10.1098/rsbl.2011.0107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Gutkowska J, Jankowski M. Oxytocin re-visited: its role in cardiovascular regulation. J Neuroendocrinol. 2011;24:599–608. doi: 10.1111/j.1365-2826.2011.02235.x. [DOI] [PubMed] [Google Scholar]
- 78.Danalache BA, Gutkowska J, Slusarz MJ, Berezowska I, Jankowski M. Oxytocin-Gly-Lys-Arg: a novel cardiomyogenic peptide. PLoS ONE. 2010;5:e13643. doi: 10.1371/journal.pone.0013643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Sawyer WH, Acosta M, Balaspiri L, Judd J, Manning M. Structural changes in the arginine vasopressin, molecule that enhance antidiuretic activity and specificity. Endocrinology. 1974;94:1106–1115. doi: 10.1210/endo-94-4-1106. [DOI] [PubMed] [Google Scholar]
- 80.Zaoral M, Kolc J, Sorm F. Amino acids and peptides. LXXI. Synthesis of 1-deamino-8-D-γ-aminobutyrine-vasopressin. 1-deamino-8-D-lysine-vasopressin, and 1-deamino-8-D-arginine-vasopressin. Coll Czech Chem Comm. 1967;32:1250–1257. [Google Scholar]
- 81.Huguenin RL. Synthèse de la Phe2-Orn8-ocytocine, deux analogues de la vasopressine douse d’une activité pressorique selective. Helv Chim Acta. 1964;47:1934–1941. [Google Scholar]
- 82.Aurell C-J, Bengtsson B, Ekholm K, Kasprzykowska R, Nilson A, Person R, Trojnar J, Abbe M, Melin P. Development of vasopressor specific analogues with prolonged effects. In: Giralt E, Andreu D, editors. Peptides 1990. Leiden: ESCOM Science Publishers; 1991. pp. 671–673. Proc of the 21European Peptide Symposium. [Google Scholar]
- 83.Wisniewski K, Galyean R, Tariga H, Alagarsamy S, Croston G, Heitzmann J, Kohan A, Wisniewska H, Laporte R, Rivière PJ, Schteingart CD. New, potent, selective, and short-acting peptidic V1a receptor agonists. J Med Chem. 2011;54:4388–4398. doi: 10.1021/jm200278m. [DOI] [PubMed] [Google Scholar]
- 84.Manning M, Coy EJ, Sawyer WH, Acosta M. Solid-phase synthesis and some pharmacological properties of 4-threonine analogs of vasopressins and vasotocin and of arginine-vasopressin and arginine-vasotocin. J Med Chem. 1973;16:463–466. doi: 10.1021/jm00263a009. [DOI] [PubMed] [Google Scholar]
- 85.Boissonnas RA, Guttmann S. Synthese d’analogues de l’oxytocine et de la lysine-vasopressine contenant de la phenylalanine ou de la tyrosine en position 2 et 3. Helv Chim Acta. 1960;43:190–200. [Google Scholar]
- 86.Guillon G, Murat B, Pena A, Trueba M, Ventura MA, Wo NC, Szeto HH, Stoev S, Cheng LL, Manning M. Selective agonists for human and rat vasopressin V1b receptors. In: Rolka K, Rekowski P, Silberring J, editors. Peptides 2006. Geneva: Kenes International; 2007. pp. 742–743. [Google Scholar]
- 87.Dyckes DF, Ferger MF, du Vigneaud V. Synthesis and some of the pharmacological properties of (4-leucine)-8-lysine-vasopressin and (1-deamino,4-leucine)-8-lysine-vasopressin. J Med Chem. 1973;16:843–847. doi: 10.1021/jm00265a022. [DOI] [PubMed] [Google Scholar]
- 88.Saito M, Sugimoto T, Tahara A, Kawashima H. Molecular cloning and characterization of rat V1b vasopressin receptor: evidence for its expression in extra-pituitary tissues. Biochem Biophys Res Commun. 1995;26:751–757. doi: 10.1006/bbrc.1995.2033. [DOI] [PubMed] [Google Scholar]
- 89.Griffante C, Green A, Curcuruto O, Haslam CP, Dickinson BA, Arban R. Selectivity of d[Cha4]AVP and SSR149415 at human vasopressin and oxytocin receptors: evidence that SSR149415 is a mixed vasopressin V1b/oxytocin receptor antagonist. Br J Pharmacol. 2005;146:744–751. doi: 10.1038/sj.bjp.0706383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Roper JA, O’Carroll A-M, Young WS, III, Lolait SJ. The vasopressin Avprib receptor: molecular and pharmacological studies. Stress. 2011;14:98–115. doi: 10.3109/10253890.2010.512376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Kruszynski M, Lammek B, Manning M, Seto J, Haldar J, Sawyer WH. [1-beta-Mercapto-beta,beta-cyclopentamethylenepropionic acid), 2-(O-methyl)tyrosine] arginine-vasopressin and [1-beta-mercapto-beta,beta-cyclopentamethylenepropionic acid)]argine-vasopressine, two highly potent antagonists of the vasopressor response to arginine-vasopressin. J Med Chem. 1980;23:364–368. doi: 10.1021/jm00178a003. [DOI] [PubMed] [Google Scholar]
- 92.Chan WY, Wo NC, Cheng LL, Manning M. Isosteric substitution of Asn5 in antagonists of oxytocin and vasopressin leads to highly selective and potent oxytocin and V1a receptor antagonists: new approaches for the design of potential tocolytics for preterm labor. J Pharmacol Exp Ther. 1996;227:999–1003. [PubMed] [Google Scholar]
- 93.Manning M, Przybylski JP, Olma A, Klis WA, Kruszynski M, Wo NC, Pelton GH, Sawyer WH. No requirement of cyclic conformation of antagonists in binding to vasopressin receptors. Nature. 1987;329:839–840. doi: 10.1038/329839a0. [DOI] [PubMed] [Google Scholar]
- 94.Manning M, Nawrocka E, Misicka A, Olma A, Klis WA, Seto J, Sawyer WH. Potent and selective antagonists of the antidiuretic responses to arginine-vasopressin based on modifications of [1-(beta-mercapto-β,β-pentamethylenepropionic acid),2-D-isoleucine,4-valine]arginine-vasopressin at position 4. J Med Chem. 1984;27:423–429. doi: 10.1021/jm00370a002. [DOI] [PubMed] [Google Scholar]
- 95.Manning M, Klis WA, Kruszynski M, Przybylski JP, Olma A, Wo NC, Pelton GH, Sawyer WH. Novel linear antagonists of the antidiuretic (V2) and vasopressor (V1) responses to vasopressin. Int J Pept Protein Res. 1988;32:455–467. doi: 10.1111/j.1399-3011.1988.tb01376.x. [DOI] [PubMed] [Google Scholar]
- 96.Manning M, Bankowski K, Barberis C, Jard S, Elands J, Chan WY. Novel approach to the design of synthetic radioiodinated linear V1a receptor antagonists of vasopressin. Int J Pept Protein Res. 1992;40:261–267. doi: 10.1111/j.1399-3011.1992.tb00300.x. [DOI] [PubMed] [Google Scholar]
- 97.Barberis C, Balestre MN, Jard S, Tribollet E, Arsenijevic Y, Dreifuss JJ, Bankowski K, Manning M, Chan WY, Schlosser Y, Holsboer F, Elands Y. Characterization of a novel, linear radioiodinated vasopressin antagonist: an excellent radioligand for vasopressin V1a receptors. Neuroendocrinology. 1995;62:135–146. doi: 10.1159/000126998. [DOI] [PubMed] [Google Scholar]
- 98.Manning M, Olma A, Klis WA, Kolodziejczyk AM, Seto J, Sawyer WH. Design of more potent antagonists of the antidiuretic responses to arginine-vasopressin. J Med Chem. 1982;25:45–50. doi: 10.1021/jm00343a009. [DOI] [PubMed] [Google Scholar]
- 99.Manning M, Misicka A, Olma A, Klis WA, Bankowski K, Nawrocka E, Kruszynski M, Kolodziejczyk A, Cheng LL, Seto J, Wo NC, Sawyer WH. C-terminal deletions in agonistic and antagonistic analogues of vasopressin that improve their specificities for antidiuretic (V2) and vasopressor (V1) receptors. J Med Chem. 1987;30:2245–2252. doi: 10.1021/jm00395a012. [DOI] [PubMed] [Google Scholar]
- 100.Sawyer WH, Bankowski K, Misicka A, Nawrocka E, Kruszynski M, Stoev S, Klis WA, Przybylski JP, Manning M. Potent V2 vasopressin antagonists with structural changes at their C-terminals. Peptides. 1988;9:157–163. doi: 10.1016/0196-9781(88)90022-8. [DOI] [PubMed] [Google Scholar]
- 101.Manning M, Przybylski J, Grzonka Z, Nawrocka E, Lammek B, Misicka A, Cheng LL, Chan WY, Wo NC, Sawyer WH. Potent V2/V1a vasopressin antagonists with C-terminal ethylenediamine-linked retro-amino acids. J Med Chem. 1992;35:3895–3904. doi: 10.1021/jm00099a018. [DOI] [PubMed] [Google Scholar]
- 102.Akerlund M. Targeting the oxytocin receptor to relax the myometrium. Expert Opin Ther Targets. 2006;10:423–427. doi: 10.1517/14728222.10.3.423. [DOI] [PubMed] [Google Scholar]
- 103.Bankowski K, Manning M, Seto J, Haldar J, Sawyer WH. Design and synthesis of potent in vivo antagonists of oxytocin. Int J Pept Prot Res. 1980;16:382–391. doi: 10.1111/j.1399-3011.1980.tb02962.x. [DOI] [PubMed] [Google Scholar]
- 104.Manning M, Miteva K, Pancheva S, Stoev S, Wo NC, Chan WY. Design and synthesis of highly selective in vitro and in vivo uterine receptor antagonists of oxytocin: comparisons with atosiban. Int J Peptide Protein Res. 1995;46:244–252. doi: 10.1111/j.1399-3011.1995.tb00596.x. [DOI] [PubMed] [Google Scholar]
- 105.Elands J, Barberis C, Jard S, Tribollet E, Dreifuss J-J, Bankowski K, Manning M, Sawyer WH. 125I-labelled d(CH25[Tyr(Me)2,Thr4,Tyr-NH29]OVT: a selective oxytocin receptor ligand. Eur J Pharmacol. 1988;147:197–207. doi: 10.1016/0014-2999(88)90778-9. [DOI] [PubMed] [Google Scholar]
- 106.Melin P, Trojnar J, Johansson B, Vilhardt H, Akerlund M. Synthetic antagonists of the myometrial response to vasopressin and oxytocin. J Endocrinol. 1986;111:125–131. doi: 10.1677/joe.0.1110125. [DOI] [PubMed] [Google Scholar]
- 107.Yamamura Y, Ogawa H, Chihara T, Kondo K, Onogawa T, Nakamura S, Mori T, Tominaga M, Yabuuchi Y. OPC-21268, as an orally effective, nonpeptide vasopressin V1 receptor antagonist. Science. 1991;252:572–574. doi: 10.1126/science.1850553. [DOI] [PubMed] [Google Scholar]
- 108.Yamamura Y, Nakamura S, Itoh S, Hirano T, Onogawa T, Yamashita T, Yamada Y, Tsujimae K, Aoyama M, Kotosai K, Ogawa H, Yamashita H, Kondo K, Tominaga M, Tsujimoto G, Mori T. OPC-41061, a highly potent human vasopressin V2-receptor antagonist: pharmacological profile and aquaretic effect by single and multiple oral dosing in rats. J Pharmacol Exp Ther. 1998;287:860–867. [PubMed] [Google Scholar]
- 109.Schrier RW, Gross P, Gheorghiade M, Berl T, Verbalis JG, Czerwiec FS, Orlandi C SALT Investigators. Tolvaptan, a selective oral vasopressin V2-receptor antagonist, for hyponatremia. N Engl J Med. 2006;355:2099–2112. doi: 10.1056/NEJMoa065181. [DOI] [PubMed] [Google Scholar]
- 110.Tahara A, Tomura Y, Wade K-I, Kusayama T, Tsukada J, Takanashi M, Yatsu T, Uchida W, Tanaka A. A pharmacological profile of YM087, a novel potent nonpeptide vasopressin V1a and V2 receptor antagonist, in vitro and in vivo. J Pharmacol Exp Ther. 1997;282:301–308. [PubMed] [Google Scholar]
- 111.Ghali JK, Tam SW. The critical link of hypervolemia and hyponatremia in heart failure and the potential role of arginine vasopressin antagonists. J Card Fail. 2010;16:419–431. doi: 10.1016/j.cardfail.2009.12.021. [DOI] [PubMed] [Google Scholar]
- 112.Robertson GL. Vaptans for the treatment of hyponatremia. Nat Rev Endocrinol. 2011;7:151–161. doi: 10.1038/nrendo.2010.229. [DOI] [PubMed] [Google Scholar]
- 113.Gross PA, Wagner A, Decaux G. Vaptans are not the mainstay of treatment in hyponatremia: perhaps not yet. Kidney Int. 2011;80:594–600. doi: 10.1038/ki.2011.78. [DOI] [PubMed] [Google Scholar]
- 114.Verbalis JG, Adler S, Schrier RW, Berl T, Zhao Q, Czerwiec FS SALT Investigators. Efficacy and safety of oral tolvaptan therapy in patients with the syndrome of inappropriate antidiuretic hormone secretion. Eur J Endocrinol. 2011;164:725–732. doi: 10.1530/EJE-10-1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Ferguson-Myrthil N. Novel agents for the treatment of hyponatremia: a review of conivaptan and tolvaptan. Cardiol Rev. 2010;18:313–321. doi: 10.1097/CRD.0b013e3181f5b3b7. [DOI] [PubMed] [Google Scholar]
- 116.Serradeil-Le Gal C, Wagnon J, Garcia C, Lacour C, Guiraudou P, Christophe B, Villanova G, Nisato D, Maffrand JP, Le Fur G. Biochemical and pharmacological properties of SR 49059, a new, potent, nonpeptide antagonist of rat and human vasopressin V1a receptors. J Clin Invest. 1993;92:224–231. doi: 10.1172/JCI116554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Manaenko A, Fathali N, Khatibi NH, Lekic T, Hasegawa Y, Martin R, Tang J, Zhang JH. Arginine-vasopressin V1a receptor inhibition improves neurologic outcomes following an intracerebral hemorrhagic brain injury. Neurochem Int. 2011;58:542–548. doi: 10.1016/j.neuint.2011.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Russell R, Doyle R, Turner J, Attkins N, Ramsey S, Weibley L, Bateman L, Bictash M, Neal-Morgan S, Ivarsson M, Pullen N. In vitro and in vivo pharmacological characterisation of the potent and selective vasopressin V(1a) receptor antagonist 4-[4-(4-chloro-phenyl)-5-[1,2,3]triazol-2-ylmethyl-4H-[1,2,4]triazol-3-yl]-piperidin-1-yl-(3,5-difluoro-phenyl) methanone (PF-00738245) Eur J Pharmacol. 2011;670:347–355. doi: 10.1016/j.ejphar.2011.09.034. [DOI] [PubMed] [Google Scholar]
- 119.Schönberger M, Leggett C, Kim SW, Hooker JM. Synthesis of [11C]SSR149415 and preliminary imaging studies using positron emission tomography. Bioorg Med Chem Lett. 2010;20:3103–3106. doi: 10.1016/j.bmcl.2010.03.108. [DOI] [PubMed] [Google Scholar]
- 120.Litvin Y, Murakami G, Pfaff DW. Effects of chronic social defeat on behavioral and neural correlates of sociality: vasopressin, oxytocin and the vasopressinergic V1b receptor. Physiol Behav. 2011;103:393–403. doi: 10.1016/j.physbeh.2011.03.007. [DOI] [PubMed] [Google Scholar]
- 121.Zmily HD, Daifallah S, Ghali JK. Tolvaptan, hyponatremia, and heart failure. Int J Nephrol Renovasc Dis. 2011;4:57–71. doi: 10.2147/IJNRD.S7032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Pang PS, Gheorghiade M, Dihu J, Swedberg K, Khan S, Maggioni AP, Grinfeld L, Zannad F, Burnett JC, Jr, Ouyang J, Udelson JE, Konstam MA. Effects of tolvaptan on physician-assessed symptoms and signs in patients hospitalized with acute heart failure syndromes: analysis from the efficacy of vasopressin antagonism in heart failure outcome study with tolvaptan (EVEREST) trials. Am Heart J. 2011;161:1067–1072. doi: 10.1016/j.ahj.2011.02.027. [DOI] [PubMed] [Google Scholar]
- 123.Gassanov N, Semmo N, Semmo M, Nia AM, Fuhr U, Er F. Arginine vasopressin (AVP) and treatment with arginine vasopressin receptor antagonists (vaptans) in congestive heart failure, liver cirrhosis and syndrome of inappropriate antidiuretic hormone secretion (SIADH) Eur J Clin Pharmacol. 2011;67:333–346. doi: 10.1007/s00228-011-1006-7. [DOI] [PubMed] [Google Scholar]
- 124.Serradeil-Le Gal C, Lacour C, Valette G, Garcia G, Foulon L, Galindo G, Bankir L, Pouzet B, Guillon G, Barberis C, Chicot D, Jard S, Vilain P, Garcia C, Marty E, Raufaste D, Brossard G, Nisato D, Maffrand JP, Le Fur G. Characterization of SR 121463A, a highly potent and selective, orally active vasopressin V2 receptor antagonist. J Clin Invest. 1996;98:2729–2738. doi: 10.1172/JCI119098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Rochdi MD, Vargas GA, Carpentier E, Oligny-Longpré G, Chen S, Kovoor A, Gitelman SE, Rosenthal SM, von Zastrow M, Bouvier M. Functional characterization of vasopressin type 2 receptor substitutions (R137H/C/L) leading to nephrogenic diabetes insipidus and nephrogenic syndrome of inappropriate antidiuresis: implications for treatments. Mol Pharmacol. 2010;77:836–845. doi: 10.1124/mol.109.061804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Ranadive SA, Ersoy B, Favre H, Cheung CC, Rosenthal SM, Miller WL, Vaisse C. Identification, characterization and rescue of a novel vasopressin-2 receptor mutation causing nephrogenic diabetes insipidus. Clin Endocrinol (Oxf) 2009;71:388–393. doi: 10.1111/j.1365-2265.2008.03513.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Yamamura Y, Ogawa H, Yamashita H, Chihara T, Miyamoto H, Nakamura S, Onogawa T, Yamashita T, Hosokawa T, Mori T. Characterization of a novel aquaretic agent, OPC-31260, as an orally effective, nonpeptide vasopressin V2 receptor antagonist. Br J Pharmacol. 1992;105:787–791. doi: 10.1111/j.1476-5381.1992.tb09058.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Naidech AM, Paparello J, Liebling SM, Bassin SL, Levasseur K, Alberts MJ, Bernstein RA, Muro K. Use of Conivaptan (Vaprisol) for hyponatremic neuro-ICU patients. Neurocrit Care. 2010;13:57–61. doi: 10.1007/s12028-010-9379-5. Erratum in: Neurocrit Care 2011; 15: 210. [DOI] [PubMed] [Google Scholar]
- 129.Velez JC, Dopson SJ, Sanders DS, Delay TA, Arthur JM. Intravenous conivaptan for the treatment of hyponatraemia caused by the syndrome of inappropriate secretion of antidiuretic hormone in hospitalized patients: a single-centre experience. Nephrol Dial Transplant. 2010;25:1524–1531. doi: 10.1093/ndt/gfp731. [DOI] [PubMed] [Google Scholar]
- 130.Williams PD, Clineschmidt BV, Erb JM, Freidinger RM, Guidotti MT, Lis EV, Pawluczyk JM, Pettibone DJ, Reiss DR, Veber DF, Woyden CY. 1-(1-[(N-acetyl-4-piperidinyl)oxy]-2-methoxybenzoyl]piperidin-4-yl)-4H-3,1-benzoxazin-2(1H)-one (L-371-257): a new, orally bioavailable, non-peptide oxytocin antagonist. J Med Chem. 1995;38:4634–4636. doi: 10.1021/jm00023a002. [DOI] [PubMed] [Google Scholar]
- 131.Ring RH, Malberg JE, Potestio L, Ping J, Boikess S, Luo B, Schechter LE, Rizzo S, Rahman Z, Rosenzweig-Lipson S. Anxiolytic-like activity of oxytocin in male mice: behavioral and autonomic evidence, therapeutic implications. Psychopharmacology. 2006;185:218–225. doi: 10.1007/s00213-005-0293-z. [DOI] [PubMed] [Google Scholar]
- 132.Liddle J, Allen MJ, Borthwick AD, Brooks DP, Davies DE, Edwards RM, Exall AM, Hamlett C, Irving WR, Mason AM, McCafferty GP, Nerozzi F, Peace S, Philp J, Pollard D, Pullen MA, Shabbir SS, Sollis SL, Westfall TD, Woollard PM, Wu C, Hickey DM. The discovery of GSK221149A: a potent and selective oxytocin antagonist. Bioorg Med Chem Lett. 2008;18:90–94. doi: 10.1016/j.bmcl.2007.11.008. [DOI] [PubMed] [Google Scholar]
- 133.Borthwick AD. Oral oxytocin antagonists. J Med Chem. 2010;53:6525–6538. doi: 10.1021/jm901812z. [DOI] [PubMed] [Google Scholar]
- 134.Wootten DL, Simms J, Massoura AJ, Trim JE, Wheatley M. Agonist-specific requirement for a glutamate in transmembrane helix 1 of the oxytocin receptor. Mol Cell Endocrinol. 2011;333:20–27. doi: 10.1016/j.mce.2010.11.029. [DOI] [PubMed] [Google Scholar]
- 135.Smith AS, Agmo A, Birnie AK, French JA. Manipulation of the oxytocin system alters social behavior and attraction in pair-bonding primates, Callithrix penicillata. Horm Behav. 2010;57:255–262. doi: 10.1016/j.yhbeh.2009.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Olszewski PK, Klockars A, Olszewska AM, Fredriksson R, Schiöth HB, Levine AS. Molecular, immunohistochemical, and pharmacological evidence of oxytocin’s role as inhibitor of carbohydrate but not fat intake. Endocrinology. 2010;151:4736–4744. doi: 10.1210/en.2010-0151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Frantz MC, Rodrigo J, Boudier L, Durroux T, Mouillac B, Hibert M. Subtlety of the structure–affinity and structure–efficacy relationships around a nonpeptide oxytocin receptor agonist. J Med Chem. 2010;53:1546–1562. doi: 10.1021/jm901084f. [DOI] [PubMed] [Google Scholar]
- 138.Ring RH, Schechter LE, Leonard SK, Dwyer JM, Platt BJ, Graf R, Grauer S, Pulicicchio C, Resnick L, Rahman Z, Sukoff Rizzo SJ, Luo B, Beyer CE, Logue SF, Marquis KL, Hughes ZA, Rosenzweig-Lipson S. Receptor and behavioral pharmacology of WAY-267464, a non-peptide oxytocin receptor agonist. Neuropharmacology. 2010;58:69–77. doi: 10.1016/j.neuropharm.2009.07.016. [DOI] [PubMed] [Google Scholar]
- 139.Insel TR. The challenge of translation in social neuroscience: a review of oxytocin, vasopressin, and affiliative behavior. Neuron. 2010;65:768–779. doi: 10.1016/j.neuron.2010.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Rahman Z, Resnick L, Rosenzweig-Lipton SJ, Ring RH. Methods of treatment using oxytocin receptor agonists. US Patent 20070117794, Kind Code: A1, Publication date 10/24/2007.
- 141.Beyer CE, Dwyer JM, Platt BJ, Neal S, Luo B, Ling HP, Lin Q, Mark RJ, Rosenzweig-Lipson S, Schechter LE. Angiotensin IV elevates oxytocin levels in the rat amygdala and produces anxiolytic-like activity through subsequent oxytocin receptor activation. Psychopharmacology. 2010;209:303–311. doi: 10.1007/s00213-010-1791-1. [DOI] [PubMed] [Google Scholar]
- 142.Jard S, Gaillard RC, Guillon G, Marie J, Schoenenberg P, Muller AF, Manning M, Sawyer WH. Vasopressin antagonists allow demonstration of a novel type of vasopressin receptor in the rat adenohypophysis. Mol Pharmacol. 1986;30:171–177. [PubMed] [Google Scholar]
- 143.Pradelles P, Morgat JL, Fromageot P, Camier M, Bonne D, Cohen P, Bockaert J, Jard S. Tritium labelling of 8-lysine vasopressin and its purification by affinity chromatography on sepharose bound neurophysins. FEBS Lett. 1972;26:189–192. doi: 10.1016/0014-5793(72)80570-2. [DOI] [PubMed] [Google Scholar]
- 144.Berridge MJ, Irvine RF. Inositol trisphosphate, a novel second messenger in cellular signal transduction. Nature. 1984;312:315–321. doi: 10.1038/312315a0. [DOI] [PubMed] [Google Scholar]
- 145.Bockaert J, Roy C, Rajerison R, Jard S. Specific binding of (3H)lysine-vasopressin to pig kidney plasma membranes. Relationship of receptoroccupancy to adenylate cyclase activation. J Biol Chem. 1973;248:5922–5931. [PubMed] [Google Scholar]
- 146.Terillon S, Cheng LL, Stoev S, Mouillac B, Barberis C, Manning M, Durroux T. Synthesis and characterization of fluorescent antagonists and agonists for human oxytocin and vasopressin V1a receptors. J Med Chem. 2002;45:2579–2588. doi: 10.1021/jm010526+. [DOI] [PubMed] [Google Scholar]
- 147.Serradeil-Le Gal C, Raufaste D, Derick S, Blankenstein J, Allen J, Pouzet B, Pascal M, Wagnon J, Ventura MA. Biological characterization of rodent and human vasopressin V1b receptors using SSR-149415, a nonpeptide V1b receptor ligand. Am J Physiol Regul Integr Comp Physiol. 2007;293:R938–R949. doi: 10.1152/ajpregu.00062.2007. [DOI] [PubMed] [Google Scholar]
- 148.Thibonnier M, Preston JA, Dulin N, Wilkins PL, Berti-Mattera LN, Mattera R. The human V3 pituitary vasopressin receptor: ligand binding profile and density-dependent signaling pathways. Endocrinology. 1997;138:4109–4122. doi: 10.1210/endo.138.10.5432. [DOI] [PubMed] [Google Scholar]
- 149.Mouillac B, Chini B, Balestre MN, Elands J, Trumpp-Kallmeyer S, Hoflack J, Hibert M, Jard S, Barberis C. The binding site of neuropeptide vasopressin V1a receptor. Evidence for a major localization within transmembrane regions. J Biol Chem. 1995;270:25771–25777. doi: 10.1074/jbc.270.43.25771. [DOI] [PubMed] [Google Scholar]
- 150.Oshikawa S, Tanoue A, Koshimizu TA, Kitagawa Y, Tsujimoto G. Vasopressin stimulates insulin release from islet cells through V1b receptors: a combined pharmacological/knockout approach. Mol Pharmacol. 2004;65:623–629. doi: 10.1124/mol.65.3.623. [DOI] [PubMed] [Google Scholar]
- 151.Chini B, Mouillac B, Balestre MN, Trumpp-Kallmeyer S, Hoflack J, Hibert M, Andriolo M, Pupier S, Jard S, Barberis C. Two aromatic residues regulate the response of the human oxytocin receptor to the partial agonist arginine vasopressin. FEBS Lett. 1996;397:201–206. doi: 10.1016/s0014-5793(96)01135-0. [DOI] [PubMed] [Google Scholar]
- 152.Tahara A, Saito M, Sugimoto T, Tomura Y, Wada K, Kusayama T, Tsukada J, Ishii N, Yatsu T, Uchida W, Tanaka A. Pharmacological characterization of the human vasopressin receptor subtypes stably expressed in Chinese hamster ovary cells. Br J Pharmacol. 1998;125:1463–1470. doi: 10.1038/sj.bjp.0702220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Elands J, Barberis C, Jard S. [3H]-[Thr4,Gly7]OT: a highly selective ligand for central and peripheral OT receptors. Am J Physiol. 1988;254:E31–E38. doi: 10.1152/ajpendo.1988.254.1.E31. [DOI] [PubMed] [Google Scholar]
- 154.Chini B, Mouillac B, Ala Y, Balestre MN, Trumpp-Kallmeyer S, Hoflack J, Elands J, Hibert M, Manning M, Jard S. Tyr115 is the key residue for determining agonist selectivity in the V1a vasopressin receptor. EMBO J. 1995;14:2176–2182. doi: 10.1002/j.1460-2075.1995.tb07211.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Andrés M, Peña A, Derick S, Raufaste D, Trojnar J, Wisniewski K, Trueba M, Serradeil-Le Gal C, Guillon G. Comparative pharmacology of bovine, human and rat vasopressin receptor isoforms. Eur J Pharmacol. 2004;501:59–69. doi: 10.1016/j.ejphar.2004.08.026. [DOI] [PubMed] [Google Scholar]
- 156.Chini B, Manning M. Agonist selectivity in the oxytocin/vasopressin receptor family: new insights and challenges. Biochem Soc Trans. 2007;35:737–741. doi: 10.1042/BST0350737. [DOI] [PubMed] [Google Scholar]
- 157.Chini B, Manning M, Guillon G. Affinity and efficacy of selective agonists and antagonists for vasopressin and oxytocin receptors: an ‘easy guide’ to receptor pharmacology. Prog Brain Res. 2008;170:513–517. doi: 10.1016/S0079-6123(08)00438-X. [DOI] [PubMed] [Google Scholar]
- 158.Sala M, Braida D, Lentini D, Busnelli M, Bulgheroni E, Capurro V, Finardi A, Donzelli A, Pattini L, Rubino T, Parolaro D, Nishimori K, Parenti M, Chini B. Pharmacologic rescue of impaired cognitive flexibility, social deficits, increased aggression, and seizure susceptibility in oxytocin receptor null mice: a neurobehavioral model of autism. Biol Psychiatry. 2011;69:875–882. doi: 10.1016/j.biopsych.2010.12.022. [DOI] [PubMed] [Google Scholar]
- 159.Mimoun MB, Derick S, Andres M, Guillon G, Wo NC, Chan WY, Stoev S, Cheng LL, Manning M. Vasopressin V2 agonists: affinities for human and rat V2 and V1a receptors reveal surprising species differences. In: Martinez J, Fehrentz JA, editors. Peptides 2000. Paris: EDK, Editions Medicales et Scientifiques; 2001. pp. 589–590. [Google Scholar]
- 160.Manning M, Cheng LL, Stoev S, Wo NC, Chan WY, Szeto HH, Durroux T, Mouillac B, Barberis C. Design of peptide oxytocin antagonists with strikingly higher affinities and selectivities for the human oxytocin receptor than atosiban. J Pept Sci. 2005;11:593–608. doi: 10.1002/psc.667. [DOI] [PubMed] [Google Scholar]
- 161.Serradeil-Le Gal C, Valette G, Foulon L, Germain G, Advenier C, Naline E, Bardou M, Martinolle JP, Pouzet B, Raufaste D, Garcia C, Double-Cazanave E, Pauly M, Pascal M, Barbier A, Scatton B, Maffrand JP, Le Fur G. SSR126768A (4-chloro-3-[(3R)-(+)-5-chloro-1-(2,4-dimethoxybenzyl)-3-methyl-2-oxo-2,3-dihydro-1H-indol-3-yl]-N-ethyl-N-(3-pyridylmethyl)-benzamide, hydrochloride): a new selective and orally active oxytocin receptor antagonist for the prevention of preterm labor. J Pharmacol Exp Ther. 2004;309:414–424. doi: 10.1124/jpet.103.061200. [DOI] [PubMed] [Google Scholar]
- 162.Schmidt A, Audigier S, Barberis C, Jard S, Manning M, Kolodziejczyk AS, Sawyer WH. A radioidinated linear vasopresin antagonist. FEBS. 1991;282:77–81. doi: 10.1016/0014-5793(91)80448-c. [DOI] [PubMed] [Google Scholar]
- 163.Jasper JR, Harrell CM, O’Brien JA, Pettibone DJ. Characterization of the human oxytocin receptor stably expressed in 293 human embryonic kidney cells. Life Sci. 1995;57:2253–2261. doi: 10.1016/0024-3205(95)02218-8. [DOI] [PubMed] [Google Scholar]
- 164.Serradeil-Le Gal C, Wagnon J, Simiand J, Griebel G, Lacour C, Guillon G, Barberis C, Brossard G, Soubrié P, Nisato D, Pascal M, Pruss R, Scatton B, Maffrand JP, Le Fur G. Characterization of (2S,4R)-1-[5-chloro-1-[(2,4-dimethoxyphenyl)sulfonyl]-3-(2-methoxy-phenyl)-2-oxo-2,3-dihydro-1H-indol-3yl]-4-hydroxy-N,N-dimethyl-2-pyrrolidine carboxamide (SSR149415), a selective and orally active vasopressin V1b receptor antagonist. J Pharmacol Exp Ther. 2002;300:1122–1130. doi: 10.1124/jpet.300.3.1122. [DOI] [PubMed] [Google Scholar]
- 165.Peters MF, Vaillancourt F, Heroux M, Valiquette M, Scott CW. Comparing label-free biosensors for pharmacological screening with cell-based functional assays. Assay Drug Dev Technol. 2010;8:219–227. doi: 10.1089/adt.2009.0232. [DOI] [PubMed] [Google Scholar]
- 166.Busnelli M, Sauliere A, Manning M, Bouvier M, Gales C, Chini B. Functional selective oxytocin-derived agonists discriminate between individual G protein family subtypes. J Biol Chem. 2012;287:3617–3629. doi: 10.1074/jbc.M111.277178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Caldwell HK, Lee H-J, Macbeth AH, Young WS., 3rd Vasopressin: behavioral roles of an original neuropeptide. Prog in Neurobiol. 2008;84:1–24. doi: 10.1016/j.pneurobio.2007.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Kelly JM, Abrahams JM, Philips PA, Mendelsohn FA, Grzonka Z, Johnston CI. [125I]-[d(CH25,Sar7]AVP: a selective radioligand for V1 vasopressin receptors. J Recept Res. 1989;9:27–41. doi: 10.3109/10799898909066043. [DOI] [PubMed] [Google Scholar]
- 169.Tribollet E, Goumaz M, Raggenbass M, Dreifuss JJ. Appearance and transient expression of vasopressin and oxytocin receptors in the rat brain. J Recept Res. 1991;11:333–346. doi: 10.3109/10799899109066412. [DOI] [PubMed] [Google Scholar]
- 170.Hernando F, Schoots O, Lolait SJ, Burbach JPH. Immunohistochemical localisation of the vasopressin V1b receptor in the rat brain and pituitary gland: anatomical support for its involvement in the central effects of vasopressin. Endocrinology. 2001;142:1659–1668. doi: 10.1210/endo.142.4.8067. [DOI] [PubMed] [Google Scholar]
- 171.Hurbin A, Boissin-Agasse L, Orcel H, Rabié A, Joux N, Desarménien MG, Richard P, Moos FC. The V1a and V1b, but not V2, vasopressin receptor genes are expressed in the supraoptic nucleus of the rat hypothalamus, and the transcripts are essentially colocalized in the vasopressinergic magnocellular neurons. Endocrinology. 1998;139:4701–4707. doi: 10.1210/endo.139.11.6320. [DOI] [PubMed] [Google Scholar]
- 172.Hurbin A, Dubrez L, Coll JL, Favrot MC. The vasopressin receptors colocalize with vasopressin in the magnocellular neurons of the rat supraoptic nucleus and are modulated by water balance. Endocrinology. 2002;143:456–466. doi: 10.1210/endo.143.2.8643. [DOI] [PubMed] [Google Scholar]
- 173.Young WS, Li J, Wersinger SR, Palkovits M. The vasopressin 1b receptor is prominent in the hippocampal area CA2 where it is unaffected by restraint stress or adrenalectomy. Neuroscience. 2006;143:1031–1039. doi: 10.1016/j.neuroscience.2006.08.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Mouillac B, Manning M, Durroux T. Fluorescent agonists and antagonists for vasopressin/oxytocin G protein-coupled receptors: usefulness in ligand screening assays and receptor studies. Mini Rev Med Chem. 2008;8:996–1005. doi: 10.2174/138955708785740607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Taki M, Hohsaka T, Murakami H, Taira K, Sisido M. A non-natural amino acid for efficient incorporation into proteins as a sensitive fluorescent probe. FEBS Lett. 2001;507:35–38. doi: 10.1016/s0014-5793(01)02935-0. [DOI] [PubMed] [Google Scholar]
- 176.Panchuk-Voloshina N, Haugland RP, Bishop-Stewart J, Bhalgat MK, Millard PJ, Mao F, Leung W-Y, Huagland R. Alexa dyes, a series of new fluorescent dyes that yield exceptionally bright, photostable conjugates. J Histochem Cytochem. 1999;47:1179–1188. doi: 10.1177/002215549904700910. [DOI] [PubMed] [Google Scholar]
- 177.Ballard JL, Peeva VK, deSilva CJ, Lynch JL, Swanson NR. Comparison of Alexa Fluor and Cy Dye for practical DNA microarray use. Mol Biotechnol. 2007;36:175–183. doi: 10.1007/s12033-007-0006-4. [DOI] [PubMed] [Google Scholar]
- 178.Durroux T, Peter M, Turcatti G, Cholet A, Balestre MN, Barberis C, Seyer R. Fluorescent pseudo-peptide linear vasopressin antagonists: design, synthesis and applications. J Med Chem. 2009;42:1312–1319. doi: 10.1021/jm9804782. [DOI] [PubMed] [Google Scholar]
- 179.Kirk KL, Buku A, Eggena P. Cell specificity of vasopressin binding in renal collecting duct: computer-enhanced imaging of a fluorescent hormone analog. Proc Natl Acad Sci USA. 1987;84:6000–6004. doi: 10.1073/pnas.84.16.6000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Bonnet D, Riché S, Loison S, Dagher R, Frantz MC, Boudier L, Rameh R, Mouillac B, Haiech J, Hibert M. Solid-phase organic tagging resins for labeling biomolecules by 1,3-dipolar cycloaddition: application to the synthesis of a fluorescent non-peptidic vasopressin recptor ligand. Chemistry. 2008;14:6247–6254. doi: 10.1002/chem.200800273. [DOI] [PubMed] [Google Scholar]
- 181.Maurel D, Kniazeff J, Mathis G, Trinquet E, Pin JP, Ansanay H. Cell surface detection of membrane protein interaction with homogeneous time-resolved fluorescence resonance energy transfer technology. Anal Biochem. 2004;329:253–262. doi: 10.1016/j.ab.2004.02.013. [DOI] [PubMed] [Google Scholar]
- 182.Milligan G, Bouvier M. Methods to monitor the quaternary structure of G protein-coupled receptors. FEBS J. 2005;272:2914–2925. doi: 10.1111/j.1742-4658.2005.04731.x. [DOI] [PubMed] [Google Scholar]
- 183.Albizu L, Balestre MN, Breton C, Pin JP, Manning M, Mouillac B, Barberis C, Durroux T. Probing the existence of G protein-coupled recptor dimers by positive and negative ligand-dependent cooperative binding. Mol Pharmacol. 2006;70:1783–1791. doi: 10.1124/mol.106.025684. [DOI] [PubMed] [Google Scholar]
- 184.Orcel H, Albizu L, Perkovska S, Durroux T, Mendre C, Ansanay H, Mouillac B, Rabié A. Differential coupling of the vasopressin V1b receptor through compartmentalization within the plasma membrane. Mol Pharmacol. 2009;75:637–647. doi: 10.1124/mol.108.049031. [DOI] [PubMed] [Google Scholar]
- 185.Terrillon S, Durroux T, Mouillac B, Breit A, Ayoub MA, Taulan M, Jockers R, Barberis C, Bouvier M. Oxytocin and vasopressin V1a and V2 receptors form constitutive homo- and heterodimers during biosynthesis. Mol Endocrinol. 2003;17:677–691. doi: 10.1210/me.2002-0222. [DOI] [PubMed] [Google Scholar]
- 186.Devost D, Zingg HH. Homo- and hetero-dimeric complex formations of the human oxytocin receptor. J Neuroendocrinol. 2004;16:372–377. doi: 10.1111/j.0953-8194.2004.01188.x. [DOI] [PubMed] [Google Scholar]
- 187.Pin JP, Maurel D, Comps-Agrar L, Monnier C, Rives A-L, Doumazane E, Rondard P, Durroux T, Prezeau L, Trinquet E. Time-resolved FRET approaches to study GPCRs complexes. In: Siehler S, Milligan G, editors. G Protein-Coupled Receptors; Structure, Signaling and Physiology. Cambridge: Cambridge University Press; 2011. pp. 67–90. [Google Scholar]
- 188.Albizu L, Teppaz G, Seyer R, Bazin H, Ansanay H, Manning M, Mouillac B, Durroux T. Toward efficient drug screening by homogeneous assays based on the development of new fluorescent vasopressin and oxytocin receptor ligands. J Med Chem. 2007;50:4976–4985. doi: 10.1021/jm061404q. [DOI] [PubMed] [Google Scholar]
- 189.Zwier JM, Roux T, Cottet M, Durroux T, Douzon S, Bdioui S, Gregor N, Bourrier E, Oueslati N, Nicolas L, Tinel N, Boisseau C, Yverneau P, Charrier-Savournin F, Fink M, Trinquet E. A fluorescent ligand-binding alternative using Tag-lite(R) Technology. J Biomol Screen. 2010;15:1248–1259. doi: 10.1177/1087057110384611. [DOI] [PubMed] [Google Scholar]
- 190.Cottet M, Faklaris O, Zwier JM, Trinquet E, Pin J-P, Durroux T. Original fluorescent ligand-based assays open new perspectives in G-protein coupled receptor drug screening. Pharmaceuticals. 2011;4:202–214. [Google Scholar]
- 191.Deblon N, Veyrat-Durebex C, Bourgoin L, Caillon A, Bussier AL, Petrosino S, Piscitelli F, Legros JJ, Geenen V, Foti M, Wahli W, Di Marzo V, Rohner-Jeanrenaud F. Mechanisms of the anti-obesity effects of oxytocin in diet-induced obese rats. PLoS ONE. 2011;6:e25565. doi: 10.1371/journal.pone.0025565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Sterns R, Hix J. Hyponatremia: vasopressin antagonists in hyponatremia: more data needed. Nat Rev Nephrol. 2011;7:132–133. doi: 10.1038/nrneph.2010.173. [DOI] [PubMed] [Google Scholar]
- 193.Vlieghe P, Lisowski V, Martinez J, Khrestchatisky JM. Synthetic therapeutic peptides: science and market. Drug Discovery Today. 2010;15:40–56. doi: 10.1016/j.drudis.2009.10.009. [DOI] [PubMed] [Google Scholar]
- 194.Reichert J. Development Trends for Peptide Therapeutics. Peptide Therapeutics Foundation; 2010. Report, http://www.peptidetherapeutics.org. [Google Scholar]
- 195.Assinder SJ. Oxytocin increases 5alpha-reductase activity of human prostate epithelial cells, but not stromal cells. Prostate. 2008;68:115–121. doi: 10.1002/pros.20671. [DOI] [PubMed] [Google Scholar]
- 196.Beery AK, Zucker I. Oxytocin and same-sex social behavior in female meadow voles. Neuroscience. 2010;169:665–673. doi: 10.1016/j.neuroscience.2010.05.023. [DOI] [PubMed] [Google Scholar]
- 197.Bosch OJ, Neumann ID. Both oxytocin and vasopressin are mediators of maternal care and aggression in rodents: from central release to sites of action. Horm Behav. 2011 doi: 10.1016/j.yhbeh.2011.11.002. Doi: 10.1016/j.yhbeh.2011.11.002. [Epub before print] [DOI] [PubMed] [Google Scholar]
- 198.Braida D, Donzelli A, Martucci R, Capurro V, Busnelli M, Chini B, Sala M. Neurohypophyseal hormones manipulation modulate social and anxiety-related behavior in zebrafish. Psychopharmacology. 2012;220:319–330. doi: 10.1007/s00213-011-2482-2. [DOI] [PubMed] [Google Scholar]
- 199.Breton J-D, Veinante P, Uhl-Bronner S, Vergnano AM, Freund-Mercier MJ, Schlichter R, Poisbeau P. Oxytocin-induced antinoiception in the spinal cord is mediated by subpopulation of glutamatergic neurons in lamina I-II which amplify GABAergic inhibition. Molecular Pain. 2008;4:19–31. doi: 10.1186/1744-8069-4-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Figueira RJ, Peabody MF, Lonstein JS. Oxytocin receptor activity in the ventrocaudal periaqueductal gray modulates anxiety-related behavior in postpartum rats. Behav Neurosci. 2008;122:618–628. doi: 10.1037/0735-7044.122.3.618. [DOI] [PubMed] [Google Scholar]
- 201.Gupta J, Russell R, Wayman C, Hurley D, Jackson V. Oxytocin-induced contractions within rat and rabbit ejaculatory tissues are mediated by vasopressin V1a receptors and not oxytocin receptors. Br J Pharmacol. 2008;155:118–126. doi: 10.1038/bjp.2008.226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Israel JM, Poulain DA, Oliet SH. Oxytocin-induced postinhibitory rebound firing facilitates bursting activity in oxytocin neurons. J Neurosci. 2008;28:385–394. doi: 10.1523/JNEUROSCI.5198-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Karavashkina TA, Kutina AV, Shakhmatova EI, Natochin YV. Mechanism of 1-deamino-arginine vasotocin induced natriuresis in rats. Gen Comp Endocrinol. 2011;170:460–467. doi: 10.1016/j.ygcen.2010.10.020. [DOI] [PubMed] [Google Scholar]
- 204.Karelina K, Stuller KA, Jarrett B, Zhang N, Wells J, Norman GJ, Devries AC. Oxytocin mediates social neuroprotection after cerebral ischemia. Stroke. 2011;42:3606–3611. doi: 10.1161/STROKEAHA.111.628008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Kennett JE, Poletini MO, Fitch CA, Freeman ME. Antagonism of oxytocin prevents suckling- and estradiol-induced, but not progesterone-induced, secretion of prolactin. Endocrinology. 2009;150:2292–2299. doi: 10.1210/en.2008-1611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Kim JS, Kim WB, Kim YB, Lee Y, Kim YS, Shen FY, Lee SW, Park D, Choi HJ, Hur J, Park JJ, Han HC, Colwell CS, Cho YW, Kim YI. Chronic hyperosmotic stress converts GABAergic inhibition into excitation in vasopressin and oxytocin neuronsin the rat. J Neurosci. 2011;31:13312–13322. doi: 10.1523/JNEUROSCI.1440-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Lee H-J, Macbeth AH, Pagani J, Young WS., III Oxytocin: the great facilitator of life. Prog Neurobiol. 2009;88:127–151. doi: 10.1016/j.pneurobio.2009.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Lukas M, Toth I, Reber SO, Slattery DA, Veenema AH, Neumann ID. The neuropeptide oxytocin facilitates pro-social behavior and prevents social avoidance in rats and mice. Neuropsychopharmacology. 2011;36:2159–2168. doi: 10.1038/npp.2011.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Macciò A, Madeddu C, Chessa P, Panzone F, Lissoni P, Mantovani G. Oxytocin both increases proliferative response of peripheral blood lymphomonocytes to phytohemagglutinin and reverses immunosuppressive estrogen activity. In Vivo. 2010;24:157–163. [PubMed] [Google Scholar]
- 210.Mak P, Broussard C, Vacy K, Broadbear JH. Modulation of anxiety behavior in the elevated plus maze using peptidic oxytocin and vasopressin receptor ligands in the rat. J Psychopharmacol. 2011 doi: 10.1177/0269881111416687. PubMed PMID: 21890582. [DOI] [PubMed] [Google Scholar]
- 211.Nakamura K, Fujiwara Y, Mizutani R, Sanbe A, Miyauchi N, Hiroyama M, Yamaguchi J, Yamashita T, Nakamura S, Mori T, Tsujimoto G, Tanoue A. Effects of vasopressin V1b receptor deficiency on adrenocorticotropin release from anterior pituitary cells in responce to oxytocin stimulation. Endocrinology. 2008;149:4883–4891. doi: 10.1210/en.2007-1528. [DOI] [PubMed] [Google Scholar]
- 212.Neumann ID, Veenema AH, Beiderbeck DI. Aggression and anxiety: social context and neurobiological links. Front Behav Neurosci. 2010;4:12–28. doi: 10.3389/fnbeh.2010.00012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Norman GJ, Karelina K, Morris JS, Zhang N, Cochran M, Courtney DeVries A. Social interaction prevents the development of depressive-like behavior postnerve injury in mice: a potential role for oxytocin. Psychosom Med. 2010;72:519–526. doi: 10.1097/PSY.0b013e3181de8678. [DOI] [PubMed] [Google Scholar]
- 214.Northrop LE, Erskine MS. Selective oxytocin receptor activation in the ventrolateral portion of the ventromedial hypothalamus is required for mating-induced pseudopregnancy in the female rat. Endocrinology. 2008;149:836–842. doi: 10.1210/en.2007-1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Nyuyki KD, Waldherr M, Baeuml S, Neumann ID. Yes, i am ready now: differential effects of paced versus unpaced mating on anxiety and central oxytocin release in female rats. PLoS ONE. 2011;6:e23599. doi: 10.1371/journal.pone.0023599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Oldfield RG, Hofmann HA. Neuropeptide regulation of social behavior in a monogamous cichlid fish. Physiol Behav. 2011;102:296–303. doi: 10.1016/j.physbeh.2010.11.022. [DOI] [PubMed] [Google Scholar]
- 217.Saito A, Nakamura K. Oxytocin changes primate paternal tolerance to offspring in food transfer. J Comp Physiol A Neuroethol Sens Neural Behav Physiol. 2011;197:329–337. doi: 10.1007/s00359-010-0617-2. [DOI] [PubMed] [Google Scholar]
- 218.Schorscher-Petcu A, Sotocinal S, Ciura S, Dupré A, Ritchie J, Sorge RE, Crawley JN, Hu SB, Nishimori K, Young LJ, Tribollet E, Quirion R, Mogil JS. Oxytocin-induced analgesia and scratching are mediated by the vasopressin-1A receptor in the mouse. J Neurosci. 2010;30:8274–8284. doi: 10.1523/JNEUROSCI.1594-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Schorscher-Petcu A, Dupre A, Tribollet E. Distribution of vasopressin and oxytocin binding sites in the brain and upper spinal cord of the common marmoset. Neurosci Lett. 2009;461:217–222. doi: 10.1016/j.neulet.2009.06.016. [DOI] [PubMed] [Google Scholar]
- 220.Shirley DG, Walter MF, Keeler BD, Waters NJ, Walter SJ. Selective blockade of oxytocin and vasopressin V(1a) receptors in anaesthetised rats: evidence that activation of oxytocin receptors rather than V(1a) receptors increases sodium excretion. Nephron Physiol. 2011;117:21–26. doi: 10.1159/000320290. [DOI] [PubMed] [Google Scholar]
- 221.Slattery DA, Neumann ID. Chronic icv oxytocin attenuates the pathological high anxiety state of selectively bred Wistar rats. Neuropharmacology. 2010;58:56–61. doi: 10.1016/j.neuropharm.2009.06.038. [DOI] [PubMed] [Google Scholar]
- 222.Takada M, Fujimaki-Aoba K, Hokari S. Vasotocin- and mesotocin-induced increases in short-circuit current across tree frog skin. J Comp Physiol B. 2011;181:239–248. doi: 10.1007/s00360-010-0523-5. [DOI] [PubMed] [Google Scholar]
- 223.Teruyama R, Lipschitz DL, Wang L, Ramoz GR, Crowley WR, Bealer SL, Armstrong WE. Central blockade of oxytocin receptors during mid-late gestation reduces amplitude of slow afterhyperpolarization in supraoptic oxytocin neurons. Am J Physiol Endocrinol Metab. 2008;295:E1167–E1171. doi: 10.1152/ajpendo.90620.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Timmer M, Cordero MI, Sevelinges Y, Sandi C. Evidence for a role of oxytocin receptors in the long-term establishment of dominance hierarchies. Neuropsychopharmacology. 2011;36:2349–2356. doi: 10.1038/npp.2011.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Veenema AH, Bredewold R, De Vries GJ. Vasopressin regulates social recognition in juvenile and adult rats of both sexes, but in sex- and age-specific ways. Horm Behav. 2012;61:50–56. doi: 10.1016/j.yhbeh.2011.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Viero C, Shibuya I, Kitamura N, Verkhratsky A, Fujihara H, Katoh A, Ueta Y, Zingg HH, Chvatal A, Sykova E, Dayanithi G. REVIEW: Oxytocin: crossing the bridge between basic science and pharmacotherapy. CNS Neurosci Ther. 2010;16:e138–e156. doi: 10.1111/j.1755-5949.2010.00185.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Viviani D, Charlet A, van den Burg E, Robinet C, Hurni N, Abatis M, Magara F, Stoop R. Oxytocin selectively gates fear responses through distinct outputs from the central amygdala. Science. 2011;333:104–107. doi: 10.1126/science.1201043. [DOI] [PubMed] [Google Scholar]
- 228.Vrachnis N, Malamas FM, Sifakis S, Deligeoroglou E, Iliodromiti Z. The oxytocin–oxytocin receptor system and its antagonists as tocolytic agents. Int J Endocrinol. 2011;2011:350546. doi: 10.1155/2011/350546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Waldherr M, Nyuyki K, Maloumby R, Bosch OJ, Neumann ID. Attenuation of the neuronal stress responsiveness and corticotrophin releasing hormone synthesis after sexual activity in male rats. Horm Behav. 2010;57:222–229. doi: 10.1016/j.yhbeh.2009.11.006. [DOI] [PubMed] [Google Scholar]
- 230.Wrobel LJ, Reymond-Marron I, Dupré A, Raggenbass M. Oxytocin and vasopressin enhance synaptic transmission in the hypoglossal motor nucleus of young rats by acting on distinct receptor types. Neuroscience. 2010;165:723–735. doi: 10.1016/j.neuroscience.2009.11.001. [DOI] [PubMed] [Google Scholar]
- 231.Yang J, Pan YJ, Zhao Y, Qiu PY, Lu L, Li P, Chen F, Yan XQ, Wang DX. Oxytocin in the rat caudate nucleus influences pain modulation. Peptides. 2011;32:2104–2107. doi: 10.1016/j.peptides.2011.08.021. [DOI] [PubMed] [Google Scholar]
- 232.Busnardo C, Tavares RF, Correa FM. Mechanisms involved in the pressor response to noradrenaline microinjection into the supraoptic nucleus of unanesthetized rats. Auton Neurosci. 2009;145:63–70. doi: 10.1016/j.autneu.2008.11.005. [DOI] [PubMed] [Google Scholar]
- 233.Chen J, Young S, Subburaju S, Sheppard AK, Atkinson H, Wood S, Lightman S, Serradeil-Le Gal C, Aguilera G. Vasopressin does not mediate hypersensivity of the hypothalamic pituitary adrenal axis during chronic stress. stress, neurotransmitters, and hormones. Ann NY Acad Sci. 2008;1148:349–359. doi: 10.1196/annals.1410.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Cudnoch-Jedrzejewska A, Szczepanska-Sadowska E, Dobruch J, Gomolka R, Puchalska L. Brain vasopressin V(1) receptors contribute to enhanced cardiovascular responses to acute stress in chronically stressed rats and rats with myocardial infarction. Am J Physiol Regul Integr Comp Physiol. 2010;298:R672–R680. doi: 10.1152/ajpregu.00543.2009. [DOI] [PubMed] [Google Scholar]
- 235.Dutertre S, Croker D, Daly NL, Andersson A, Muttenthaler M, Lumsden NG, Craik DJ, Alewood PF, Guillon G, Lewis RJ. Conopressin-T from Conus tulipa reveals an antagonist switch in vasopressin-like peptides. J Biol Chem. 2008;283:7100–7108. doi: 10.1074/jbc.M706477200. [DOI] [PubMed] [Google Scholar]
- 236.Goel M, Zuo CD, Schilling WP. Role of cAMP/PKA signalling cascade in vasopressin-induced trafficking of TRPC3 channels in principal cells of the collecting duct. Am J Physiol Renal Physiol. 2010;298:F988–F996. doi: 10.1152/ajprenal.00586.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Koshimizu TA, Tsujimoto G. New topics in vasopressin receptors and approach to novel drugs: vasopressin and pain perception. J Pharmacol Sci. 2009;109:33–37. doi: 10.1254/jphs.08r18fm. [DOI] [PubMed] [Google Scholar]
- 238.Kublaoui BM, Gemelli T, Tolson KP, Wang Y, Zinn AR. Oxytocin deficiency mediates hyperphagic obesity of Sim1 haploinsufficient mice. Mol Endocrinol. 2008;22:1723–1734. doi: 10.1210/me.2008-0067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Louiset E, Contesse V, Groussin L, Cartier D, Duparc C, Perraudin V, Bertherat J, Lefebvre H. Expression of vasopressin receptors in ACTH-independent macronodular bilateral adrenal hyperplasia causing Cushing’s syndrome: molecular, immunohistochemical and pharmacological correlates. J Endocrinol. 2008;196:1–9. doi: 10.1677/JOE-07-0413. [DOI] [PubMed] [Google Scholar]
- 240.Rehberg S, Ertmer C, Lange M, Morelli A, Whorton E, Dunser M, Strohhacker AK, Lipke E, Kampmeier TG, Van Aken H, Traber DL, Westphal M. Role of selective V2-receptor-antagonism in septic shock: a randomized, controlled, experimental study. Crit Care. 2010;14:R200. doi: 10.1186/cc9320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Rodriguez-Borrero E, Rivera-Escalera F, Candelas F, Montalvo J, Munoz-Miranda WJ, Walker JR, Maldonado-Vlaar CS. Arginine vasopressin gene expression changes within the nucleus accumbens during environment elicited cocaine-conditioned response in rats. Neuropharmacology. 2010;58:88–101. doi: 10.1016/j.neuropharm.2009.06.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Searcy BT, Bradford CS, Thompson RR, Fitz TM, Moore FL. Identification and characterization of mesotocin and V1a-like vasotocin receptors in a urodele amphibian, Taricha granulosa. Gen Comp Endocrinol. 2011;170:131–143. doi: 10.1016/j.ygcen.2010.09.017. [DOI] [PubMed] [Google Scholar]
- 243.Tanoue A. New topics in vasopressin receptors and approach to novel drugs: effects of vasopressin receptor on regulations of hormone secretion and metabolisms of glucose, fat, and protein. J Pharmacol Sci. 2009;109:50–52. doi: 10.1254/jphs.08r15fm. [DOI] [PubMed] [Google Scholar]
- 244.Veitenheimer B, Osborn JW. Role of spinal V1a receptors in regulation of arterial pressure during acute and chronic osmotic stress. Am J Physiol Regul Integr Comp Physiol. 2011;300:R460–R469. doi: 10.1152/ajpregu.00371.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Yang Z, Han D, Coote JH. Cardiac sympatho-exicatory action of PVN-spinal oxitocin neurones. Auton Neurosci. 2009;147:80–85. doi: 10.1016/j.autneu.2009.01.013. [DOI] [PubMed] [Google Scholar]
- 246.Yao Y, Fu LY, Zhang X, van den Pol AN. Vasopressin and oxytocin excite MCH neurons, but not other lateral hypothalamic GABA neurons. Am J Physiol Regul Integr Comp Physiol. 2012 doi: 10.1152/ajpregu.00452.2011. [Epub ahead of print] PMID:22262306. [DOI] [PMC free article] [PubMed] [Google Scholar]