

Abstract
Mutations in the OCRL gene encoding the phosphatidylinositol 4,5-bisphosphate (PI(4,5)P2) 5-phosphatase OCRL cause Lowe syndrome (LS), which is characterized by intellectual disability, cataracts and selective proximal tubulopathy. OCRL localizes membrane-bound compartments and is implicated in intracellular transport. Comprehensive analysis of clathrin-mediated endocytosis in fibroblasts of patients with LS did not reveal any difference in trafficking of epidermal growth factor, low density lipoprotein or transferrin, compared with normal fibroblasts. However, LS fibroblasts displayed reduced mannose 6-phosphate receptor (MPR)-mediated re-uptake of the lysosomal enzyme arylsulfatase B. In addition, endosome-to-trans Golgi network (TGN) transport of MPRs was decreased significantly, leading to higher levels of cell surface MPRs and their enrichment in enlarged, retromer-positive endosomes in OCRL-depleted HeLa cells. In line with the higher steady-state concentration of MPRs in the endosomal compartment in equilibrium with the cell surface, anterograde transport of the lysosomal enzyme, cathepsin D was impaired. Wild-type OCRL counteracted accumulation of MPR in endosomes in an activity-dependent manner, suggesting that PI(4,5)P2 modulates the activity state of proteins regulated by this phosphoinositide. Indeed, we detected an increased amount of the inactive, phosphorylated form of cofilin and lower levels of the active form of PAK3 upon OCRL depletion. Levels of active Rac1 and RhoA were reduced or enhanced, respectively. Overexpression of Rac1 rescued both enhanced levels of phosphorylated cofilin and MPR accumulation in enlarged endosomes. Our data suggest that PI(4,5)P2 dephosphorylation through OCRL regulates a Rac1-cofilin signalling cascade implicated in MPR trafficking from endosomes to the TGN.
INTRODUCTION
The oculocerebrorenal syndrome of Lowe [or Lowe syndrome (LS); MIM 309000] is a rare X-linked disease characterized by intellectual disability, hypotonia, congenital cataract and selective proximal tubulopathy (1–3). LS is caused by mutations in the OCRL gene encoding the ubiquitously expressed, inositol polyphosphate 5-phosphatase OCRL (4). OCRL encodes two isoforms; the longer isoform a is present in all tissues, while the shorter isoform b, which lacks eight amino acids (707EDSFLEKE714) encoded by the alternative exon 18a, is present in all tissues except the brain (5,6). OCRL preferentially hydrolyzes the phosphoinositides phosphatidylinositol 4,5-bisphosphate (PI(4,5)P2) and phosphatidylinositol 3,4,5-trisphosphate at the 5-position to phosphatidylinositol 4-phosphate and phosphatidylinositol 3,4-bisphosphate, respectively (7,8).
OCRL is a multidomain protein with an N-terminal PH (pleckstrin homology) domain (9), a central 5-phosphatase domain (IPPc: inositol polyphosphate phosphatase, catalytic), an ASH (ASPM, SPD-2, Hydin) domain (10) and a catalytically inactive, RhoGAP-like domain at the C-terminus (11,12). The intracellular localization of OCRL is mediated by its interaction with a variety of proteins. Targeting of OCRL to membranes requires binding to Rab GTPases via the ASH domain (13). In particular, interaction of OCRL with Rab5 and Rab6 targets the 5-phosphatase to endosomes and the trans Golgi network (TGN), respectively (13). The C-terminal RhoGAP domain mediates interaction with the Rho GTPases Rac1 and Cdc42 as well as Arf1 and Arf6, two members of the Arf family of small GTPases (12,14). In response to growth factor stimulation, OCRL translocates to the plasma membrane where it co-localizes with Rac1 (15). Interaction of OCRL with Rac1 and Cdc42 and OCRL's role in cell migration suggest that it is involved in regulating cellular actin dynamics (16). Indeed, OCRL was found to control actin polymerization in a PI(4,5)P2-dependent manner during cytokinesis (17,18), in early steps of Listeria monocytogenes infection (19), and at the interface of early endosomes (20).
OCRL binds to clathrin heavy chain and to the plasma membrane adaptor AP2 that causes enrichment of OCRL in clathrin-coated vesicles (9,21,22). A subpopulation of OCRL can also be found on endocytic transport intermediates (11,22). The Rab5 effector APPL1 is located together with OCRL on this endocytic platform (11), and the recently identified new interaction partners of OCRL, IPIP27A and B (also named Ses1 and 2) turned out to be key regulators of endocytic trafficking (23,24).
Patients with LS have a selective proximal tubulopathy characterized by low molecular weight proteinuria and albuminuria (3), and in most of the patients, lysosomal enzymuria was also elevated (3,25). The lysosomal enzymuria has recently been linked to abnormal membrane trafficking of the cation-independent mannose 6-phosphate receptor (CI-MPR) or 300 kDa MPR (MPR300) (26). The two distinct MPRs, MPR300, and the 46 kDa MPR [MPR46 or cation-dependent MPR (CD-MPR)] are required for delivery of newly synthesized lysosomal enzymes from the Golgi apparatus to the endosomal and lysosomal compartment (27). Increased secretion of the lysosomal hydrolases cathepsin D and N-acetyl-β-d-glucosaminidase into the urine of patients with LS has been found to be secondary to abnormal trafficking of MPR300 to the apical membrane of proximal tubule cells (26). These data are consistent with increased secretion of cathepsin D and hexosaminidase in OCRL-depleted HK-2 and HeLa cells, respectively (20,28), as well as with partial redistribution of the MPR300 from the TGN to endosomal structures in HeLa and proximal tubule cells lacking OCRL (20,22). These findings reinforced the idea that OCRL has a role in membrane trafficking between the TGN and endosomes and may also function at other stations of the endocytic pathway. Consistent with this, OCRL has recently been found to control trafficking through early endosomes of the multi-ligand receptor megalin, transferrin receptor (TfR), epidermal growth factor (EGF) receptor, and MPR (20), and OCRL isoform a participates in Tf endocytosis (29). However, other studies of megalin trafficking (28) and Tf and EGF internalization in different cell lines depleted of OCRL (16) failed to detect such defects.
Thus, we undertook a systematic approach to investigate membrane trafficking of various clathrin-dependent receptors in fibroblasts from LS-affected patients and OCRL-depleted HeLa cells. We show here that depletion of OCRL does not cause a general defect in clathrin-mediated endocytic membrane traffic; however, it leads to a strong decrease in MPR transport from endosomes to the TGN that in turn leads to higher steady-state levels of MPR in enlarged early endosomes in equilibrium with the cell surface. We show further that OCRL's 5-phosphatase activity is essential for retrograde transport of MPRs and regulates the activity of the actin modulatory protein cofilin and the RhoGTPase Rac1, both implicated in F-actin dynamics. Our data highlight a tight connection between the Rac1-cofilin signalling pathway and endosome-to-TGN trafficking paving the way to understand the pathophysiology of LS.
RESULTS
Reduced MPR300-mediated internalization of arylsulfatase B in LS fibroblasts
To elucidate the effect of OCRL deficiency on receptor-mediated endocytosis, we took advantage of six dermal skin fibroblast cell lines with OCRL mutations from patients with LS (LS 1–LS 6, Fig. 1A). Five of the six cell lines tested did not contain detectable levels of OCRL, which is detected as a 105 kDa polypeptide in cells from healthy individuals (Con 1– Con 3; Fig. 1A). In cell line LS 6, a truncated OCRL variant of ∼85 kDa was detected, in accordance with the predicted molecular mass of the mutant protein (Fig. 1A). Patient LS 6 has a microdeletion that includes exons 16–18 leading to an in-frame deletion of 513 bp of the OCRL coding region (Fig. 1A). The encoded OCRL mutant protein lacks the amino acids from position 535–705, including the last five amino acids of the IPPc domain, the complete ASH domain and the first 27 amino acids of the RhoGAP domain. Deletion of these 171 amino acids is expected to abolish binding of OCRL to Rab proteins, APPL1, and IPIP27A and B. However, impaired OCRL folding leading to a more global disruption in protein function cannot be excluded.
Figure 1.

Dermal skin fibroblasts from patients with LS show reduced internalization of the lysosomal enzyme ASB. (A) Cell lysates of dermal skin fibroblasts from six patients with LS (LS 1 to LS 6) and three healthy individuals (Con 1 to Con 3) were subjected to SDS–PAGE, and immunoblotting with an anti-OCRL antibody was performed. Equal protein loading was confirmed by probing with an anti-GAPDH antibody. OCRL mutations in patients LS 1 to LS 6 were identified by routine genetic testing in our lab and are indicated for each cell line. (B) Control and LS fibroblasts were incubated with [125I]ASB for 3h as described in Materials and Methods. Intracellular radioactivity was determined in a γ-counter and normalized to the protein content. The amount of internalized [125I]ASB in LS fibroblasts is expressed as percentage of control cells. Each bar is the mean ± SD of at least five independent experiments. ***P < 0.001 (two-tailed Student's t-test).
The MPR300-mediated uptake of purified, recombinant human arylsulfatase B (ASB) was next tested. Cell cultures were incubated with [125I]labelled ASB in the presence or absence of 10 mm mannose 6-phosphate (M6P), a competitive inhibitor for the mannose 6-phosphate-binding site of MPR300. After 3 h internalization, surface-bound ligand was removed and intracellular radioactivity determined in lysates. For all six LS cell lines, [125I]ASB uptake decreased by 35–65% (P < 0.001) compared with control fibroblasts (Fig. 1B). The uptake of [125I]ASB was specific and was completely inhibited by M6P (data not shown). To determine whether the reduced internalization of [125I]ASB in LS fibroblasts was due to a generalized defect in clathrin-mediated endocytosis, we analyzed the uptake of [125I]Tf, [125I]low density lipoprotein (LDL) and AlexaFluor488-EGF. Fibroblasts were incubated with either [125I]Tf for 5, 10 or 30 min, [125I]LDL for 4 h or with AlexaFluor488-EGF for 15 or 30 min at 37°C. Quantification of endocytosed ligands was performed by determination of intracellular radioactivity for Tf and LDL or intracellular fluorescence for EGF. We found no differences in the amount of internalized Tf, LDL or EGF between LS and control fibroblasts (Supplementary material, Fig. S1). Similar results were obtained when the distribution and the amount of intracellular AlexaFluor488-Tf and -EGF was analyzed by immunofluorescence microscopy (data not shown). Together, the data suggest no general involvement of OCRL in clathrin-mediated endocytosis of receptor-ligand complexes. However, the presence of OCRL in clathrin-coated pits and its binding to AP2 may still indicate a rate-limiting role of OCRL in clathrin-mediated trafficking in some cell types, although not in HeLa or fibroblast cells.
Depletion of OCRL increases cell surface MPR300 and enhances its internalization
The reduced internalization of [125I]ASB in LS fibroblasts could be due to decreased levels of MPR300 at the cell surface, a lower internalization rate of MPR300 or both. Therefore, we used cell surface biotinylation to determine the amount of MPR300 at the plasma membrane in OCRL-depleted cells. HeLa cells were treated for 72 h with three different OCRL siRNAs. As a control, HeLa cells were treated with GFP siRNA. As shown in Figure 2A, treatment of HeLa cells with OCRL-specific siRNAs caused a ≥90% depletion of endogenous OCRL. siRNA-treated HeLa cells were biotinylated at 4°C with sulfo-NHS-SS-biotin. After removal of unbound biotin, cells were lysed and biotinylated proteins were precipitated with streptavidin agarose followed by MPR300 immunoblotting.
Figure 2.

OCRL-depleted HeLa cells exhibit an increased plasma membrane level and internalization of MPR300. (A) HeLa cells were treated with OCRL-specific siRNAs for 72 h. GFP-specific siRNA was used as a control. Depletion was monitored by immunoblotting with an anti-OCRL antibody, and an anti-tubulin antibody was used as loading control. The blot shows a representative result obtained upon depletion with one of three different OCRL-specific siRNAs applied in this work. Depletion efficiency of all siRNAs was comparable with >90% of OCRL depleted after 72 h. (B) GFP and OCRL siRNA-treated cells were biotinylated at 4°C and subjected to streptavidin agarose as described in Materials and Methods. Aliquots of total cell lysates (2%; T) and precipitated cell surface proteins (20%; CS) were analysed by MPR300 immunoblotting. A representative immunoblot of four independent experiments is shown. (C) Levels of MPR300 were quantified by scanning densitometry, and cell surface MPR300 was normalized to total MPR300. Each bar is the mean ± SD of four independent experiments. **P = 0.003 (two-tailed Student's t-test). To exclude off-target effects, three different siRNAs against OCRL were independently applied. (D–F) GFP and OCRL siRNA-treated cells were biotinylated at 4°C and either harvested (CS) or incubated for 2 (I2) or 5 (I5) min at 37°C. Remaining biotin at the cell surface was removed using glutathione washes at 4°C, and the internalized biotinylated proteins were precipitated with streptavidin agarose followed by MPR300 immunoblotting. Levels of MPR300 were quantified by scanning densitometry and normalized to total MPR300. (D) A representative immunoblot is shown. Internalized MPR300 is expressed as percentage of cell surface MPR300 ± SD of six independent experiments. (E) Internalization rates were calculated as percentage of biotinylated cell surface receptors which are internalized per minute. Each bar is the mean ± SD of six independent experiments. (F) Internalized MPR300 in OCRL-depleted cells is expressed as percentage of control cells (white square GFP siRNA, black square OCRL siRNA). Each bar is the mean ± SD of at least six independent experiments. I2 (incubation for 2 min at 37°C): *P = 0.04; I5 (incubation for 5 min at 37°C): *P = 0.02 (two-tailed Student's t-test).
Figure 2B shows an increased amount of MPR300 at the cell surface in OCRL-depleted cells compared with control cells. Densitometric evaluation of immunoblots revealed that 2% of total MPR300 was present at the plasma membrane in GFP siRNA-treated cells, compared with 4–6% in OCRL-depleted cells (P = 0.003, Fig. 2C). Thus, the amount of plasma membrane-localized MPR300 increased by 2.5-fold in OCRL-depleted compared with control cells. Next, we measured MPR300 internalization rates. Cell surface proteins were biotinylated at 4°C and cells were either harvested (Fig. 2D, lanes 1 and 4) or were re-warmed for 2 or 5 min to 37°C to permit endocytosis (Fig. 2D, lanes 2–3 and 5–6). Then, cells were cooled to 4°C and bound biotin remaining at the cell surface was removed by glutathione treatment. Cells that were glutathione-treated directly after cell surface biotinylation at 4°C showed loss of biotinylated MPR300 and were used as control (Fig. 2D, lane 7). Following cell lysis, biotinylated proteins were precipitated with streptavidin agarose and MPR300 was detected by immunoblotting. Densitometric analysis of immunoblots revealed that after 2 min, 39 and 48% and after 5 min 61 and 68% of cell surface MPR300 were internalized in control and OCRL-depleted cells, respectively (Fig. 2D). The mean internalization rate of 13%/min in control cells versus 15%/min in OCRL-depleted cells was calculated (Fig. 2E). Thus, the internalization kinetics of MPR300 are similar in OCRL-depleted and control cells. However, when we determined the absolute amount of internalized MPR300 in OCRL-depleted cells relative to control cells, a 2-fold (P = 0.04) increase in the amount of internalized MPR300 after 2 min and a 3-fold (P = 0.02) increase after 5 min was observed (Fig. 2F). These results indicate that the increased amount of internalized MPR300 correlates well with the 2.5-fold increase in the number of MPR300 at the cell surface in OCRL-depleted cells.
The increase in cell surface-localized MPR300 in OCRL-depleted cells could be the result of increased, total cellular levels of MPR300 or instead, to an altered intracellular distribution. Immunoblot analysis revealed that the total amount of MPR300 is similar in OCRL-depleted and control cells (Supplementary material, Fig. S2A and B). These results were confirmed by the determination of the MPR300 half-life in pulse-chase experiments. RNAi-treated HeLa cells were labelled with [35S]methionine/cysteine and chased for various times. After cell lysis, MPR300 was immunoprecipitated, subjected to SDS–PAGE and analyzed by autoradiography to determine the degradation rate (Supplementary material, Fig. S2C). In the absence of OCRL, the estimated half-life of MPR300 was 20 h which was not significantly higher than in control cells with 16 h (Supplementary material, Fig. S2D). These results demonstrate that the cellular level of MPR300 is not altered in OCRL-depleted cells.
We also followed MPR trafficking in fibroblasts from healthy individuals and LS patients by determining (i) the amount of radioactively labelled cell surface and cellular MPR300 and (ii) the absolute amount of internalized MPR300 after 2 and 5 min by cell surface biotinylation. We did not detect a difference in the amounts of cell surface, total and internalized MPR300 between control and LS fibroblasts (Supplementary material, Fig. S3A–C). We therefore conclude that the increase in cell surface-localized MPR300 in OCRL-depleted HeLa cells and the reduced internalization of ASB in LS fibroblasts are likely due to altered distribution of intracellular MPR300.
We next analyzed whether RNAi-mediated depletion of OCRL influences the distribution and/or internalization kinetics of other clathrin-dependent receptors, such as the TfR. We first determined the fraction of TfR located at the plasma membrane at the steady-state level by cell surface biotinylation experiments followed by immunoblotting. No significant differences of cell surface-localized TfR were detected when control and OCRL-depleted cells were compared (15 versus 16%; Supplementary material, Fig. S4A). In addition, the absolute amounts of internalized TfR after 2 and 5 min were similar in OCRL-depleted cells relative to control cells (Supplementary material, Fig. S4B). Furthermore, the average internalization rate of 19%/min of plasma membrane-localized TfR in control cells was similar to 20%/min in cells lacking OCRL (Supplementary material, Fig. S4C). These data indicate that in contrast to MPR300, there are no significant differences in the distribution and internalization rates of TfR between control cells and cells depleted of OCRL.
Enrichment of MPRs in enlarged retromer-positive endosomes in OCRL-depleted cells
To examine whether increased amounts of MPR300 at the cell surface are due to an intracellular redistribution, we analyzed the localization of endogenous MPR300 and MPR46 by confocal laser scanning microscopy. In control cells, MPR300 and MPR46 were clustered in small vesicular structures in the perinuclear region (Fig. 3A). Upon OCRL depletion, MPR46 and MPR300 were still distributed to the perinuclear Golgi region, but there they colocalized in enlarged endosomal vesicles (Fig. 3A). Double labelling with the TGN marker TGN46 and the endosomal marker EEA1 did not reveal any difference in co-localization with MPR300 in OCRL-depleted and control cells and further confirmed enrichment of MPR300 in enlarged endosomes in the perinuclear region of OCRL-depleted HeLa cells (Fig. 3B). We estimated the size of MPR300-positive vesicular structures, subdivided them into four groups and quantified them. As shown in Figure 3C, cells depleted of OCRL contained significantly more (P < 0.001) large MPR300-positive structures than control cells. The identity of the enlarged MPR-containing vesicles in OCRL-depleted cells was determined using endosomal marker proteins. Enlarged MPR300-positive vesicles co-localized with both Rab5 and TfR, identifying them as clusters of early endosomes (Fig. 4i and ii). SNX1, a component of the retromer machinery, also decorated MPR300-positive early endosomes (Fig. 4iii). In contrast, Rab7, a late endosomal marker, showed only partial co-localization with MPR300 (Fig. 4iv).
Figure 3.

MPR46 and MPR300 colocalize in enlarged endosomal vesicles in the perinuclear region in OCRL-depleted cells. (A–C) HeLa cells were treated with OCRL- or GFP-specific siRNA for 72 h, labelled with antibodies (see below) and analyzed by confocal laser scanning microscopy. (A) RNAi-treated HeLa cells were stained with an anti-MPR300 antibody, followed by an AlexaFluor488-conjugated secondary antibody (green), and an anti-MPR46 antibody, followed by an AlexaFluor546-conjugated secondary antibody (red). Co-localization of the two MPRs is shown on the merged images. The nucleus was visualized with DAPI (blue). Arrows point to enlarged MPR-positive vesicles. Scale bar = 10 µm. To exclude off-target effects, three different siRNAs against OCRL were independently applied. (B) RNAi-treated HeLa cells were stained with an anti-MPR300 antibody, followed by an AlexaFluor488- (green) or AlexaFluor546- (red) conjugated secondary antibody, and an anti-TGN46 antibody, followed by an AlexaFluor488-conjugated secondary antibody (green) or an anti-EEA1 antibody, followed by an AlexaFluor546-conjugated antibody (red). Co-localization is shown on the merged images. Scale bar = 10 µm. (C) Quantification of MPR300-positive vesicles of various sizes was performed by the Metamorph software. To enable direct comparison, all images were taken under the same magnification and laser intensity settings. Sizes of MPR300-positive structures are given in arbitrary units (AU). The number of MPR300-decorated vesicles per AU (≤40, 41–60, 61–80 and ≥81) is given as percentage of the number of vesicles counted in total. Results are from three independent experiments with a total of 20 cells per condition counted, and are shown as the mean ± SD (white square GFP siRNA, black square OCRL siRNA). ***P < 0.001 (two-tailed Student's t-test).
Figure 4.
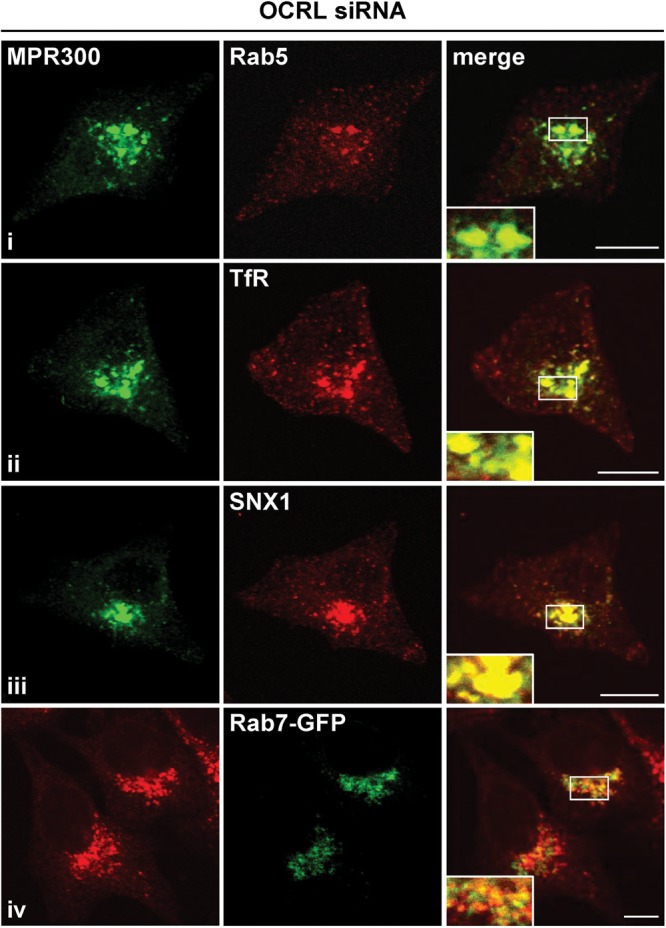
MPR300 localizes in enlarged retromer-positive early endosomes in OCRL-depleted cells. OCRL-depleted cells were labelled with an anti-MPR300 antibody [followed by an AlexaFluor488-conjugated secondary antibody (green in i–iii) or an AlexaFluor546-conjugated secondary antibody (red in iv)] and an anti-Rab5 (i, early endosome), anti-TfR (ii, early and recycling endosome) or anti-SNX1 antibody (iii, component of retromer complex), followed by an AlexaFluor546-conjugated antibody (red). To label the late endosomal compartment, OCRL-depleted cells were transfected with a construct expressing Rab7-GFP (iv, green). Co-localization is shown on the merged images. Scale bar = 10 µm.
We also investigated the intracellular localization of endogenous MPR300 in LS and control fibroblasts. The MPR300 was found to redistribute to the cell periphery and was increasingly associated with endosomal vesicles that also contained EEA1 in the perinuclear region in fibroblasts from patients with LS, when compared with control fibroblasts (Supplementary material, Fig. S3D). Together, the data demonstrate an altered MPR distribution upon loss of OCRL: in OCRL-depleted HeLa cells, MPRs are present in enlarged retromer-positive early endosomes in the perinuclear region, while in LS fibroblasts, MPRs are more scattered and localize to peripheral endosomal structures in addition to the perinuclear region.
OCRL depletion impairs retrograde transport of MPR46 to the TGN
To examine whether OCRL is implicated in regulating transport from endosomes to the Golgi, we performed a quantitative analysis of the endosome-to-TGN transport of MPRs in vivo using HEK-293 cells stably expressing a modified version of MPR46 (HMY-MPR46). This variant contains an N-terminal histidine tag and a tyrosine sulfation site derived from the cholecystokinin precursor (30). The in vivo assay takes advantage of tyrosine sulfotransferase that is exclusively located in the TGN and catalyzes tyrosine sulfation of HMY-MPR46 when it is transported from endosomes to the TGN. [35S]3′-phosphoadenosine 5′-phosphosulfate can be used as the sulfate donor by metabolic labelling of cells with [35S]sulfate. Cells were treated with OCRL- or GFP-specific siRNA and incubated in sulfate-free media containing 20 mm sodium chlorate to prevent sulfation of newly synthesized proteins including HMY-MPR46. HMY-MPR46 was chased to its steady-state localization by addition of cycloheximide to cells prior to the experiment. Retrograde transport of HMY-MPR46 from endosomes to the TGN was then measured by substituting chlorate with [35S]sulfate and incubating cells for 2 h. The amount of 35S-labelled HMY-MPR46 was quantified by Ni-NTA precipitation and scintillation counting.
As shown in Figure 5A, OCRL was almost completely depleted in HMY-MPR46-expressing HEK-293 cells after RNAi-mediated depletion of OCRL. [35S]sulfate incorporation into HMY-MPR46 was significantly reduced in cells depleted of OCRL, displaying an ∼50% (P = 0.01) inhibition when compared with control cells (Fig. 5B). These data show that transport of MPR46 from endosomes to the TGN is strongly reduced in OCRL-depleted cells, resulting in higher steady-state levels of MPR46 in enlarged early endosomes that cycle in equilibrium with the cell surface.
Figure 5.

Endosome-to-TGN transport of MPR46 is impaired in OCRL-depleted HeLa cells. (A) HEK-293 cells stably expressing HMY-MPR46 were treated with OCRL- or GFP-specific siRNA for 72 h and cultivated in sulfate-free media containing chlorate. Depletion was monitored by immunoblotting with an anti-OCRL antibody, and an anti-guanine nucleotide dissociation inhibitor (GDI) antibody was used as loading control. (B) Cyclohexamide was added to the sulfate-free media containing chlorate prior to the experiment. Chlorate was substituted by [35S]sulfate to measure retrograde transport of MPR46 to the TGN. The amount of sulfated MPR46 was determined by Ni-NTA precipitation and scintillation counting and was normalized to the protein content. For more details, see the Materials and Methods section. The amount of sulfated MPR46 in OCRL-depleted cells is expressed as percentage of control cells. Each bar is the mean ± SD of two independent experiments, each performed in duplicate. **P = 0.01 (two-tailed Student's t-test).
OCRL's 5-phosphatase activity is essential for its role in MPR retrograde transport
We have shown that depletion of OCRL prevents MPR transport from endosomes to the TGN. Thus, we next tested whether PI(4,5)P2 metabolism has a role in this process. The PI(4,5)P2 5-phosphatase activity of the yeast synaptojanin-like protein, Inp53p, is required for delivery of proteins from the TGN to endosomes (31), suggesting that PI(4,5)P2 levels may influence this trafficking step. To examine the role of the 5-phosphatase domain of OCRL in endosome-to-TGN transport, we generated several siRNA-resistant OCRL constructs, such as HA-tagged wild-type OCRL isoforms a and b (HA-OCRL a and b; Fig. 6A). The amino acid residues Arg493 and Arg500 were substituted by Ala and Thr, respectively, both in OCRL isoforms a and b (HA-OCRLR493A/R500T a and b; Fig. 6A). Arg residues at positions 493 and 500 are highly conserved in PI(4,5)P2 5-phosphatases, such as synaptojanin 2, and their substitution abolishes the 5-phosphatase activity (32). OCRL was depleted in HeLa cells followed by transient expression of the OCRL variants in these cells. We observed equal expression of siRNA-resistant HA-tagged wild-type OCRL and the 5-phosphatase-deficient OCRLR493A/R500T mutant in OCRL-depleted cells, both for isoforms a (Fig. 6B) and b (data not shown). All OCRL variants showed a perinuclear staining and punctate vesicular distribution (Fig. 6C), typical of endogenous and overexpressed OCRL (21,22,29,33,34). In OCRL-depleted cells not expressing either of the four OCRL proteins, MPR300 was localized to enlarged endosomal structures (compare Fig. 3A and B with Fig. 6C), which was rescued by re-expression of wild-type OCRL isoform a or b (Fig. 6C, asterisks). Determination of the size of MPR300-positive vesicles and their quantification revealed that cells expressing wild-type OCRL isoform a or b contained significantly lower numbers of large MPR300-positive vesicles (P < 0.001), while those expressing one of the two 5-phosphatase-deficient mutants (HA-OCRLR493A/R500T a or b) were characterized by large vesicular clusters decorated with MPR300 that were comparable with those calculated in OCRL-depleted cells (Fig. 6D). Thus, OCRL variants with a non-functional 5-phosphatase domain were unable to rescue enlarged MPR300-positive vesicles, whereas both OCRL isoforms a and b induced distribution of the receptor in small vesicles in the perinuclear region, suggesting that OCRL-mediated hydrolysis of PI(4,5)P2 is needed for retrograde transport of MPR300.
Figure 6.

The 5-phosphatase activity of OCRL is required for proper localization of MPR300. (A) Schematic view of the OCRL domain structure of isoforms a and b. The position of the siRNA-resistant rescue sequence is indicated by an arrow below the domain structure. The eight additional amino acids encoded by exon 18a in isoform a and amino acid substitutions for producing the 5-phosphatase-inactive OCRL variants are indicated above the domain structure. The first and last amino acid residues of OCRL isoforms a and b are given. PH, pleckstrin homology; IPPc, inositol polyphosphate phosphatase, catalytic; ASH, ASPM, SPD-2, hydin; RhoGAP, Rho GTPase activating protein. (B–D) HeLa cells were treated with siRNA against OCRL for 72 h, followed by transfection with siRNA-resistant constructs expressing HA-tagged wild-type OCRL isoform a or b (HA-OCRL a, HA-OCRL b) or 5-phosphatase-inactive OCRL isoform a or b (HA-OCRLR493A/R500T a, HA-OCRLR493A/R500T b), prior to cell lysis or fixation for immunofluorescence analysis. (B) After cell lysis, siRNA-resistant OCRL variants were detected by immunoblotting using an anti-HA-HRP antibody. Depletion was monitored by immunoblotting with an anti-OCRL antibody, and an anti-tubulin antibody was used as loading control. The blot shows a representative result obtained upon expression of wild-type and 5-phosphatase-deficient OCRL isoform a. The same results were obtained after expression of both OCRL isoform b variants (data not shown). (C) siRNA-resistant OCRL variants were detected by staining cells with an anti-HA antibody, followed by an AlexaFluor546-conjugated secondary antibody (red). MPR300 localization was visualized by using an anti-MPR300 antibody, followed by an AlexaFluor488-conjugated secondary antibody (green). * indicates cells transfected with an siRNA-resistant OCRL construct. The nucleus was visualized using DAPI (blue). Scale bar = 20 µm. (D) Quantification of MPR300-positive vesicles of various sizes was performed by the Metamorph software. To enable direct comparison, all images were taken under the same magnification and laser intensity settings. Sizes of MPR300-positive structures are given in arbitrary units (AU). The number of MPR300-positive vesicles per AU (≤40, 41–60, 61–80 and ≥81) is given as percentage of the number of vesicles counted in total. Results are from three independent experiments with a total of 20 cells per condition counted and are shown as the mean ± SD (black square OCRL siRNA, white square OCRL siRNA+HA-OCRL a, gray square OCRL siRNA+HA-OCRL b, square with cross lines OCRL siRNA+HA-OCRLR493A/R500T, a shaded square with cross lines OCRL siRNA+HA-OCRLR493A/R500T b). *P = 0.025; ***P < 0.001 (two-tailed Student's t-test).
OCRL depletion decreases active Rac1 and increases active RhoA
The catalytic activity of OCRL is necessary for PI(4,5)P2 dephosphorylation as elevated PI(4,5)P2 levels have been detected in different LS cell types (35) and OCRL-depleted MDCK cells (28). Although we were not able to detect any gross difference in the amount and subcellular distribution of PI(4,5)P2 between OCRL-depleted and control HeLa cells by using the PH domain of the PLC-delta protein (PLCδ-PH) which specifically binds PI(4,5)P2 (36) (Supplementary material, Fig. S5), a local increase in PI(4,5)P2 on endosomal membranes has been described in OCRL-depleted HeLa cells (20). Because the 5-phosphatase domain of OCRL is required for sorting and retrograde transport of MPRs, we explored how higher levels of PI(4,5)P2 may affect MPR trafficking. PI(4,5)P2 influences the organization of the actin cytoskeleton by modulating the activity of actin regulatory proteins such as N-WASP, cofilin and gelsolin (37). The actin cytoskeleton, however, is also regulated by small GTPases of the Rho family, such as RhoA and Rac1. Importantly, Rac1 is one of several OCRL interaction partners (11,12,14). Therefore, we determined the amount of endogenous, GTP-bound Rac1 and RhoA in OCRL-depleted and control cells by GTPase activation assays. Immunoblot quantitation revealed that the levels of activated, GTP-bound Rac1 were significantly reduced to 60% (P = 0.002) (Fig. 7A and B), while the amount of active GTP-bound RhoA was 2.7-fold (P = 0.005) increased in OCRL-depleted versus control cells (Fig. 7C and D). Although similar studies in fibroblasts could not confirm reduced levels of active Rac1 in LS cells (Supplementary material, Fig. S6A and B), our data indicate a role of OCRL in regulating the activity state of Rac1 and RhoA, at least in certain cell types.
Figure 7.

The activation status of the RhoGTPases Rac1 and RhoA is altered upon OCRL depletion in HeLa cells. (A) RNAi-treated HeLa cells were harvested and GTP-bound Rac1 was pulled down from protein lysates using the GST-PAK[PBD] fusion protein as described in Materials and Methods. Precipitated (active) and total amounts (active and inactive) of Rac1 were detected by immunoblotting using an anti-Rac1 antibody (representative immunoblots—two top panels). OCRL depletion was monitored with an anti-OCRL antibody, and an anti-tubulin antibody was used as loading control (two lower panels). (B) Activity levels of Rac1 were determined by scanning densitometry and normalization of active Rac1 to total Rac1. Rac1 activity in OCRL-depleted cells is expressed as percentage of control cells. Each bar is the mean ± SD of six independent experiments. **P = 0.002 (two-tailed Student's t-test). To exclude off-target effects, three different siRNAs against OCRL were independently applied. (C) RNAi-treated HeLa cells were stimulated with 0.1 mg/ml calpeptin for 40 min, harvested and GTP-bound RhoA was pulled down from protein lysates using GST-Rhotekin[RBD]. Precipitated (active) and total amounts (active and inactive) of RhoA were detected by immunoblotting using an anti-RhoA antibody (representative immunoblots—two top panels). OCRL depletion was monitored with an anti-OCRL antibody, and an anti-tubulin antibody was used as loading control (two lower panels). (D) Activity levels of RhoA were determined by scanning densitometry and normalization of active RhoA to total RhoA. RhoA activity in OCRL-depleted cells is expressed as percentage of control cells. Each bar is the mean ± SD of seven independent experiments. **P = 0.005 (two-tailed Student's t-test). To exclude off-target effects, three different siRNAs against OCRL were independently applied.
Elevated phospho-cofilin and reduced phospho-PAK3 in OCRL-depleted cells
As levels of active Rac1 are decreased and active RhoA increased in OCRL-depleted cells, we examined the phosphorylation status of cofilin and the serine/threonine kinase p21-activated kinase 3 (PAK3), two downstream effector proteins of these Rho GTPases (38–40). Lysates of both OCRL-depleted and control cells were subjected to western blotting to analyze levels of the total and phosphorylated form of cofilin and PAK3 (Fig. 8A and C). By densitometric evaluation followed by statistical analysis, we identified a 1.5-fold increase (P = 0.02) in the amount of phosphorylated cofilin (phospho-cofilin, Fig. 8B), while PAK3 phosphorylation levels were significantly lowered to 60% (P < 0.001, Fig. 8D) in OCRL-depleted cells compared with control cells. Quantification of indirect immunofluorescence microscopy data confirmed a 1.8-fold increase (P < 0.001) in phospho-cofilin fluorescence intensity in OCRL-depleted versus control cells (Fig. 9A and B). The increase in fluorescence intensity could be normalized to control levels by overexpression of siRNA-resistant OCRL wild-type but not the 5-phosphatase-deficient OCRLR493A/R500T mutant in OCRL-depleted HeLa cells (Fig. 9C and D). Moreover, we observed elevated levels of phospho-cofilin in LS compared with control fibroblasts (Supplementary material, Fig. S6C), indicating a generality in actin cytoskeletal-related defects in cells deficient of OCRL. Together, our results suggest that OCRL depletion alters the activation state of RhoGTPases and thereby reduces PAK3 phosphorylation. In addition, the observation that increased phospho-cofilin levels in OCRL-depleted cells can be counteracted by OCRL's 5-phosphatase activity reveals a direct role of OCRL in regulating the activity state of the actin-depolymerizing factor cofilin.
Figure 8.

OCRL depletion in HeLa cells leads to enhanced phosphorylation of cofilin, while phosphorylation of PAK3 is diminished. (A) HeLa cells were treated with OCRL- or GFP-specific siRNA, stimulated with 10 ng/ml EGF for 20 min, harvested and analysed by immunoblotting using an anti-phospho-cofilin or an anti-cofilin antibody (representative immunoblots—two upper panels). OCRL depletion was monitored with an anti-OCRL antibody, and an anti-tubulin antibody was used as loading control (two lower panels). (B) The phosphorylation state of cofilin was determined by scanning densitometry and normalization of phospho-cofilin to total cofilin. The phosphorylation level of cofilin in OCRL-depleted cells is expressed as percentage of control cells. Each bar is the mean ± SD of five independent experiments. *P = 0.02 (two-tailed Student's t-test). To exclude off-target effects, three different siRNAs against OCRL were independently applied. (C) RNAi-treated HeLa cells were stimulated with 10 ng/ml EGF for 20 min, harvested and analyzed by immunoblotting using an anti-phospho-PAK3 or an anti-PAK3 antibody (representative immunoblots—two upper panels). OCRL depletion was monitored with an anti-OCRL antibody, and an anti-tubulin antibody was used as loading control (two lower panels). (D) The phosphorylation state of PAK3 was determined by scanning densitometry and normalization of phosphorylated PAK3 to total PAK3. The phosphorylation level of PAK3 in OCRL-depleted cells is expressed as percentage of control cells. Each bar is the mean ± SD of six independent experiments. ***P < 0.001 (two-tailed Student's t-test). To exclude off-target effects, three different siRNAs against OCRL were independently applied.
Figure 9.

OCRL's 5-phosphatase activity regulates phosphorylation of cofilin. (A) HeLa cells were transfected with OCRL- or GFP-specific siRNA, stimulated with 10 ng/ml EGF for 20 min, fixed and analyzed by confocal laser scanning microscopy. Cells were stained with an anti-phospho-cofilin antibody, followed by an AlexaFluor488-conjugated secondary antibody (green). Scale bar = 10 µm. To exclude off-target effects, three different siRNAs against OCRL were independently applied. (B) Quantification of the mean fluorescence of cells stained for phospho-cofilin was performed by the Metamorph software. To enable direct comparison, all images were taken under the same magnification and laser intensity settings. The fluorescence intensity of OCRL-depleted cells is expressed as percentage of control cells. Results are from three independent experiments with 10 sections per experiment evaluated, and are shown as the mean ± SD. ***P < 0.001 (two-tailed Student's t-test). (C) HeLa cells were treated with siRNA against OCRL, followed by transfection with siRNA-resistant constructs expressing HA-tagged wild-type OCRL isoform a (HA-OCRL a) or the 5-phosphatase-inactive OCRL isoform a (HA-OCRLR493A/R500T a), prior to stimulation with 10 ng/ml EGF for 20 min, fixation and immunofluorescence analysis. OCRL variants were detected by staining cells with an anti-HA antibody, followed by an AlexaFluor546-conjugated secondary antibody (red). Phospho-cofilin was visualized using an anti-phospho-cofilin antibody, followed by an AlexaFluor488-conjugated secondary antibody (green). * indicates cells transfected with an siRNA-resistant OCRL construct. Scale bar = 10 µm. (D) Quantification of the mean fluorescence of phospho-cofilin-stained cells was performed by the Metamorph software. To enable direct comparison, all images were taken under the same magnification and laser intensity settings. The fluorescence intensity of OCRL-depleted cells expressing either HA-OCRL a or HA-OCRLR493A/R500T a is expressed as percentage of cells depleted of OCRL without OCRL re-expression. Results are from 10 different sections per condition, and are shown as the mean ± SD. ***P < 0.001 (two-tailed Student's t-test).
Active Rac1 rescues enhanced cofilin phosphorylation in OCRL-depleted cells
We reasoned that if the increase in phospho-cofilin is a consequence of lower levels of active Rac1, then overexpression of Rac1 should reset phospho-cofilin to normal levels. To achieve this possible rescue, we expressed myc-tagged wild-type Rac1 or a dominant-negative Rac1 with amino acid substitution Thr17Asn (RacN17) in OCRL-depleted and control HeLa cells, followed by immunostaining of phospho-cofilin and Rac1. Overexpression of wild-type Rac1 drastically decreased the levels of phospho-cofilin in the vast majority of OCRL-depleted cells (Fig. 10A). In contrast, we found no significant difference in the fluorescence intensity of phospho-cofilin in cells depleted of OCRL and overexpressing RacN17 compared with OCRL-depleted cells alone (Fig. 10A). These observations were confirmed by quantitative measurement of the fluorescence intensity: a lowered level of phospho-cofilin to 60% (P = 0.003) was observed in OCRL-depleted cells expressing wild-type Rac1 compared with cells depleted of OCRL (Fig. 10B). This level of phospho-cofilin corresponds well to that found in cells treated with GFP siRNA (Fig. 9B). Our results demonstrate that increased phosphorylation of cofilin is a consequence of reduced Rac1 activation upon OCRL depletion.
Figure 10.
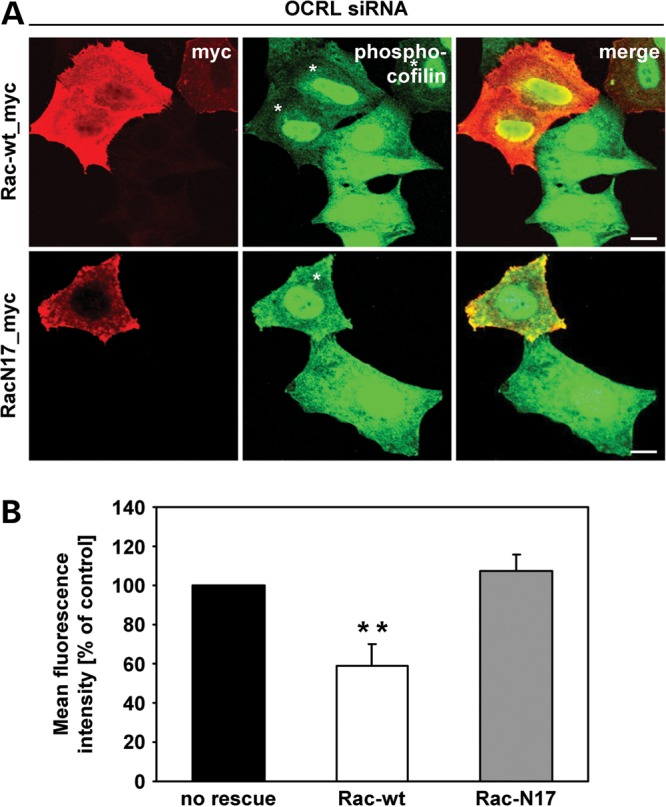
Expression of wild-type Rac1 in OCRL-depleted cells rescues enhanced phosphorylation levels of cofilin. (A) HeLa cells were treated with siRNA against OCRL, followed by transfection with constructs expressing myc-tagged Rac-wt or RacN17, prior to stimulation with 10 ng/ml EGF for 20 min, fixation and immunofluorescence analysis. Rac1 variants were detected by staining cells with an anti-myc antibody, followed by an AlexaFluor546-conjugated secondary antibody (red). Phospho-cofilin was visualized using an anti-phospho-cofilin antibody, followed by an AlexaFluor488-conjugated secondary antibody (green). * indicates cells transfected with a Rac1 expression vector. Scale bar = 10 µm. To exclude off-target effects, three different siRNAs against OCRL were independently applied. (B) Quantification of the mean fluorescence of phospho-cofilin-stained cells was performed by the Metamorph software. To enable direct comparison, all images were taken under the same magnification and laser intensity settings. The fluorescence intensity of OCRL-depleted cells expressing either Rac-wt or RacN17 is expressed as percentage of cells depleted of OCRL without Rac1 re-expression. Results are from three independent experiments with 10 sections per condition of each experiment evaluated and are shown as the mean ± SD. **P = 0.003 (two-tailed Student's t-test).
Active Rac1 acts downstream of OCRL in MPR sorting
We next asked whether impaired endosome-to-Golgi transport and enhanced levels of phospho-cofilin are causally linked in OCRL-depleted cells. If so, then expression of Rac1 should not only rescue high levels of phospho-cofilin but also accumulation of MPR in enlarged perinuclear endosomes in OCRL-depleted cells. We reasoned that OCRL may function in endosome-to-TGN trafficking of MPR, at least to a certain extent, by regulating the activity state of Rac1. We thus expressed myc-tagged wild-type or dominant-negative Rac1 in OCRL-depleted HeLa cells, followed by immunostaining of MPR300 and Rac1. Cells depleted of OCRL and expressing wild-type Rac1 regained the perinuclear MPR distribution in small vesicles, as seen in control cells (compare Fig. 3A and B with Fig. 11A), while MPR300 was enriched in enlarged vesicular structures in OCRL-depleted cells expressing RacN17 or no Rac1 variant (compare Fig. 3A and B, Fig. 6C and Fig. 11A). Determination of the size of MPR300-positive vesicles and their quantification revealed a significant decrease in the number of large MPR300-positive vesicles in OCRL-depleted cells that received the wild-type Rac1 plasmid (**P = 0.008; ***P < 0.001) in comparison with OCRL-depleted cells (Fig. 11B). In contrast, expression of dominant-negative Rac1 was accompanied by the same number of large vesicular clusters decorated with MPR300 compared with cells depleted of OCRL alone (Fig. 11B). These results suggest that (i) Rac1 acts downstream of OCRL and (ii) the amount of active Rac1 is critical for proper MPR trafficking.
Figure 11.
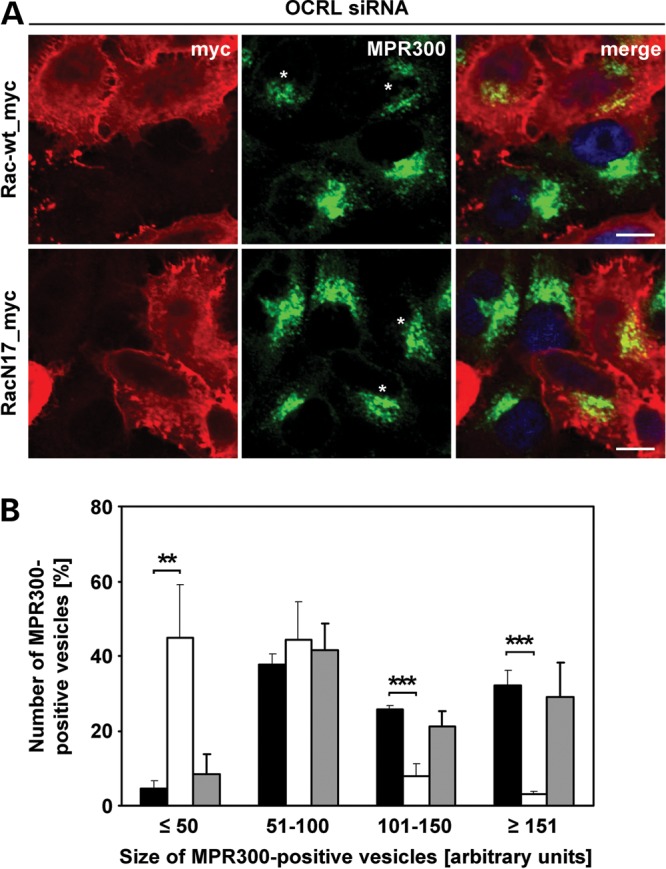
Wild-type Rac1 counteracts accumulation of MPR300 in enlarged endosomal vesicles. (A) HeLa cells were treated with OCRL-specific siRNA, followed by transfection with constructs expressing myc-tagged Rac-wt or RacN17, prior to fixation and immunofluorescence analysis. Rac1 variants were detected by staining cells with an anti-myc antibody, followed by an AlexaFluor546-conjugated secondary antibody (red). MPR300 localization was visualized by using an anti-MPR300 antibody, followed by an AlexaFluor488-conjugated secondary antibody (green). * indicates cells transfected with a Rac1 expression vector. The nucleus was visualized using DAPI (blue). Scale bar = 10 µm. (B) Quantification of MPR300-positive vesicles of various sizes was performed by the Metamorph software. To enable direct comparison, all images were taken under the same magnification and laser intensity settings. Sizes of MPR300-positive structures are given in arbitrary units (AU). The number of MPR300-positive vesicles per AU (≤50, 51–100, 101–150 and ≥151) is given as percentage of the number of vesicles counted in total. Results are from three independent experiments with 10 sections per condition of each experiment evaluated, and are shown as the mean ± SD (black square OCRL siRNA, white square OCRL siRNA+Rac-wt, gray square OCRL siRNA+RacN17). **P = 0.008; ***P < 0.001 (two-tailed Student's t-test).
OCRL depletion impairs cathepsin D maturation and increases Lamp1 levels
We have shown that OCRL is needed for retrograde trafficking of MPRs from endosomes to the TGN, likely via a pathway that depends on GTP-bound Rac1. We hypothesized that enrichment of MPRs in large perinuclear endosomal structures in OCRL-depleted cells could interfere with anterograde transport of lysosomal enzymes from the TGN to endosomes. The lysosomal enzyme cathepsin D is synthesized as pre-pro-cathepsin D, which is converted into inactive pro-cathepsin D after removal of the signal peptide in the endoplasmic reticulum. Pro-cathepsin D (52 kDa) is then transported to endosomes. In the late endosomal compartment, pro-cathepsin D is processed into an intermediate form (48 kDa) and finally into mature cathepsin D (34 kDa) in the lysosome (41). To follow cathepsin D maturation, lysates of OCRL-depleted and control cells were subjected to western blotting and incubated with an anti-cathepsin D antibody to detect the three forms of cathepsin D. In control cells, the intermediate cathepsin D 48 kDa band was barely visible on blot, whereas this form was very abundant in OCRL-depleted cells (Fig. 12A). A similar trend was observed for the pro-cathepsin D 52 kDa band in cells depleted of OCRL (Fig. 12A). Densitometric evaluation revealed a 2-fold increase (P = 0.024) in the amount of the 48 kDa form of cathepsin D in OCRL-depleted cells, and the 52 kDa form was about 1.5-fold more abundant (Fig. 12B). Although the levels of mature cathepsin D were the same in OCRL-depleted and control cells, the total amount of cathepsin D was higher in cells depleted of OCRL compared with control cells (Fig. 12B). In summary, upon depletion of OCRL, the relative amount of the two immature cathepsin D forms was increased, and the total amount was higher than in control cells. These data show that depletion of OCRL impairs cathepsin D maturation, suggesting that the transport of cathepsin D from endosomes to lysosomes is altered.
Figure 12.

Depletion of OCRL results in increased amounts of the cathepsin D intermediate form and the lysosomal protein Lamp1. (A) RNAi-treated HeLa cells were harvested and analyzed by immunoblotting using an anti-cathepsin D (catD) antibody (representative immunoblot—top panel). OCRL depletion was monitored by immunoblotting with an anti-OCRL antibody (middle panel), and an anti-tubulin antibody was used as loading control (lower panel). (B) Amounts of pro-cathepsin D (52 kDa), cathepsin D intermediate (48 kDa) and mature cathepsin D (34 kDa) were determined by scanning densitometry and normalization to tubulin. The amount of each cathepsin D form in OCRL-depleted cells is expressed as percentage of control cells (white square GFP siRNA, black square OCRL siRNA). Each bar is the mean ± SD of four independent experiments. *P = 0.024 (two-tailed Student's t-test). (C) HeLa cells were treated with OCRL- or GFP-specific siRNA, fixed and analyzed by confocal laser scanning microscopy. Cells were stained with an anti-Lamp1 antibody, followed by an AlexaFluor488-conjugated secondary antibody (green). The nucleus was visualized with DAPI (blue). Scale bar = 10 µm. (D) RNAi-treated HeLa cells were harvested and analyzed by immunoblotting using an anti-Lamp1 antibody (representative immunoblot—top panel). Depletion of OCRL was monitored by immunoblotting with an anti-OCRL antibody (middle panel), and an anti-tubulin antibody was used as loading control (lower panel). (E) The Lamp1 amount was determined by scanning densitometry and normalization to tubulin. The amount of Lamp1 in OCRL-depleted cells is expressed as percentage of control cells. Each bar is the mean ± SD of seven independent experiments. *P = 0.025 (two-tailed Student's t-test).
Disturbance of the anterograde transport of lysosomal enzymes may cause a delay in lysosome-dependent protein degradation in OCRL-depleted cells. Indeed, impaired EGF degradation and enlarged Lamp1-positive lysosomes have been found in cells depleted of IPIP27A or IPIP27B, two OCRL interaction partners implicated in endocytic trafficking (23). To analyze if lysosomal morphology is affected by OCRL depletion, we immunostained for Lamp1 in control and OCRL-depleted cells. Confocal laser scanning microscopy revealed a very similar distribution of Lamp1 in OCRL-depleted and control cells (Fig. 12C); however, the intensity of Lamp1 fluorescence was drastically increased in OCRL-depleted HeLa cells (Fig. 12C). In addition, lysosomes appeared much larger in cells depleted of OCRL than in GFP-treated cells (Fig. 12C). We confirmed a 2-fold increase (P = 0.025) in the amount of Lamp1 in cells depleted of OCRL by immunoblotting and densitometric evaluation (Fig. 12D and E). Upregulation of soluble and membrane-bound lysosomal enzymes, and defects in lysosomal enzyme processing have been previously seen in cells which are defective in retrograde transport of MPRs (42).
DISCUSSION
Interaction of OCRL with proteins that participate in endocytic membrane trafficking and OCRL's localization to different compartments of the endocytic pathway have implied a possible role for OCRL in receptor-mediated endocytosis (9,11,29). We undertook a systematic approach to study clathrin-mediated endocytosis in fibroblasts of patients with LS and OCRL-depleted HeLa cells. INPP5B, an inositol polyphosphate 5-phosphatase homologous to OCRL, is present in low levels in HeLa cells (data not shown and 16,20,43) and can compensate for loss of OCRL (44). However, how this compensation occurs is unclear, and our data suggest that INPP5B is involved in trafficking routes distinct from those in which OCRL participates in HeLa cells. We found that Tf, LDL and EGF uptake was normal in LS fibroblasts. In addition, cell surface levels and the rate of internalization of Tf receptors were similar in OCRL-depleted and control cells.
Our study provides evidence for a specific function of OCRL in membrane trafficking of MPRs. First, MPR-mediated uptake of the lysosomal enzyme ASB was significantly decreased in LS fibroblasts. Second, in OCRL-depleted HeLa cells, we detected dramatically increased levels of MPR at the cell surface, accompanied by an increase in MPR internalization. These effects were not due to changes in the total cellular content of MPRs or their half-life. MPRs located at the cell surface are in equilibrium with intracellular receptors localizing to the TGN and endosomes (45). We showed that higher levels of MPR at the cell surface in OCRL-depleted HeLa cells and reduced ASB internalization in LS fibroblasts are likely the consequence of altered intracellular MPR sorting. Indeed, MPRs accumulate in the early endosomal compartment in HeLa cells depleted of OCRL and redistribute to peripheral endosomes in cells of LS-affected patients (our data and 20). We extended this finding by (i) demonstrating MPR enrichment in enlarged, perinuclear early endosomes containing Rab5, the Tf receptor and the retromer component SNX1 and (ii) identifying a 50% reduction in endosome-to-TGN transport of MPRs in OCRL-depleted cells. Our failure to find a general defect in receptor-mediated endocytosis suggests that disturbed trafficking of MPR from the endosomal compartment to the TGN is the likely primary defect caused by OCRL depletion. Impaired retrograde transport has been shown to be compensated by higher levels of MPRs at the cell surface (46) and an increase in their internalization rate (47). We propose that the cellular equilibrium of MPRs shifts to enhanced plasma membrane-to-endosome trafficking upon OCRL depletion in HeLa cells, most likely due to disturbed MPR recycling back to the TGN. Although the decrease in ASB re-uptake and redistribution of MPRs to more scattered perinuclear and peripheral endosomes in LS fibroblasts were not found to be associated with any detectable alteration of cell surface, internalized and cellular MPR levels, the data suggest that recycling of the MPR from the endosomes to the Golgi complex is also impaired in LS fibroblasts, as has been described in renal proximal tubule cells of patients with LS (20). Nevertheless, the cellular consequences of defective MPR trafficking in cells of LS patients seem to be far more complex and difficult to uncover.
Others have reported reduced HA-antibody internalization via a megalin receptor variant in proximal tubule and MDCK cells lacking OCRL and decreased Tf uptake in OCRL-depleted HeLa cells (20). Vicinanza et al. (20) followed internalization of an antibody or ligand by immunofluorescence to quantify the amount of the internalized receptor. In contrast, we used radioactive-labelling methods, FACS and biotinylation assays to obtain quantitative determination of internalized ligands and receptors. In our biotinylation experiments, the receptor itself is quantified, which provides a precise measurement of the internalized amount of receptors. We detected reduced internalization of ASB and an enhanced amount of cell surface MPRs, demonstrating that data from ligand and receptor internalization assays must be evaluated with great care. Consistent with our findings, Coon et al. (16) also found no evidence of altered clathrin-mediated endocytosis in LS fibroblasts, and OCRL depletion did not affect megalin trafficking in MDCK and proximal tubule cells (28). Our data and previously published work together suggest that the endocytic subpopulation of OCRL does not play a major role in modulating clathrin-mediated receptor endocytosis, at least not in certain cell types.
Reduced uptake of lysosomal enzymes and slightly impaired anterograde trafficking of cathepsin D to lysosomes may result in lower levels of hydrolases in lysosomes. In line with this possibility, we observed increased staining and a higher cellular amount of the lysosomal membrane protein Lamp1 in OCRL-depleted cells. Together with reduced EGF receptor degradation upon OCRL depletion (20), these data suggest impaired lysosomal protein degradation in OCRL-depleted cells. Increased lysosomal hydrolase secretion in patients with LS and cells depleted of OCRL (3,20,25,26,28) is often a consequence of a block in retrograde trafficking (46,48,49).
We have shown that only catalytically active OCRL can counteract accumulation of MPR in enlarged vesicular structures induced by OCRL depletion. These data suggest that increased cellular PI(4,5)P2 levels are detrimental to early endosome function and local PI(4,5)P2 dephosphorylation seems to be an important trigger for MPR trafficking along the endosome-to-TGN transport route. Indeed, in the absence of OCRL, certain cellular pools of PI(4,5)P2 increase (17–20). PI(4,5)P2 is an important regulator of actin dynamics by modulating the activation status of a variety of actin cytoskeletal components (37). OCRL deficiency has previously been associated with changes in the organization of the actin cytoskeleton (50), enhanced formation of actin comets (28,51,52), decreased levels of actin stress fibres (20,50) and altered remodelling of F-actin during the last step of cell division (18). This leads to the question whether OCRL acts in regulating F-actin dynamics through modulation of PI(4,5)P2 levels. The finding of increased subcellular PI(4,5)P2 pools that are accompanied by enhanced F-actin levels strongly supports this conclusion (18–20). The actin-depolymerizing protein cofilin is a likely candidate to be regulated by local PI(4,5)P2 amounts and mediating F-actin dynamics upon PI(4,5)P2 dephosphorylation. Indeed, its actin-severing activity is inhibited by binding to membrane-associated PI(4,5)P2 or by phosphorylation (53,54). We have shown here that cofilin phosphorylation is dramatically increased in OCRL-depleted HeLa cells and LS fibroblasts, suggesting that cofilin activity is negatively regulated by both high PI(4,5)P2 levels and enhanced phosphorylation upon loss of OCRL. Indeed, we found OCRL's 5-phosphatase activity to be required for resetting enhanced levels of phospho-cofilin to control levels, and expression of the constitutively active form of cofilin caused a reduction in the PI(4,5)P2-dependent increase in actin polymerization on early endosomes (20). These data suggest that OCRL controls cofilin activation, both by balancing PI(4,5)P2 levels and regulating its phosphorylation status.
How could OCRL affect cofilin phosphorylation? We have demonstrated here that Rho GTPases which interact with OCRL (11,14) and are key regulators of the actin cytoskeleton are broadly dysregulated when OCRL is downregulated: while the amount of active, GTP-bound RhoA is increased, that of Rac1 is drastically decreased. Active RhoA mediates cofilin phosphorylation via the ROCK-LIM kinase pathway (38), and GTP-bound Rac1 is required for cofilin Ser3 dephosphorylation in certain cell types (40,55,56). These findings suggest that both elevated levels of active RhoA and decreased levels of active Rac1 account for enhanced cofilin phosphorylation upon OCRL depletion. Our observation that overexpressed wild-type Rac1 but not the dominant-negative form has the ability to reduce the amount of phosphorylated cofilin in OCRL-depleted cells to control levels corroborates this conclusion. Furthermore, elevated levels of phosphorylated cofilin were accompanied by defects in cell migration and wound healing (40) that have also been described in LS fibroblasts (16). How OCRL regulates the activity of RhoA and Rac1 remains to be determined. Although OCRL possesses a Rho GAP domain, it is catalytically inactive (11,12). However, OCRL is able to bind both Rac1 and Cdc42 (14) and may modulate their activity. Alternatively, enhanced levels of PI(4,5)P2 induced by OCRL depletion could alter recruitment and activation of GTPase activating proteins and/or guanine nucleotide exchange factors to membranes, leading to changes in the amount of GTP- versus GDP-bound forms of RhoGTPases (57–59). In addition, the activity states of Rho GTPases themselves can be regulated by the availability of PI(4,5)P2 (60,61), and PI(4,5)P2 also promotes recruitment of Rho GTPases to the plasma membrane (62). Thus, whether deficient interaction of OCRL with Rho GTPases and/or altered activation or membrane recruitment of these small GTPases or their modulators due to high PI(4,5)P2 levels trigger changes in their activity state in cells depleted of OCRL needs to be clarified in the future.
We have shown here that expression of wild-type Rac1, but not its dominant-negative form, can rescue the accumulation of MPRs in enlarged endosomal vesicles in OCRL-depleted cells. This study makes Rac1 a likely candidate to act as downstream factor of OCRL in retrograde transport, probably by mediating cofilin-dependent actin turnover. Indeed, a Rac1-cofilin-actin signalling module has been linked to retrograde transport in NGF-stimulated sympathetic neurons (56). Here, Rac1 localizes to TrkA-positive endosomes, where it recruits and probably activates cofilin. Active cofilin in turn mediates actin depolymerisation to allow retrograde transport (56). The authors conclude that NGF/TrkA endosomes use a signalling pathway composed of Rac1 and cofilin to promote the maturation of retrograde-competent endosomes. Thus, F-actin breakdown on endosomes through actin modulatory proteins such as cofilin or other actin-binding and -severing proteins is important for retrograde transport (56). PAK3, a downstream effector of Rac1 (39,63), has recently been shown to regulate the F-actin binding capacity of the actin nucleation-promoting factor cortactin by phosphorylation (64). Our finding of reduced levels of phosphorylated PAK3 in OCRL-depleted cells suggests that the activity of cortactin and/or other actin-modulatory proteins may be altered upon OCRL depletion leading to changes in F-actin severing and polymerization activities essential for endosome-to-TGN trafficking. The relevance of actin remodelling for endosomal trafficking processes, such as protein trafficking at the TGN, mobilization and translocation of secretory vesicles to the plasma membrane, and early-to-late endosome transport has recently been demonstrated (65–67). Furthermore, the actin-severing activity of cofilin plays a key role in regulating actin patches which serve as secretory cargo sorting domains at the TGN (68–70). Which of these molecular processes are regulated by the OCRL-dependent Rac1-cofilin cascade will be important to determine. Nevertheless, our data establish a functional link between the Rac1-cofilin signalling module and endosome-to-TGN trafficking through the control of the 5-phosphatase OCRL and sheds important new light on the pathophysiological defects in LS.
MATERIALS AND METHODS
Plasmids
Human Rab7 cDNA in pGreen Lantern-1 was a kind gift from Stefan Linder (University Medical Center Hamburg-Eppendorf, Hamburg, Germany). Human Rac1 (Rac-wt) and dominant negative Rac1 (RacN17) cDNA in pRK5-myc was a kind gift from Alan Hall (Memorial Sloan-Kettering Cancer Center, New York, NY, USA). Human PLCδ-PH cDNA in pEGFP_N1 was a kind gift from Tamas Balla (NICHD, National Institutes of Health, Bethesda, MD, USA). We amplified the coding region of wild-type OCRL for the generation of an expression construct by using OCRL-specific PCR primers and OCRL cDNAs [transcript variant 1 encoding isoform a (Genbank accession no. NM_000276) and transcript variant 2 encoding isoform b (Genbank accession no. NM_001587)] as templates. OCRL cDNA inserts with mutations resulting in amino acid substitutions R493A and R500T were established by PCR-mediated mutagenesis (71). Purified PCR products were cloned into pENTR3C (Invitrogen, Karlsruhe, Germany) via restriction and ligation according to the protocol provided. Constructs were sequenced for integrity and used for cloning wild-type and mutated OCRL coding regions into plasmid pMT2SM-HA-DEST (N-terminal HA epitope) via left–right reaction following the manufacturer's instructions. Conversion of plasmid pMT2SM-HA into the GATEWAY compatible vector pMT2SM-HA-DEST has been described elsewhere (72). siRNA-resistant wild-type and mutated OCRL cDNA constructs were established by site-directed mutagenesis using a QuikChange® kit (Stratagene, Agilent Technologies, Waldbronn, Germany) according to the manufacturer's instructions.
Cell culture and transfection
Skin biopsies from six patients with LS (LS 1–6) were obtained with informed consent from their legal guardians. Mutations in the OCRL gene in the six patients with LS were identified by sequencing the 24 coding exons and adjacent intronic regions (Genbank accession no. NM_000276). Control fibroblasts were derived from one healthy child (Con 1) and two healthy adult individuals (Con 2 and Con 3). Primary cells were cultivated in Dulbecco's modified Eagle medium (DMEM; Invitrogen) supplemented with 10% (v/v) fetal bovine serum (FBS; PAA Laboratories, Cölbe, Germany) and penicillin–streptomycin (P/S; 100 U/ml and 100 µg/ml, respectively; Invitrogen) and incubated at 37°C in a humidified atmosphere with 5% CO2. All experiments were performed between cell passages 3 and 10. HeLa and HEK-293 cells stably expressing a modified MPR46 were cultured in 10 cm dishes at 37°C and 5% CO2 in DMEM supplemented with 7.5% (v/v) FBS plus 1% (v/v) P/S and α-MEM (Invitrogen) supplemented with 7.5% (v/v) FBS, 1% (v/v) P/S and 250 µg/ml G418 (Invitrogen). For plasmid transfections, cells grown on 12-well plates were transfected with pMT2SM-HA-DEST-OCRL a and b, pMT2SM-HA-DEST-OCRLR493A/R500T a and b, and pRK5-myc-Rac-wt, pRK5-myc-RacN17, pEGFP-N1-PLCδ-PH and pGreen Lantern-1-Rab7, respectively, using Lipofectamine 2000 Reagent (Invitrogen) according to the manufacturer's protocol and incubated overnight.
RNA interference was applied for down-regulation of OCRL expression. We used three different OCRL-specific siRNAs to exclude off-target effects (target sequences: 5-GGGTGAAGGTTGTGGATGA-3; 5-TGGCAAAGCGAGAGAAAGATT-3; 5-GGGCAATATGAGTTAATAATT-3; Dharmacon, Thermo Scientific, Lafayette, CO, USA). Cells were transfected on 10 cm dishes using 35 µl Lipofectamine 2000 Reagent and 700 pmol OCRL-specific siRNA (for HeLa cells) or 40 µl Lipofectamine 2000 Reagent and 900 pmol OCRL-specific siRNA (for modified HEK-293 cells) and cultured for 72 h. GFP Duplex I (Dharmacon) was used as a negative control.
Immunoblotting
For immunoblots, cells were washed with ice-cold 1× PBS 72 h after siRNA transfection and harvested in modified RIPA buffer [150 mm NaCl, 50 mm Tris–HCl, pH 8.0, 0.5% sodium desoxycholate (w/v), 1% NP-40 (v/v), 0.1% sodium dodecyl sulfate (w/v)] containing protease inhibitor cocktail (Roche, Mannheim, Germany) and if applicable phosphatase inhibitor cocktail (Roche) for 10 min at 4°C. For EGF stimulation, cells were serum-starved overnight and stimulated with 10 ng/ml EGF (Sigma-Aldrich, Taufkirchen, Germany) for 20 min. After harvesting cells, cell debris was sedimented by centrifugation at 14.000 rpm for 10 min at 4°C. Proteins from each sample were separated on SDS–polyacrylamide gels and analyzed by immunoblotting. As primary antibodies, goat anti-cathepsin D antibody (1:200 dilution, Santa Cruz Biotechnology, Heidelberg, Germany), goat anti-PAK3 antibody (1:200 dilution, Santa Cruz Biotechnology), mouse anti-Lamp1 antibody (H4A3) (1:500 dilution, developed by J. Thomas August and James E. K. Hildreth, Johns Hopkins University School of Medicine, Baltimore, MD, USA; obtained from the Developmental Studies Hybridoma Bank developed under the auspices of the NICHD and maintained by the University of Iowa, Department of Biology, Iowa City, IA, USA), mouse anti-OCRL antibody (1:1000 dilution, a kind gift from Robert L. Nussbaum, University of California San Francisco School of Medicine, San Francisco, CA, USA), mouse anti-Rac1 antibody (1:2000 dilution; Millipore, Schwalbach, Germany), mouse anti-RhoA antibody (1:90 dilution; Santa Cruz Biotechnology), mouse anti-TfR antibody (1:500 dilution, Invitrogen), rabbit anti-Cofilin antibody (1:500 dilution, Cytoskeleton, Offenbach, Germany), rabbit anti-phospho-cofilin antibody (1:200 dilution, Santa Cruz Biotechnology), rabbit anti-Lamp1 antibody (1:900 dilution, Abcam, Cambridge, UK), rabbit anti-MPR300 antibody (1:2000 dilution, a kind gift from Stefan Höning, University of Cologne, Cologne, Germany) and rabbit anti-pPAK3 antibody (1:500 dilution, Abcam) were used. As loading controls, mouse anti-GAPDH (1:10 000 dilution, Abcam), mouse anti-tubulin antibody (1:7500 dilution, Sigma-Aldrich) and rabbit anti-GDI-β antibody (1:1000 dilution, a kind gift from Suzanne R. Pfeffer) were used. The following secondary antibodies were used: horseradish peroxidase-conjugated (HRP) anti-mouse or anti-rabbit antibodies (1:5000–1:30 000 dilution, GE Healthcare, Munich, Germany), HRP-conjugated anti-goat antibody (1:25 000 dilution; Santa Cruz Biotechnology) and HRP-conjugated anti-HA antibody (1:4000 dilution; Roche). Immunoreactive proteins were visualized using the Immobilon Western Chemiluminescent HRP Substrate (Millipore). Scanning densitometry was performed using the ImageJ software.
Immunofluorescence
Cells grown on cover slips were washed with 1× PBS+/+ (0.9 mm CaCl2, 0.52 mm MgCl2, 0.16 mm MgSO4 in 1× PBS), followed by fixation with 4% (w/v in 1× PBS+/+) paraformaldehyde at room temperature for 10–15 min. For EGF stimulation, cells were serum-starved overnight and stimulated with 10 ng/ml EGF (Sigma-Aldrich) for 20 min prior to fixation. After washing three times with 1× PBS+/+ for 10 min, cells were incubated in blocking solution [2% bovine serum albumin (BSA) (w/v), 3% goat serum (v/v), 0.5% NP-40 (v/v) in 1× PBS+/+] for 45 min. Subsequently, cells were covered with 50 µl antibody solution [3% goat serum (v/v), 0.1% NP-40 (v/v) in 1× PBS+/+] containing diluted primary antibodies on parafilm for 2 h at room temperature. The following primary antibodies were used: mouse anti-EEA1 (1:500 dilution, Becton Dickinson Biosciences, Heidelberg, Germany), mouse anti-HA antibody (1:1000 dilution, Covance, Munich, Germany), mouse anti-Lamp1 antibody (H4A3) (1:150 dilution, DSHB), mouse anti-MPR46 antibody (22d4) (1:150 dilution, developed by Donald Messner, Bastyr University, Kenmore, WA, USA; DSHB), mouse anti-myc antibody (1:100 dilution, Sigma-Aldrich), mouse anti-Rab5 antibody (1:500 dilution, Becton Dickinson Biosciences), mouse anti-SNX1 antibody (1:100 dilution, Becton Dickinson Biosciences), mouse anti-TfR antibody (1:500 dilution, Invitrogen), rabbit anti-phospho-cofilin antibody (1:100 dilution, Santa Cruz Biotechnology), rabbit anti-MPR300 antibody (1:200 dilution, a kind gift from Stefan Höning) and sheep anti-TGN46 antibody (1:100 dilution, AbD Serotec, Düsseldorf, Germany). Cells were washed twice with 1× PBS+/+ containing 0.5 m NaCl and twice with 1× PBS+/+ for 10 min. Cells were then covered with 50 µl antibody solution containing AlexaFluor488- and/or AlexaFluor546-conjugated anti-mouse, anti-rabbit and anti-sheep secondary antibodies (1:1000 dilution, Invitrogen) and 4′,6-diamidino-2-phenylindole (DAPI, 1 µg/ml, Serva, Heidelberg, Germany) (in some experiments) on parafilm. After an incubation for 1 h at room temperature, cells were washed three times with 1× PBS+/+ for 10 min at room temperature, mounted in Mowiol 4–88 (Merck, Darmstadt, Germany) and allowed to dry overnight at 4°C. Images were acquired on a Leica DMIRE2 digital scanning confocal microscope and processed using Corel Paint Shop ProX software. Quantification of vesicle sizes was performed using MetaMorph® software.
Internalization assays
Internalization of radioactively labelled ligands
Recombinant human ASB was kindly provided by Dr M. Vellard (BIOMARIN, Novato, CA, USA) and iodinated with Iodination Reagent (Pierce, Thermo Fisher Scientific, Bonn, Germany) and sodium [125I] (Hartmann Analytic, Braunschweig, Germany) to a specific activity of 6 μCi/μg as described previously (73). Human holo-Tf (AppliChem GmbH, Darmstadt, Germany) was iodinated with Iodination Beads (Pierce) and sodium [125I] (Hartmann Analytic) to a specific activity of 1.1 μCi/μg as described previously (74). LDL was labelled with sodium [125I] (Hartmann Analytic) by the iodine monochloride method (75) to a specific activity of 0.05 μCi/μg as described previously (76). Equal numbers of dermal skin fibroblasts were grown on 35 mm dishes (ASB) or 12-well plates (Tf, LDL) for 3–4 days prior to the experiment. Cells were washed with 1× PBS and pre-incubated in starvation medium (DMEM containing 1% P/S and 0.1% BSA) for 1 h (ASB) or overnight (Tf, LDL) at 37°C. For ASB internalization, 0.14 µCi [125I]ASB in 700 µl starvation medium was added and cells were incubated in the presence or absence of 10 mm M6P for 3 h at 37°C. Cells were transferred on ice, washed five times with ice-cold 1× PBS, two times with ice-cold 1× PBS containing 2 mm M6P and additional three times with ice-cold 1× PBS. Cells were harvested in 1× PBS, counted and analyzed by SDS–PAGE and autoradiography. To determine the amount of internalized ASB, intracellular radioactivity was normalized to protein content which was determined by the procedure of Bradford (77). For Tf and LDL internalization, 28 µCi [125I]Tf in 500 µl starvation medium was added to the cells for 5, 10 or 30 min at 37°C and 1.3 µCi [125I]LDL in 500 µl starvation medium was added for 4 h, respectively. Cells were transferred on ice and washed with ice-cold 1× PBS. For Tf internalization, cells were washed with ice-cold acid wash (0.5 m NaCl, 0.2 m acetic acid, pH 2.5). For LDL internalization, cells were washed two times with 1× PBS + heparin (100 U/ml). Then, cells were washed with ice-cold 1× PBS and lysed in 0.1 m NaOH for 30 min on ice. Internalized Tf or LDL was determined by gamma counting and normalization to protein content which was quantitated by the procedure of Lowry (78).
Internalization of fluorescently labelled EGF
Equal numbers of dermal skin fibroblasts were grown on six-well plates for 2–3 days prior to the experiment. Cells were washed with 1× PBS and pre-incubated in starvation medium overnight at 37°C. One microgram per millilitre AlexaFluor488-labelled EGF (Invitrogen) in 700 µl starvation medium was added and cells were incubated for 1 h at 4°C to saturate surface receptors. Cells were then shifted to 37°C and incubated for 15 or 30 min. After transferring plates on ice, cells were washed twice with ice-cold acid wash for 5 min and three times with ice-cold 1× PBS. To detach cells from plates, TripLE™ Express (Invitrogen) was added. After 5 min, cells were taken up in 1× PBS, washed three times with 1× PBS and analyzed by FACS (FACS Calibur, BD, Heidelberg, Germany) with the CellQuest® Pro software.
Determination of radioactively labelled cell surface and cellular MPR300
The monoclonal antibody 2C2 against human liver MPR was prepared and iodinated as described previously (79). Equal numbers of dermal skin fibroblasts were grown on 35 mm dishes for 3–4 days prior to the experiment. Cells were washed three times with 1× PBS and pre-incubated in starvation medium (DMEM containing 1% P/S and 0.1% BSA) for 1 h at 37°C. For detection of cell surface MPR300, cells were washed three times with ice-cold 1× PBS and 0.14 µCi [125I]2C2 in 700 µl starvation medium containing 20 mm HEPES, pH 7.4 was added. After incubation for 4 h at 4°C, cells were washed five times with ice-cold 1× PBS. To determine the amount of MPR300-bound 2C2, cells were washed three times for 5 min with ice-cold acid wash (0.5 m NaCl, 0.2 m acetic acid, pH 2.5) and the collected radioactivity was determined by gamma counting. Then, the cells were additionally washed two times with 1× PBS, harvested in 1 ml 1 m NaOH and cell-associated radioactivity was determined by gamma counting. Cell surface MPR300 was calculated by normalization of the detected radioactivity to protein content which was quantitated by the procedure of Bradford (77). To determine cellular MPR300, cells were pre-incubated in starvation medium containing 20 mm HEPES, pH 7.4 and 0.1% saponin (w/v) for 45 min at 4°C, prior to incubation with 0.14 µCi [125I]2C2 in 700 µl starvation medium containing 20 mm HEPES, pH 7.4 and 0.1% saponin (w/v) for 2 h at 4°C. Cells were washed five times with 1× PBS containing 0.1% saponin (w/v) and harvested in 1 ml 1 m NaOH. Cellular MPR300 was calculated by gamma counting and normalization to protein content which was quantitated by the procedure of Bradford (77).
In vivo endosome-to-TGN transport assay
The assay was modified from that described previously (80). Human embryonic kidney (HEK-293) cells stably expressing a modified MPR46 containing an N-terminal His tag and a tyrosine sulfation site (30) were employed. Cells were transfected with the indicated siRNA on 10 cm dishes 72 h prior to the assay and cultured in α-MEM supplemented with 7.5% FBS and 20 mm sodium chlorate 6 h after transfection. Culture medium was replaced daily. Prior to labelling, 10 µg/ml cycloheximide was added. After 2 h incubation, medium was replaced by α-MEM containing FBS, cycloheximide and 400 mCi sodium [35S]sulfate (Perkin Elmer, Rodgau, Germany) for 2 h. Cells were transferred on ice, washed three times with ice-cold 1× PBS and lysed in 1 ml ice-cold RIPA buffer plus protease inhibitors for 15 min. After centrifugation at 55 000 rpm for 15 min at 4°C, supernatants were mixed with 20 µl RIPA-washed Ni-NTA agarose beads for 45 min at room temperature, followed by centrifugation at 6000 rpm for 1 min. Supernatants were kept to determine protein concentrations and depletion levels. The beads were washed three times in RIPA and incubated with 125 µl RIPA containing 25 mm EDTA for 5 min at room temperature to elute MPRs from beads. [35S] incorporation was quantified by scintillation counting and normalization to protein content.
Metabolic labelling and immunoprecipitation of MPR300
HeLa cells were transfected with the indicated siRNA and equal numbers of cells were split into six-well plates after 24 h. Metabolic labelling was started 48 h after siRNA transfection and performed as described (81). Briefly, cells were washed twice with TD buffer (137 mm NaCl, 5 mm KCl, 0.7 mm Na2HPO4, 25 mm Tris–HCl, pH 7.4) and incubated for 30 min in medium lacking methionine and cysteine plus 7.5% dialyzed FBS. One hundred µCi of Tran[35S]-label (MP Biomedicals, Solon, OH, USA) were added and cells were incubated for 1 h at 37°C, washed twice with growth medium (DMEM plus 7.5% FBS and P/S) containing methionine and cysteine and chased for the indicated times with the same medium. Cells were washed three times with ice-cold 1×PBS on ice, followed by cell lysis in 500 µl RIPA buffer plus protease inhibitors for 10 min. After centrifugation at 55 000 rpm for 15 min at 4°C, supernatants were incubated with 20 µl RIPA-washed protein A-agarose for 15 min at room temperature, followed by centrifugation at 12 000 rpm for 1 min. MPR300 was immunoprecipitated from pre-cleared supernatants by incubation with 1 µl rabbit anti-MPR300 antibody (a kind gift of Suzanne R. Pfeffer) for 1.5 h at room temperature. To isolate immune complexes, 20 µl RIPA-washed protein A-agarose was added, followed by incubation for 30 min at room temperature and centrifugation at 12 000 rpm for 1 min. Pellets were washed four times in RIPA and resuspended in SDS-sample buffer. Samples were subjected to non-reducing SDS–PAGE. The SDS gel was dried and analyzed by PhosphorImager. Visualized radiolabelled MPR300 was subjected to scanning densitometry for quantification.
GTPase activation assay
GST-PAK[PBD] and GST-Rhotekin[RBD] fusion proteins were isolated from Escherichia coli strain BL21 transformed with pGEX-2TK-PAK[PBD] and pGEX-2TK-Rhotekin[RBD], respectively, by snap freezing in liquid nitrogen. Lysis was achieved by incubation in 20 ml lysis buffer component 1 (50 mm Tris–HCl, pH 8.0; 40 mm EDTA; 25% [w/v] sucrose plus protease inhibitors) for 10 min at 4°C, followed by addition of 8 ml lysis buffer component 2 (50 mm Tris–HCl, pH 8.0; 100 mm MgCl2; 0.2% Triton X-100 plus protease inhibitors) and incubation for 10 min at 4°C. After sonification, cell debris was sedimented by centrifugation at 10 000 rpm for 60 min. Subsequently, fusion proteins were coupled with glutathione-bound agarose beads by incubation with 1 ml bead solution (Novagen, Darmstadt, Germany) for 1 h at 4°C, washed four times in wash buffer (50 mm Tris–HCl, pH 8.0; 50 mm NaCl; 5 mm MgCl2) and finally diluted to 65% slurry with wash buffer/glycerol. For cell lysis, equal numbers of HeLa cells transfected with the indicated siRNA and cultivated for 72 h or dermal skin fibroblasts from LS patients and healthy individuals were washed with PBS, and lysed in ice-cold lysis buffer (50 mm Tris–HCl, pH 8.0; 150 mm NaCl; 1% NP-40 plus protease inhibitors). For detection of RhoA activity, HeLa cells were serum-starved overnight and stimulated with 0.1 mg/ml calpeptin (Merck) for 40 min prior to cell lysis. After lysates were cleared by centrifugation at 14 000 rpm for 10 min at 4°C, 75 µl samples were withdrawn to assess total GTPase levels. The remaining lysates were incubated with 20–30 µl of the GST-fusion protein solution for 45 min at 4°C to trap the active form of Rac1 or RhoA. Beads were precipitated by centrifugation at 2500 rpm and 4°C and washed three times with lysis buffer. Total cell lysates and precipitates were separated on SDS–polyacrylamide gels, transferred to PVDF membranes and subjected to immunodetection.
Biotinylation assays
Cell surface biotinylation
Biotinylation experiments were modified from those described previously (47). Briefly, HeLa cells transfected with the indicated siRNA for 60 h or fibroblasts from LS patients and controls were serum-starved overnight, transferred on ice and washed three times with ice-cold HBSS (Invitrogen). Subsequently, cells were incubated with 0.5 mg/ml sulfo-NHS-SS biotin (Pierce, Thermo Scientific, Bonn, Germany) in HBSS for 20 min (HeLa cells) or 30 min (fibroblasts) at 4°C. Unbound biotin was removed by washing with ice-cold HBSS containing 5 mm Tris–HCl (pH 7.4) followed by rinsing with HBSS.
Detection of cell surface receptors
Subsequent to biotinylation, cells were harvested in ice-cold RIPA buffer containing protease inhibitor cocktail. After the cell debris was removed by centrifugation, protein concentrations were determined using the BCA Protein Assay Kit (Pierce, Thermo Scientific) and matched by adding RIPA buffer. Aliquots of the total cell lysates were kept for immunoblotting and the remaining supernatants were subjected to precipitation with streptavidin agarose (Sigma-Aldrich) overnight at 4°C. Probes were centrifuged for 1 min at 8700 rpm at 4°C, and agarose pellets were washed with the RIPA buffer. Precipitates and total cell lysates were separated on SDS–polyacrylamide gels, transferred to PVDF membranes and subjected to immunodetection. For detection of MPR300, non-reducing SDS-sample buffer was used.
Internalization assay
After surface biotinylation, cells were incubated with pre-warmed DMEM containing 10% FBS and P/S for 2 or 5 min at 37°C to induce receptor internalization. As a control, a parallel culture was left untreated. To stop endocytosis and remove remaining cell surface-bound biotin, cells were transferred on ice and immediately covered and washed with ice-cold glutathione-containing buffer [50 mm glutathione, 1 mm EDTA, 75 mm NaCl, 10% FBS (v/v), 75 mm NaOH]. Cells were scraped off in modified RIPA buffer and biotinylated proteins were precipitated using streptavidin-conjugated agarose beads. After washing precipitates with the RIPA buffer, both total cell lysates and precipitates were separated by SDS–PAGE and subjected to immunoblotting. For quantification, levels of MPR300 and TfR in the biotin-streptavidin precipitates were normalized relative to the levels of total MPR300 and TfR, respectively.
Statistical analysis
Two-tailed unpaired Student's t-test was used to determine the statistical significance. Values are presented as the mean ± standard deviation (SD) and were considered significant at P (P-value) < 0.05.
SUPPLEMENTARY MATERIAL
FUNDING
This work was supported by the Deutsche Forschungsgemeinschaft (GRK1459) and the Gemeinnütziger Verein Lowe-Syndrom e. V. J.N. received a fellowship from the Werner-Otto Stiftung. Retrograde transport experiments were supported by a grant to S.R.P. from the US NIH DK37332.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Georg Rosenberger, Fanny Kortüm, Stephan Storch and the members of the Pfeffer lab, especially Eric J. Espinosa, for insightful discussions and experimental assistance, Verena Kochan for generating OCRL wild-type constructs and Stefanie Meien and Sandra Ehret for excellent technical assistance.
Conflict of Interest statement. None declared.
REFERENCES
- 1.Lowe C.U., Terrey M., Mac L.E. Organic-aciduria, decreased renal ammonia production, hydrophthalmos, and mental retardation; a clinical entity. AMA Am. J. Dis. Child. 1952;83:164–184. doi: 10.1001/archpedi.1952.02040060030004. [DOI] [PubMed] [Google Scholar]
- 2.Loi M. Lowe syndrome. Orphanet J. Rare Dis. 2006;1:16. doi: 10.1186/1750-1172-1-16. doi:10.1186/1750-1172-1-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bockenhauer D., Bokenkamp A., van't Hoff W., Levtchenko E., Kist-van Holthe J.E., Tasic V., Ludwig M. Renal phenotype in Lowe Syndrome: a selective proximal tubular dysfunction. Clin. J. Am. Soc. Nephrol. 2008;3:1430–1436. doi: 10.2215/CJN.00520108. doi:10.2215/CJN.00520108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Attree O., Olivos I.M., Okabe I., Bailey L.C., Nelson D.L., Lewis R.A., McInnes R.R., Nussbaum R.L. The Lowe's oculocerebrorenal syndrome gene encodes a protein highly homologous to inositol polyphosphate-5-phosphatase. Nature. 1992;358:239–242. doi: 10.1038/358239a0. doi:10.1038/358239a0. [DOI] [PubMed] [Google Scholar]
- 5.Nussbaum R.L., Orrison B.M., Janne P.A., Charnas L., Chinault A.C. Physical mapping and genomic structure of the Lowe syndrome gene OCRL1. Hum. Genet. 1997;99:145–150. doi: 10.1007/s004390050329. doi:10.1007/s004390050329. [DOI] [PubMed] [Google Scholar]
- 6.Johnson J.M., Castle J., Garrett-Engele P., Kan Z., Loerch P.M., Armour C.D., Santos R., Schadt E.E., Stoughton R., Shoemaker D.D. Genome-wide survey of human alternative pre-mRNA splicing with exon junction microarrays. Science. 2003;302:2141–2144. doi: 10.1126/science.1090100. doi:10.1126/science.1090100. [DOI] [PubMed] [Google Scholar]
- 7.Schmid A.C., Wise H.M., Mitchell C.A., Nussbaum R., Woscholski R. Type II phosphoinositide 5-phosphatases have unique sensitivities towards fatty acid composition and head group phosphorylation. FEBS Lett. 2004;576:9–13. doi: 10.1016/j.febslet.2004.08.052. doi:10.1016/j.febslet.2004.08.052. [DOI] [PubMed] [Google Scholar]
- 8.Zhang X., Jefferson A.B., Auethavekiat V., Majerus P.W. The protein deficient in Lowe syndrome is a phosphatidylinositol-4,5-bisphosphate 5-phosphatase. Proc. Natl Acad. Sci. USA. 1995;92:4853–4856. doi: 10.1073/pnas.92.11.4853. doi:10.1073/pnas.92.11.4853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mao Y., Balkin D.M., Zoncu R., Erdmann K.S., Tomasini L., Hu F., Jin M.M., Hodsdon M.E., De Camilli P. A PH domain within OCRL bridges clathrin-mediated membrane trafficking to phosphoinositide metabolism. EMBO J. 2009;28:1831–1842. doi: 10.1038/emboj.2009.155. doi:10.1038/emboj.2009.155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ponting C.P. A novel domain suggests a ciliary function for ASPM, a brain size determining gene. Bioinformatics. 2006;22:1031–1035. doi: 10.1093/bioinformatics/btl022. doi:10.1093/bioinformatics/btl022. [DOI] [PubMed] [Google Scholar]
- 11.Erdmann K.S., Mao Y., McCrea H.J., Zoncu R., Lee S., Paradise S., Modregger J., Biemesderfer D., Toomre D., De Camilli P. A role of the Lowe syndrome protein OCRL in early steps of the endocytic pathway. Dev. Cell. 2007;13:377–390. doi: 10.1016/j.devcel.2007.08.004. doi:10.1016/j.devcel.2007.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lichter-Konecki U., Farber L.W., Cronin J.S., Suchy S.F., Nussbaum R.L. The effect of missense mutations in the RhoGAP-homology domain on ocrl1 function. Mol. Genet. Metab. 2006;89:121–128. doi: 10.1016/j.ymgme.2006.04.005. doi:10.1016/j.ymgme.2006.04.005. [DOI] [PubMed] [Google Scholar]
- 13.Hyvola N., Diao A., McKenzie E., Skippen A., Cockcroft S., Lowe M. Membrane targeting and activation of the Lowe syndrome protein OCRL1 by rab GTPases. EMBO J. 2006;25:3750–3761. doi: 10.1038/sj.emboj.7601274. doi:10.1038/sj.emboj.7601274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Faucherre A., Desbois P., Satre V., Lunardi J., Dorseuil O., Gacon G. Lowe syndrome protein OCRL1 interacts with Rac GTPase in the trans-Golgi network. Hum. Mol. Genet. 2003;12:2449–2456. doi: 10.1093/hmg/ddg250. doi:10.1093/hmg/ddg250. [DOI] [PubMed] [Google Scholar]
- 15.Faucherre A., Desbois P., Nagano F., Satre V., Lunardi J., Gacon G., Dorseuil O. Lowe syndrome protein Ocrl1 is translocated to membrane ruffles upon Rac GTPase activation: a new perspective on Lowe syndrome pathophysiology. Hum. Mol. Genet. 2005;14:1441–1448. doi: 10.1093/hmg/ddi153. doi:10.1093/hmg/ddi153. [DOI] [PubMed] [Google Scholar]
- 16.Coon B.G., Mukherjee D., Hanna C.B., Riese D.J., 2nd, Lowe M., Aguilar R.C. Lowe syndrome patient fibroblasts display Ocrl1-specific cell migration defects that cannot be rescued by the homologous Inpp5b phosphatase. Hum. Mol. Genet. 2009;18:4478–4491. doi: 10.1093/hmg/ddp407. doi:10.1093/hmg/ddp407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ben El Kadhi K., Roubinet C., Solinet S., Emery G., Carreno S. The inositol 5-phosphatase dOCRL controls PI(4,5)P2 homeostasis and is necessary for cytokinesis. Curr. Biol. 2011;21:1074–1079. doi: 10.1016/j.cub.2011.05.030. doi:10.1016/j.cub.2011.05.030. [DOI] [PubMed] [Google Scholar]
- 18.Dambournet D., Machicoane M., Chesneau L., Sachse M., Rocancourt M., El Marjou A., Formstecher E., Salomon R., Goud B., Echard A. Rab35 GTPase and OCRL phosphatase remodel lipids and F-actin for successful cytokinesis. Nat. Cell Biol. 2011;13:981–988. doi: 10.1038/ncb2279. doi:10.1038/ncb2279. [DOI] [PubMed] [Google Scholar]
- 19.Kühbacher A., Dambournet D., Echard A., Cossart P., Pizarro-Cerda J. The phosphatidylinositol 5-phosphatase oculocerebrorenal syndrome of Lowe protein (OCRL) controls actin dynamics during early steps of Listeria monocytogenes infection. J. Biol. Chem. 2012;287:13128–13136. doi: 10.1074/jbc.M111.315788. doi:10.1074/jbc.M111.315788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vicinanza M., Di Campli A., Polishchuk E., Santoro M., Di Tullio G., Godi A., Levtchenko E., De Leo M.G., Polishchuk R., Sandoval L., et al. OCRL controls trafficking through early endosomes via PtdIns4,5P-dependent regulation of endosomal actin. EMBO J. 2011;30:4970–4985. doi: 10.1038/emboj.2011.354. doi:10.1038/emboj.2011.354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ungewickell A., Ward M.E., Ungewickell E., Majerus P.W. The inositol polyphosphate 5-phosphatase Ocrl associates with endosomes that are partially coated with clathrin. Proc. Natl Acad. Sci. USA. 2004;101:13501–13506. doi: 10.1073/pnas.0405664101. doi:10.1073/pnas.0405664101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Choudhury R., Diao A., Zhang F., Eisenberg E., Saint-Pol A., Williams C., Konstantakopoulos A., Lucocq J., Johannes L., Rabouille C., et al. Lowe syndrome protein OCRL1 interacts with clathrin and regulates protein trafficking between endosomes and the trans-Golgi network. Mol. Biol. Cell. 2005;16:3467–3479. doi: 10.1091/mbc.E05-02-0120. doi:10.1091/mbc.E05-02-0120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Noakes C.J., Lee G., Lowe M. The PH domain proteins IPIP27A and B link OCRL1 to receptor recycling in the endocytic pathway. Mol. Biol. Cell. 2011;22:606–623. doi: 10.1091/mbc.E10-08-0730. doi:10.1091/mbc.E10-08-0730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Swan L.E., Tomasini L., Pirruccello M., Lunardi J., De Camilli P. Two closely related endocytic proteins that share a common OCRL-binding motif with APPL1. Proc. Natl Acad. Sci. USA. 2010;107:3511–3516. doi: 10.1073/pnas.0914658107. doi:10.1073/pnas.0914658107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ungewickell A.J., Majerus P.W. Increased levels of plasma lysosomal enzymes in patients with Lowe syndrome. Proc. Natl Acad. Sci. USA. 1999;96:13342–13344. doi: 10.1073/pnas.96.23.13342. doi:10.1073/pnas.96.23.13342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Norden A.G., Gardner S.C., Van't Hoff W., Unwin R.J. Lysosomal enzymuria is a feature of hereditary Fanconi syndrome and is related to elevated CI-mannose-6-P-receptor excretion. Nephrol. Dial. Transplant. 2008;23:2795–2803. doi: 10.1093/ndt/gfm898. doi:10.1093/ndt/gfm898. [DOI] [PubMed] [Google Scholar]
- 27.Braulke T., Bonifacino J.S. Sorting of lysosomal proteins. Biochim. Biophys. Acta. 2009;1793:605–614. doi: 10.1016/j.bbamcr.2008.10.016. doi:10.1016/j.bbamcr.2008.10.016. [DOI] [PubMed] [Google Scholar]
- 28.Cui S., Guerriero C.J., Szalinski C.M., Kinlough C.L., Hughey R.P., Weisz O.A. OCRL1 function in renal epithelial membrane traffic. Am. J. Physiol. Renal Physiol. 2010;298:F335–F345. doi: 10.1152/ajprenal.00453.2009. doi:10.1152/ajprenal.00453.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Choudhury R., Noakes C.J., McKenzie E., Kox C., Lowe M. Differential clathrin binding and subcellular localization of OCRL1 splice isoforms. J. Biol. Chem. 2009;284:9965–9973. doi: 10.1074/jbc.M807442200. doi:10.1074/jbc.M807442200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Itin C., Rancano C., Nakajima Y., Pfeffer S.R. A novel assay reveals a role for soluble N-ethylmaleimide-sensitive fusion attachment protein in mannose 6-phosphate receptor transport from endosomes to the trans Golgi network. J. Biol. Chem. 1997;272:27737–27744. doi: 10.1074/jbc.272.44.27737. doi:10.1074/jbc.272.44.27737. [DOI] [PubMed] [Google Scholar]
- 31.Ha S.A., Torabinejad J., DeWald D.B., Wenk M.R., Lucast L., De Camilli P., Newitt R.A., Aebersold R., Nothwehr S.F. The synaptojanin-like protein Inp53/Sjl3 functions with clathrin in a yeast TGN-to-endosome pathway distinct from the GGA protein-dependent pathway. Mol. Biol. Cell. 2003;14:1319–1333. doi: 10.1091/mbc.E02-10-0686. doi:10.1091/mbc.E02-10-0686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Malecz N., McCabe P.C., Spaargaren C., Qiu R., Chuang Y., Symons M. Synaptojanin 2, a novel Rac1 effector that regulates clathrin-mediated endocytosis. Curr. Biol. 2000;10:1383–1386. doi: 10.1016/s0960-9822(00)00778-8. doi:10.1016/S0960-9822(00)00778-8. [DOI] [PubMed] [Google Scholar]
- 33.Olivos-Glander I.M., Janne P.A., Nussbaum R.L. The oculocerebrorenal syndrome gene product is a 105-kD protein localized to the Golgi complex. Am. J. Hum. Genet. 1995;57:817–823. [PMC free article] [PubMed] [Google Scholar]
- 34.Dressman M.A., Olivos-Glander I.M., Nussbaum R.L., Suchy S.F. Ocrl1, a PtdIns(4,5)P(2) 5-phosphatase, is localized to the trans-Golgi network of fibroblasts and epithelial cells. J. Histochem. Cytochem. 2000;48:179–190. doi: 10.1177/002215540004800203. doi:10.1177/002215540004800203. [DOI] [PubMed] [Google Scholar]
- 35.Zhang X., Hartz P.A., Philip E., Racusen L.C., Majerus P.W. Cell lines from kidney proximal tubules of a patient with Lowe syndrome lack OCRL inositol polyphosphate 5-phosphatase and accumulate phosphatidylinositol 4,5-bisphosphate. J. Biol. Chem. 1998;273:1574–1582. doi: 10.1074/jbc.273.3.1574. doi:10.1074/jbc.273.3.1574. [DOI] [PubMed] [Google Scholar]
- 36.Garcia P., Gupta R., Shah S., Morris A.J., Rudge S.A., Scarlata S., Petrova V., McLaughlin S., Rebecchi M.J. The pleckstrin homology domain of phospholipase C-delta 1 binds with high affinity to phosphatidylinositol 4,5-bisphosphate in bilayer membranes. Biochemistry. 1995;34:16228–16234. doi: 10.1021/bi00049a039. doi:10.1021/bi00049a039. [DOI] [PubMed] [Google Scholar]
- 37.Yin H.L., Janmey P.A. Phosphoinositide regulation of the actin cytoskeleton. Annu. Rev. Physiol. 2003;65:761–789. doi: 10.1146/annurev.physiol.65.092101.142517. doi:10.1146/annurev.physiol.65.092101.142517. [DOI] [PubMed] [Google Scholar]
- 38.Maekawa M., Ishizaki T., Boku S., Watanabe N., Fujita A., Iwamatsu A., Obinata T., Ohashi K., Mizuno K., Narumiya S. Signaling from Rho to the actin cytoskeleton through protein kinases ROCK and LIM-kinase. Science. 1999;285:895–898. doi: 10.1126/science.285.5429.895. doi:10.1126/science.285.5429.895. [DOI] [PubMed] [Google Scholar]
- 39.Manser E., Leung T., Salihuddin H., Zhao Z.S., Lim L. A brain serine/threonine protein kinase activated by Cdc42 and Rac1. Nature. 1994;367:40–46. doi: 10.1038/367040a0. doi:10.1038/367040a0. [DOI] [PubMed] [Google Scholar]
- 40.Kligys K., Claiborne J.N., DeBiase P.J., Hopkinson S.B., Wu Y., Mizuno K., Jones J.C. The slingshot family of phosphatases mediates Rac1 regulation of cofilin phosphorylation, laminin-332 organization, and motility behavior of keratinocytes. J. Biol. Chem. 2007;282:32520–32528. doi: 10.1074/jbc.M707041200. doi:10.1074/jbc.M707041200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zaidi N., Maurer A., Nieke S., Kalbacher H. Cathepsin D: a cellular roadmap. Biochem. Biophys. Res. Commun. 2008;376:5–9. doi: 10.1016/j.bbrc.2008.08.099. doi:10.1016/j.bbrc.2008.08.099. [DOI] [PubMed] [Google Scholar]
- 42.Riederer M.A., Soldati T., Shapiro A.D., Lin J., Pfeffer S.R. Lysosome biogenesis requires Rab9 function and receptor recycling from endosomes to the trans-Golgi network. J. Cell Biol. 1994;125:573–582. doi: 10.1083/jcb.125.3.573. doi:10.1083/jcb.125.3.573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Williams C., Choudhury R., McKenzie E., Lowe M. Targeting of the type II inositol polyphosphate 5-phosphatase INPP5B to the early secretory pathway. J. Cell Sci. 2007;120:3941–3951. doi: 10.1242/jcs.014423. doi:10.1242/jcs.014423. [DOI] [PubMed] [Google Scholar]
- 44.Janne P.A., Suchy S.F., Bernard D., MacDonald M., Crawley J., Grinberg A., Wynshaw-Boris A., Westphal H., Nussbaum R.L. Functional overlap between murine Inpp5b and Ocrl1 may explain why deficiency of the murine ortholog for OCRL1 does not cause Lowe syndrome in mice. J. Clin. Invest. 1998;101:2042–2053. doi: 10.1172/JCI2414. doi:10.1172/JCI2414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sahagian G.G., Neufeld E.F. Biosynthesis and turnover of the mannose 6-phosphate receptor in cultured Chinese hamster ovary cells. J. Biol. Chem. 1983;258:7121–7128. [PubMed] [Google Scholar]
- 46.Seaman M.N. Cargo-selective endosomal sorting for retrieval to the Golgi requires retromer. J. Cell Biol. 2004;165:111–122. doi: 10.1083/jcb.200312034. doi:10.1083/jcb.200312034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Meyer C., Eskelinen E.L., Guruprasad M.R., von Figura K., Schu P. Mu 1A deficiency induces a profound increase in MPR300/IGF-II receptor internalization rate. J. Cell Sci. 2001;114:4469–4476. doi: 10.1242/jcs.114.24.4469. [DOI] [PubMed] [Google Scholar]
- 48.Ganley I.G., Espinosa E., Pfeffer S.R. A syntaxin 10-SNARE complex distinguishes two distinct transport routes from endosomes to the trans-Golgi in human cells. J. Cell Biol. 2008;180:159–172. doi: 10.1083/jcb.200707136. doi:10.1083/jcb.200707136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Progida C., Cogli L., Piro F., De Luca A., Bakke O., Bucci C. Rab7b controls trafficking from endosomes to the TGN. J. Cell Sci. 2010;123:1480–1491. doi: 10.1242/jcs.051474. doi:10.1242/jcs.051474. [DOI] [PubMed] [Google Scholar]
- 50.Suchy S.F., Nussbaum R.L. The deficiency of PIP2 5-phosphatase in Lowe syndrome affects actin polymerization. Am. J. Hum. Genet. 2002;71:1420–1427. doi: 10.1086/344517. doi:10.1086/344517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Allen P.G. Actin filament uncapping localizes to ruffling lamellae and rocketing vesicles. Nat. Cell Biol. 2003;5:972–979. doi: 10.1038/ncb1059. doi:10.1038/ncb1059. [DOI] [PubMed] [Google Scholar]
- 52.Guerriero C.J., Weixel K.M., Bruns J.R., Weisz O.A. Phosphatidylinositol 5-kinase stimulates apical biosynthetic delivery via an Arp2/3-dependent mechanism. J. Biol. Chem. 2006;281:15376–15384. doi: 10.1074/jbc.M601239200. doi:10.1074/jbc.M601239200. [DOI] [PubMed] [Google Scholar]
- 53.Arber S., Barbayannis F.A., Hanser H., Schneider C., Stanyon C.A., Bernard O., Caroni P. Regulation of actin dynamics through phosphorylation of cofilin by LIM-kinase. Nature. 1998;393:805–809. doi: 10.1038/31729. doi:10.1038/31729. [DOI] [PubMed] [Google Scholar]
- 54.Gorbatyuk V.Y., Nosworthy N.J., Robson S.A., Bains N.P., Maciejewski M.W., Dos Remedios C.G., King G.F. Mapping the phosphoinositide-binding site on chick cofilin explains how PIP2 regulates the cofilin-actin interaction. Mol. Cell. 2006;24:511–522. doi: 10.1016/j.molcel.2006.10.007. doi:10.1016/j.molcel.2006.10.007. [DOI] [PubMed] [Google Scholar]
- 55.Pandey D., Goyal P., Dwivedi S., Siess W. Unraveling a novel Rac1-mediated signaling pathway that regulates cofilin dephosphorylation and secretion in thrombin-stimulated platelets. Blood. 2009;114:415–424. doi: 10.1182/blood-2008-10-183582. doi:10.1182/blood-2008-10-183582. [DOI] [PubMed] [Google Scholar]
- 56.Harrington A.W., St Hillaire C., Zweifel L.S., Glebova N.O., Philippidou P., Halegoua S., Ginty D.D. Recruitment of actin modifiers to TrkA endosomes governs retrograde NGF signaling and survival. Cell. 2011;146:421–434. doi: 10.1016/j.cell.2011.07.008. doi:10.1016/j.cell.2011.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Erlmann P., Schmid S., Horenkamp F.A., Geyer M., Pomorski T.G., Olayioye M.A. DLC1 activation requires lipid interaction through a polybasic region preceding the RhoGAP domain. Mol. Biol. Cell. 2009;20:4400–4411. doi: 10.1091/mbc.E09-03-0247. doi:10.1091/mbc.E09-03-0247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Crompton A.M., Foley L.H., Wood A., Roscoe W., Stokoe D., McCormick F., Symons M., Bollag G. Regulation of Tiam1 nucleotide exchange activity by pleckstrin domain binding ligands. J. Biol. Chem. 2000;275:25751–25759. doi: 10.1074/jbc.M002050200. doi:10.1074/jbc.M002050200. [DOI] [PubMed] [Google Scholar]
- 59.Bollag G., McCormick F. Differential regulation of rasGAP and neurofibromatosis gene product activities. Nature. 1991;351:576–579. doi: 10.1038/351576a0. doi:10.1038/351576a0. [DOI] [PubMed] [Google Scholar]
- 60.Chuang T.H., Bohl B.P., Bokoch G.M. Biologically active lipids are regulators of Rac.GDI complexation. J. Biol. Chem. 1993;268:26206–26211. [PubMed] [Google Scholar]
- 61.Zheng Y., Glaven J.A., Wu W.J., Cerione R.A. Phosphatidylinositol 4,5-bisphosphate provides an alternative to guanine nucleotide exchange factors by stimulating the dissociation of GDP from Cdc42Hs. J. Biol. Chem. 1996;271:23815–23819. doi: 10.1074/jbc.271.39.23815. doi:10.1074/jbc.271.39.23815. [DOI] [PubMed] [Google Scholar]
- 62.Heo W.D., Inoue T., Park W.S., Kim M.L., Park B.O., Wandless T.J., Meyer T. PI(3,4,5)P3 and PI(4,5)P2 lipids target proteins with polybasic clusters to the plasma membrane. Science. 2006;314:1458–1461. doi: 10.1126/science.1134389. doi:10.1126/science.1134389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Jaffer Z.M., Chernoff J. p21-activated kinases: three more join the Pak. Int. J. Biochem. Cell Biol. 2002;34:713–717. doi: 10.1016/s1357-2725(01)00158-3. doi:10.1016/S1357-2725(01)00158-3. [DOI] [PubMed] [Google Scholar]
- 64.Webb B.A., Zhou S., Eves R., Shen L., Jia L., Mak A.S. Phosphorylation of cortactin by p21-activated kinase. Arch. Biochem. Biophys. 2006;456:183–193. doi: 10.1016/j.abb.2006.06.011. doi:10.1016/j.abb.2006.06.011. [DOI] [PubMed] [Google Scholar]
- 65.Poupon V., Girard M., Legendre-Guillemin V., Thomas S., Bourbonniere L., Philie J., Bright N.A., McPherson P.S. Clathrin light chains function in mannose phosphate receptor trafficking via regulation of actin assembly. Proc. Natl Acad. Sci. USA. 2008;105:168–173. doi: 10.1073/pnas.0707269105. doi:10.1073/pnas.0707269105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wen P.J., Osborne S.L., Zanin M., Low P.C., Wang H.T., Schoenwaelder S.M., Jackson S.P., Wedlich-Soldner R., Vanhaesebroeck B., Keating D.J., et al. Phosphatidylinositol(4,5)bisphosphate coordinates actin-mediated mobilization and translocation of secretory vesicles to the plasma membrane of chromaffin cells. Nat. Commun. 2011;2:491. doi: 10.1038/ncomms1500. doi:10.1038/ncomms1500. [DOI] [PubMed] [Google Scholar]
- 67.Morel E., Parton R.G., Gruenberg J. Annexin A2-dependent polymerization of actin mediates endosome biogenesis. Dev. Cell. 2009;16:445–457. doi: 10.1016/j.devcel.2009.01.007. doi:10.1016/j.devcel.2009.01.007. [DOI] [PubMed] [Google Scholar]
- 68.Salvarezza S.B., Deborde S., Schreiner R., Campagne F., Kessels M.M., Qualmann B., Caceres A., Kreitzer G., Rodriguez-Boulan E. LIM kinase 1 and cofilin regulate actin filament population required for dynamin-dependent apical carrier fission from the trans-Golgi network. Mol. Biol. Cell. 2009;20:438–451. doi: 10.1091/mbc.E08-08-0891. doi:10.1091/mbc.E08-08-0891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.von Blume J., Alleaume A.M., Cantero-Recasens G., Curwin A., Carreras-Sureda A., Zimmermann T., van Galen J., Wakana Y., Valverde M.A., Malhotra V. ADF/cofilin regulates secretory cargo sorting at the TGN via the Ca2+ ATPase SPCA1. Dev. Cell. 2011;20:652–662. doi: 10.1016/j.devcel.2011.03.014. doi:10.1016/j.devcel.2011.03.014. [DOI] [PubMed] [Google Scholar]
- 70.von Blume J., Duran J.M., Forlanelli E., Alleaume A.M., Egorov M., Polishchuk R., Molina H., Malhotra V. Actin remodeling by ADF/cofilin is required for cargo sorting at the trans-Golgi network. J. Cell Biol. 2009;187:1055–1069. doi: 10.1083/jcb.200908040. doi:10.1083/jcb.200908040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ito W., Ishiguro H., Kurosawa Y. A general method for introducing a series of mutations into cloned DNA using the polymerase chain reaction. Gene. 1991;102:67–70. doi: 10.1016/0378-1119(91)90539-n. doi:10.1016/0378-1119(91)90539-N. [DOI] [PubMed] [Google Scholar]
- 72.Wimplinger I., Morleo M., Rosenberger G., Iaconis D., Orth U., Meinecke P., Lerer I., Ballabio A., Gal A., Franco B., et al. Mutations of the mitochondrial holocytochrome c-type synthase in X-linked dominant microphthalmia with linear skin defects syndrome. Am. J. Hum. Genet. 2006;79:878–889. doi: 10.1086/508474. doi:10.1086/508474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Marschner K., Kollmann K., Schweizer M., Braulke T., Pohl S. A key enzyme in the biogenesis of lysosomes is a protease that regulates cholesterol metabolism. Science. 2011;333:87–90. doi: 10.1126/science.1205677. doi:10.1126/science.1205677. [DOI] [PubMed] [Google Scholar]
- 74.Stoorvogel W., Geuze H.J., Strous G.J. Sorting of endocytosed transferrin and asialoglycoprotein occurs immediately after internalization in HepG2 cells. J. Cell Biol. 1987;104:1261–1268. doi: 10.1083/jcb.104.5.1261. doi:10.1083/jcb.104.5.1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.McFarlane A.S. Efficient trace-labelling of proteins with iodine. Nature. 1958;182:53. doi: 10.1038/182053a0. doi:10.1038/182053a0. [DOI] [PubMed] [Google Scholar]
- 76.Heeren J., Weber W., Beisiegel U. Intracellular processing of endocytosed triglyceride-rich lipoproteins comprises both recycling and degradation. J. Cell Sci. 1999;112:349–359. doi: 10.1242/jcs.112.3.349. [DOI] [PubMed] [Google Scholar]
- 77.Bradford M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. doi:10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 78.Lowry O.H., Rosebrough N.J., Farr A.L., Randall R.J. Protein measurement with the Folin phenol reagent. J. Biol. Chem. 1951;193:265–275. [PubMed] [Google Scholar]
- 79.Braulke T., Gartung C., Hasilik A., von Figura K. Is movement of mannose 6-phosphate-specific receptor triggered by binding of lysosomal enzymes? J. Cell Biol. 1987;104:1735–1742. doi: 10.1083/jcb.104.6.1735. doi:10.1083/jcb.104.6.1735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Sincock P.M., Ganley I.G., Krise J.P., Diederichs S., Sivars U., O'Connor B., Ding L., Pfeffer S.R. Self-assembly is important for TIP47 function in mannose 6-phosphate receptor transport. Traffic. 2003;4:18–25. doi: 10.1034/j.1600-0854.2003.40104.x. doi:10.1034/j.1600-0854.2003.40104.x. [DOI] [PubMed] [Google Scholar]
- 81.Ganley I.G., Carroll K., Bittova L., Pfeffer S. Rab9 GTPase regulates late endosome size and requires effector interaction for its stability. Mol. Biol. Cell. 2004;15:5420–5430. doi: 10.1091/mbc.E04-08-0747. doi:10.1091/mbc.E04-08-0747. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.