

Abstract
Heterozygous mutations in Myoclonin1/EFHC1 cause juvenile myoclonic epilepsy (JME), the most common form of genetic generalized epilepsies, while homozygous F229L mutation is associated with primary intractable epilepsy in infancy. Heterozygous mutations in adolescent JME patients produce subtle malformations of cortical and subcortical architecture, whereas homozygous F229L mutation in infancy induces severe brain pathology and death. However, the underlying pathological mechanisms for these observations remain unknown. We had previously demonstrated that EFHC1 is a microtubule-associated protein (MAP) involved in cell division and radial migration during cerebral corticogenesis. Here, we show that JME mutations, including F229L, do not alter the ability of EFHC1 to colocalize with the centrosome and the mitotic spindle, but act in a dominant-negative manner to impair mitotic spindle organization. We also found that mutants EFHC1 expression disrupted radial and tangential migration by affecting the morphology of radial glia and migrating neurons. These results show how Myoclonin1/EFHC1 mutations disrupt brain development and potentially produce structural brain abnormalities on which epileptogenesis is established.
INTRODUCTION
Juvenile myoclonic epilepsy (JME) is the most common form of genetic generalized epilepsies and represents 10–30% of all epilepsies (1,2). JME typically starts during adolescence as symmetric and asymmetric myoclonic jerks, clonic tonic–clonic seizures and sometimes absence seizures (1,2). A genetic contribution to JME has long been established. Twenty-two chromosomal loci have been genetically linked with JME (3), although mutations have been identified only in five Mendelian genes (CACNB4, CASR, GABRA1, GABRD and Myoclonin1/EFHC1. http://omim.org and http://www.ncbi.nlm.nih.gov/omim/). Three susceptibility single-nucleotide polymorphisms in BRD2, Cx-36 and ME2 have also been genetically associated with and contribute to the risk of JME (2,3).
So far, most mutations discovered in the coding sequence of EFHC1 are heterozygous missense mutations (4–9). Rarely, nonsense, deletions and deletions/frameshifts were also observed (8). Heterozygous mutations in Myoclonin1/EFHC1 are to date the most frequent, reported in several unrelated families around the world, causing 3–9% of JME including autosomal dominant, singletons and sporadic cases (4–8). More recently, homozygous F229L mutation in Myoclonin1/EFHC1 was shown to produce severe drug-resistant epilepsy in infancy starting 12–16h after birth (10). While heterozygous mutations in adolescent JME patients produce subtle malformations of cortical and subcortical architecture (2,11), homozygous F229L mutation in infancy induces severe brain pathology and death at 6 months to 3 years of age (10).
EFHC1 encodes a 70 kDa protein with three DM10 domains of unknown function and a single EF-hand motif with a Ca2+-binding domain (4). We have previously demonstrated that EFHC1 is a microtubule-associated protein (MAP) that localized to the centrosome and the mitotic spindle through its N-terminus (12,13). We have shown that EFHC1 plays a role in cell division as its loss of function disrupts mitotic spindles organization. Moreover, in the developing neocortex (NCx), an acute EFHC1 deficiency impairs radial migration of projection neurons (13).
The development of the cerebral cortex is a very complex process, which follows strictly regulated and tightly linked sequences of proliferation, cell cycle exit, cell migration to specific cell layers and neuronal differentiation. Based on their origin, there are two major types of neurons in the developing brain, excitatory projection neurons and inhibitory interneurons; both types being important to maintain the balance between excitation and inhibition in the cortex. Projection neurons are generated in the dorsal telecephalon directly from radial glial cells in the proliferative germinal ventricular zone (VZ) (14–16). Radial glial cells undergo rapid proliferative divisions to expand the progenitor pool before they exit the cell cycle. Newborn neurons move radially to the subventricular zone (SVZ)-intermediate zone (IZ) where they pause and adopt a multipolar morphology before converting to a bipolar cell and recommencing their radial migration along radial glial processes to reach their final position in the cortical plate (CP) (16). Interneurons, on the other hand, originate from ganglionic eminences of the ventral forebrain and reach the cortex by tangential migration (15,17). Unlike radially migrating neurons, they are independent of radial glia for their migration (18). Cells from both types move forward by means of nucleokinesis, a saltatory process that involves forward extension of the leading process and somal translocation to keep up (19,20).
Here, we investigate the impact of the first four JME-causing mutations discovered (D210N, R221H, F229L and D253Y) and two coding polymorphisms (R159W and I619L), used as control (4) on EFHC1 function during the cell division and cortical development. We show that, in HEK293 cells, EFHC1 mutations do not alter the ability of the protein to colocalize with centrosomes and mitotic spindles but induce mitotic spindle defects. Moreover, we present evidence that mutants EFHC1 deleteriously impact radial and tangential migration during brain development by affecting the radial glia scaffold organization and the morphology of radially and tangentially migrating neurons. Overall, our results demonstrate a dominant-negative effect of mutations on EFHC1 properties and identify multiple distinct roles for EFHC1 in corticogenesis. These findings clarify the observed microdysgenesis produced by heterozygous mutations, the severe brain pathology created by homozygous mutations and hyperexcitable circuits in both JME and severe epilepsy of infancy.
RESULTS
The subcellular localization of mutants EFHC1 is similar to wild-type
As previously described for wild-type EFHC1 (12) (Fig. 1), we examined by fluorescence imaging the subcellular distribution in HEK293 cells of the first four mutations that have been associated with JME by Suzuki et al. (4), i.e. D210N, R221H, F229L and D253Y. In interphase cells, EGFP-tagged mutants EFHC1 were observed all over the cytoplasm and in the nucleus, except the nucleoli (Fig. 1A). Moreover, mutant proteins colocalized with the centrosome, as shown by the γ-tubulin staining (Fig. 1A). During mitosis, all proteins associated with the mitotic spindle (Fig. 1B). Between prophase to late telophase, the staining was specifically located at spindle poles (Fig. 1B and supplementary Material, S1A). It was also observed at the spindle midzone from late anaphase to telophase and at the midbody during cytokinesis (Supplementary Material, Fig. S1A). We found the same results in the stably transfected HEK Flp-In T-Rex-293 cell line (not shown). In addition, we analyzed the subcellular localization of some EFHC1 coding polymorphisms (R159W and I619L) that were also present in healthy control population (4), and found no difference with the wild-type protein (Fig. 1 and Supplementary Material, Fig. S1B). Therefore, these data showed that all mutated and polymorphic forms of EFHC1 studied present the same expression pattern as the wild-type protein (12), indicating that they did not impair the subcellular targeting of EFHC1.
Figure 1.
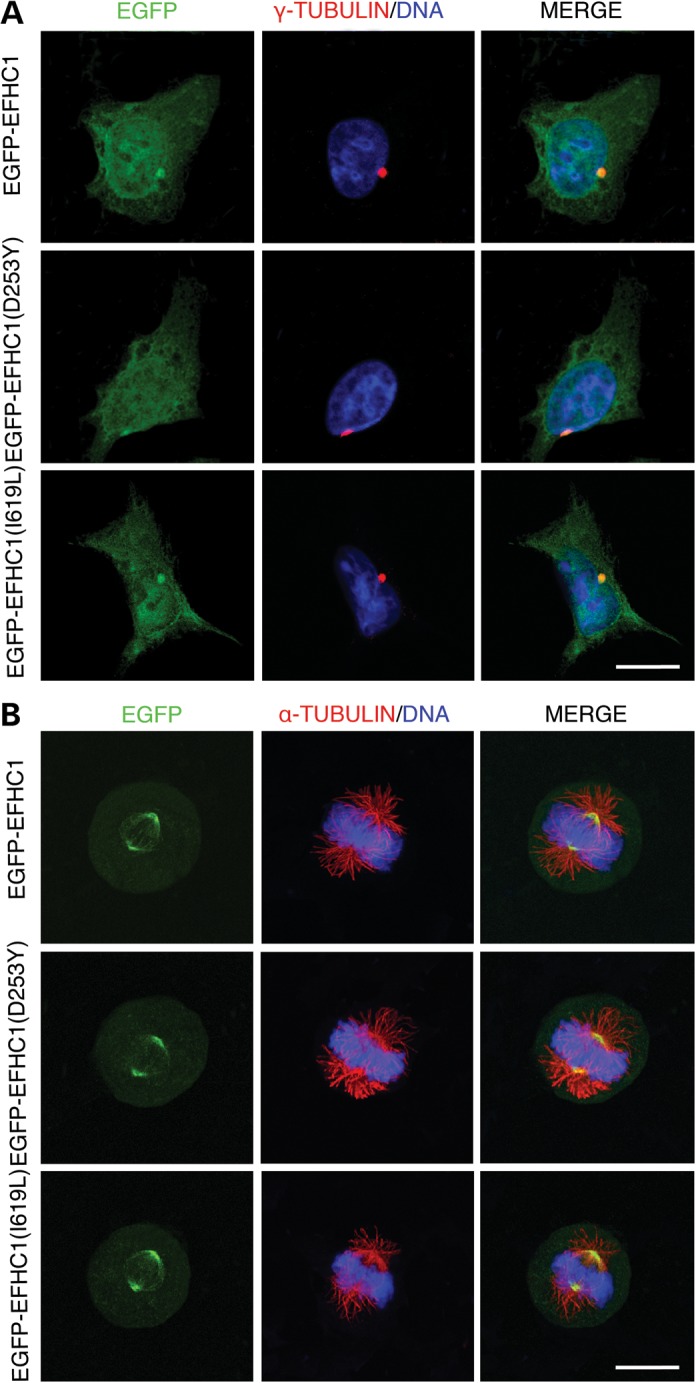
Subcellular localization of mutant and polymorphic EFHC1 proteins. HEK293 cells expressing EGFP-tagged wild-type (EGFP-EFHC1), EGFP-EFHC1(D253Y) or EGFP-EFHC1(I619L) (green). EGFP-EFHC1(D253Y) and EGFP-EFHC1(I619L) images are representative of mutants and polymorphisms, respectively. (A) Interphase cells were stained for γ-tubulin (red) to visualize centrosomes. DNA displayed in blue. Merge panels show that all EGFP-tagged proteins colocalized with the centrosome. (B) Mitotic cells were stained for α-tubulin (red) to visualize microtubules. DNA displayed in blue. Merge panels show that all EGFP-tagged proteins associate with the mitotic spindle. Scale bars represent 20 μm.
JME mutations induce mitotic spindle defects
Based on our previous study reporting that EFHC1 loss of function leads to abnormal mitotic spindles (13), we further investigated the impact of EFHC1 mutations on mitosis. We, therefore, analyzed spindle morphology in HEK293 cells transfected with wild-type, mutants or polymorphics EGFP-EFHC1 after α-tubulin staining (Fig. 2). We found that overexpression of mutated forms resulted in mitotic spindle defects, including monopolar spindles and chromosomes alignment failures during metaphase (chromosomes congression abnormalities) (Fig. 2A). We quantified their occurrence in cultures overexpressing different EGFP-tagged EFHC1 proteins (Fig. 2B) and found that the percentage of abnormal spindles was significantly higher for mutant proteins (65.7 ± 2.2% for D210N, 57.8 ± 2.9% for R221H, 67.2 ± 2.3% for F229L and 71.3 ± 4.6% for D253Y) compared with EGFP-EFHC1 (32.5 ± 5.9%) or polymorphisms (36.4 ± 5.6% for R159W and 37.7 ± 4.5% for I619L). We found the same results in the stably transfected HEK Flp-In T-Rex-293 cell line (not shown). These results suggest that mutant forms of EFHC1 could act as dominant-negative proteins that impair the normal function of wild-type protein.
Figure 2.

EFHC1 mutations induce mitotic spindle defects. (A) Representative images of mitotic spindle defects observed in HEK293 cells expressing mutants EGFP-EFHC1 (green). Monopolar spindle and chromosome alignment failure on metaphase plate (congression abnormality, arrows) are shown. The cells were stained for α-tubulin (red) and DNA (blue). Scale bar represents 20 μm. (B) Quantification of abnormal spindles in cultures 2 days after transfections with EGFP, EGFP-tagged wild-type, mutants or polymorphics EFHC1. (C) Quantification of the mitotic index 2 days after transfections with EGFP, EGFP-EFHC1, mutant EGFP-EFHC1(D253Y) or polymorphic EGFP-EFHC1(I619L). Error bars show SEM. **P < 0.01.
Because we have previously demonstrated that mitotic spindle defects led to an accumulation of mitotic cells (13), we quantified the number of mitosis in cultures expressing EGFP, EGFP-EFHC1, EGFP-EFHC1(D253Y) (the mutant form that gave the more important alteration of mitosis) or EGFP-EFHC1(I619L) (Fig. 2C). We found no differences in the mitotic index between the different above conditions.
Mutants EFHC1 impair radial migration of cortical projection neurons
Since EFHC1 is essential for radial migration of projection neurons during cortical development (13), we examine the effect of its mutated forms on this process. We introduced EGFP or EGFP-tagged wild-type, mutant and polymorphic EFHC1 proteins into neural progenitor cells in mouse NCx by in utero electroporation at embryonic day (E) 14.5. Three days later, we analyzed the distribution of EGFP-positive cells in the VZ/SVZ, IZ and CP (Fig. 3A). As previously described (13), no obvious differences in the spatial distribution between EGFP and EGFP-EFHC1 transfected brains were observed (Fig. 3A and B). However, expression of EFHC1 mutations impaired radial migration as it resulted in the accumulation of electroporated cells in the IZ (Fig. 3B, 45.5 ± 3.4% for D210N, 51.8 ± 2.9% for R221H, 45.9 ± 3.6% for F229L and 48.9 ± 2.8% for D253Y versus 38.6 ± 2.2% for EFHC1 and 42.6 ± 3.7% for EGFP) together with a reduced percentage of projection neurons in the CP (Fig. 3B, 21.8 ± 4.3% for D210N, 17.5 ± 3.9% for R221H, 22.8 ± 2.2% for F229L and 18.1 ± 1.9% for D253Y versus 29.8 ± 1.8% for EFHC1 and 27.1 ± 2.8% for EGFP). In contrast, polymorphic proteins had no significant effect on radial migration (Fig. 3A and B). As experiments performed on cell lines (Fig. 2), these results point out a dominant-negative effect of mutant proteins. To verify this hypothesis, we analyzed the migration of cells electroporated with a dominant-negative form of EFHC1 containing only the microtubule-binding domain of the protein (EGFP-N45; (13)). We found that EGFP-N45 construct interfered with radial migration in the same way as mutants EFHC1 (Supplementary Material, Fig. S2), confirming the dominant-negative effect of mutant proteins.
Figure 3.

Acute expression of EFHC1 mutations alters radial migration of projection neurons in the cerebral cortex. (A) Distribution of EGFP-positive cells in different cortical regions (VZ/SVZ, IZ and CP) 3 days after in utero electroporation at E14.5 of EGFP, EGFP-EFHC1, EGFP-EFHC1(D253Y) or EGFP-EFHC1(I619L) (green). EGFP-EFHC1(D253Y) and EGFP-EFHC1(I619L) images are representative of mutants and polymorphisms, respectively. EGFP-EFHC1(D253Y) panel shows radial migration defect in the CP. Scale bar represents 100 μm. (B) Quantification of EGFP-positive cells in different cortical regions (VZ/SVZ, IZ and CP) 3 days after in utero electroporation at E14.5 under different above conditions. Error bars show SEM. *P < 0.05, **P < 0.01.
Mutated forms of EFHC1 does not influence progenitor cell proliferation and cell cycle exit
Because cortical progenitor cell division occurs before and is tightly coupled to neuronal migration, we decided to study the influence of EFHC1 mutations on mitosis, proliferation and cell cycle exit of cortical progenitors by in utero electroporation at E14.5. As the disruption of radial migration was similar for all mutant and polymorphic proteins, we selected the same mutant (EGFP-EFHC1(D253Y)) and polymorphism (EGFP-EFHC1(I619L)) as previously for further analysis. Sections from electroporated brains collected 2 days after electroporation were subjected to immunofluorescence to phospho-Histone H3 (PHH3), an M-phase marker (Fig. 4A). Similarly to cell cultures (Fig. 2C), the quantification of the mitotic index revealed that the percentage of mitotic cells remained unchanged after mutant protein expression compared with EGFP or EGFP-EFHC1 (Fig. 4B).
Figure 4.

Mutants EFHC1 do not influence proliferation and cell cycle exit of cortical progenitors. (A) Immunolabeling for PHH3 (red), a mitotic marker, of VZ/SVZ cells in brains 2 days after in utero electroporation at E14.5 with EGFP, EGFP-EFHC1, EGFP-EFHC1(D253Y) or EGFP-EFHC1(I619L) (green). (B) Quantification of the mitotic index under different above conditions. (C) Immunolabeling for BrdU (red) and Ki67 (blue), of VZ/SVZ cells in cortices 2 days after in utero electroporation at E14.5 with EGFP, EGFP-EFHC1, EGFP-EFHC1(D253Y) or EGFP-EFHC1(I619L) (green). (D) Quantification of the cell cycle exit index in the different above conditions. Scale bars represent 50 μm. Error bars show SEM.
We next explored whether mutants EFHC1 influence the rate of cell cycle exit of cortical progenitors. For this purpose, embryos were electroporated in utero at E14.5 with EGFP, EGFP-EFHC1, EGFP-EFHC1(D253Y) or EGFP-EFHC1(I619L), pulse labeled by a single injection of BrdU at E15.5 and sacrificed at E16.5. Triple immunolabeling with antibodies directed against green fluorescent protein (GFP), BrdU and Ki67 (Fig. 4C) allowed us to establish the cell cycle exit index. We found no difference in the percentage of cells that had exited the cell cycle between all conditions tested (Fig. 4D). Altogether, our results demonstrate that mutations of EFHC1 do not influence the proliferation and the cell cycle exit of cortical progenitors.
Mutants EFHC1 affect the morphology of radial glia and migrating projection neurons
To better understand the observed defect in radial migration and the accumulation of cells in the IZ induced by mutants EFHC1, we analyzed the morphology of radial glia and migrating cells within the IZ and CP (Fig. 5) 3 days after in utero electroporation at E14.5. In these experiments, again, we studied one representative mutation (EGFP-EFHC1(D253Y)) and one polymorphism (EGFP-EFHC1(I619L)) together with EGFP and EGFP-EFHC1 used as control.
Figure 5.

Mutants EFHC1 affect the morphology of radial glia and radially migrating cells. (A) Immunolabeling for BLBP (red), a marker for radial glia, in cortices 3 days after in utero electroporation at E14.5 with EGFP, EGFP-EFHC1, EGFP-EFHC1(D253Y) or EGFP-EFHC1(I619L). EGFP-EFHC1(D253Y) panel shows a disruption of radial glia scaffold. (B) Morphology of cells in the IZ of brains 3 days after in utero electroporation at E14.5 with EGFP, EGFP-EFHC1, EGFP-EFHC1(D253Y) or EGFP-EFHC1(I619L) (green). Many cells expressing EGFP, EGFP-EFHC1 or EGFP-EFHC1(I619L) are bipolar (arrows), whereas most EGFP-EFHC1(D253Y) transfected cells remained with a multipolar morphology (arrow heads). (C) Quantification of bipolar and multipolar cells within the IZ under different above conditions. (D) Morphology of cells in the CP of brains 3 days after in utero electroporation at E14.5 with EGFP, EGFP-EFHC1, EGFP-EFHC1(D253Y) or EGFP-EFHC1(I619L) (green). Cells electroporated with EGFP, EGFP-EFHC1 or EGFP-EFHC1(I619L) present long leading processes, though EGFP-EFHC1(D253Y) transfected cells display abnormal leading processes: short and twisted (white arrows), harboring several bulges (white arrow heads), round-shaped cell lacking a leading process (red arrow heads). (E) Quantification of leading processes length of migrating cells within the CP under different above conditions. Scale bars represent 100 μm. Error bars show SEM. *P < 0.05, **P < 0.01.
Immunohistochemistry using an antibody to brain lipid-binding protein (BLBP), a marker of radial glia, revealed a disruption of normal radial glia architecture in the presence of mutant protein (Fig. 5A). Brains expressing EGFP, EGFP-EFHC1 or EGFP-EFHC1 (I619L) presented long parallel radial glial fibers that terminate with tight pial end-feet. In contrast, cortices electroporated with EGFP-EFHC1(D253Y) showed a reduction in the extent and number of radial fibers with few pial end-feet.
In the IZ, we quantified the number of multipolar (Fig. 5B, arrow heads) and bipolar (Fig. 5B, arrows) cells. We showed that the expression of EGFP-EFHC1(D253Y) leads to an accumulation of multipolar cells (Fig. 5C, 76.7 ± 4.54%) compared with EGFP (33.1 ± 5.4%), EGFP-EFHC1 (37.1 ± 5.3%) and EGFP-EFHC1(I619L) (44.7 ± 7.4%), suggesting a defect in the multipolar–bipolar transition.
In the CP of brains transfected with EGFP, EGFP-EFHC1 and EGFP-EFHC1(I619L), labeled cells were bipolar with an elongated cell body and long leading processes orientated in roughly the same direction (Fig. 5D). Though, in the presence of EGFP-EFHC1(D253Y), the leading processes seemed to be shorter, twisted (Fig. 5D, white arrows) or harbored several bulges (Fig. 5D, white arrow heads). Also some cells with round-shaped cell body lacking a leading process could be observed (Fig. 5D, red arrow heads). The quantification of leading processes length of migrating neurons in the CP revealed that leading processes are smaller in the presence of mutant EFHC1 [Fig. 5E, 44.1 ± 4.1 µm for EGFP, 47.3 ± 4.9 µm for EGFP-EFHC1, 39.2 ± 4.7 µm for EGFP-EFHC1(I619L) versus 28.6 ± 2.4 µm for EGFP-EFHC1(D253Y)]. These results highlight that mutants EFHC1 interfere with leading processes extension, and thus perturb nucleokinesis of migrating neurons. All together, our data suggest that mutants EFHC1 alter the morphology of radial glia and projection neurons at different steps of radial migration, resulting in their accumulation in the IZ and impairment of their correct migration in the CP.
Mutants EFHC1 disrupt tangential migration of interneurons
During cortical development, radial and tangential migrations set up the arsenal of cortical cell controlling the balance between synaptic excitation and inhibition, whose integrity is impaired in epileptic phenomena. As we showed a role of EFHC1 in radial migration, we therefore studied the impact of EFHC1 and its mutations on the tangential migration of interneurons. For that purpose, we performed focal electroporation directly into the median ganglionic eminence (MGE) of rat organotypic slice cultures at E17. Three days after electroporation of EGFP, EGFP-tagged wild-type, mutant and polymorphic EFHC1 proteins, the position of electroporated cells was noted (Fig. 6A) and the proportion of EGFP-positive cells that had crossed the corticostriatal junction (CSJ) to reach the NCx was quantified (Fig. 6B). The percentage of EGFP-EFHC1 cells in the cortex was not significantly different from EGFP (Fig. 6B, 33.09 ± 1.56% versus 36.08 ± 1.57%, respectively), indicating that EFHC1 overexpression has no effect on the tangential migration of interneurons in the developing brain. On the contrary, the expression of mutants EFHC1 induced a significant reduction of EGFP+ cells in the cortex compared with wild-type and polymorphics EFHC1 proteins or EGFP (Fig. 6B, 13.1 ± 3.6% for D210N, 15.2 ± 2.9% for R221H, 15.8 ± 4.5% for F229L and 12.3 ± 2.4% for D253Y).
Figure 6.

Expression of mutants EFHC1 interferes with tangential migration of interneurons. (A) Distribution of EGFP-positive cells in coronal brain slices 3 days after focal electroporation at E17 of EGFP, EGFP-EFHC1, EGFP-EFHC1(D253Y) or EGFP-EFHC1(I619L) (green). EGFP-EFHC1(D253Y) and EGFP-EFHC1(I619L) images are representative of mutants and polymorphisms, respectively. EGFP-EFHC1(D253Y) panel shows tangential migration defect in the cortex. Scale bar represents 200 μm. (B) Quantification of EGFP-positive cells in the cortex 3 days after focal electroporation at E17 under different above conditions. (C) Morphology of migrating interneurons in brain slices 3 days after focal electroporation at E17 of EGFP, EGFP-EFHC1, EGFP-EFHC1(D253Y) or EGFP-EFHC1(I619L) (green). Most EGFP-EFHC1(D253Y) expressing cells appear with a short and twisted leading process. Scale bar represents 20 μm. (D) Quantification of leading processes length of migrating cells within the cortex under different above conditions. Error bars show SEM. *P < 0.05, **P < 0.01.
We next examined the morphology of cells that had migrated in the cortex (Fig. 6C). We found that, similarly to projection neurons, most interneurons expressing mutants EFHC1 displayed shorter and twisted leading processes. [Fig. 6D, 40.6 ± 4.1 µm for EGFP; 37.7 ± 4.5 µm for EGFP-EFHC1; 38.8 ± 4.4 µm for EGFP-EFHC1(I619L) versus 25.5 ± 4.3 µm for EGFP-EFHC1(D253Y)]. Taken altogether, these results indicate that EFHC1 is necessary for tangential migration of interneurons, possibly by regulating cell morphology and process formation during migration.
DISCUSSION
The principal questions in the pathophysiology of JME caused by heterozygous mutations of EFHC1 and intractable epilepsy of infancy due to homozygous mutations of EFHC1 are—which function(s) of myoclonin1/EFHC1 (i) cause(s) the characteristic JME myoclonic and clonic tonic–clonic seizures, (ii) facilitate(s) the establishment of chronic epilepsy (epileptogenesis), (iii) produce(s) the subtle ‘microdysgenesis’ malformations of cortical and subcortical architecture in its adolescent syndrome and severe intractable epilepsy, brain pathology and death in its syndrome of infancy? Several, but not mutually exclusive, hypotheses have been proposed for the functions of EFHC1. Suzuki et al. showed that overexpression of EFHC1 in mouse hippocampal primary culture neurons induced apoptosis by enhancing R-type voltage-dependent Ca2+-channel currents, an effect that is reduced by mutations (4). More recently, Katano et al. reported a molecular and functional interaction between EFHC1 and TRPM2, a Ca2+-permeable channel that mediates susceptibility to cell death (21). These data suggest that EFHC1 controls cellular calcium homeostasis and could explain how seizures are triggered and how impaired apoptosis result. Additionally, EFHC1 was shown to be expressed in tissues containing motile cilia and flagella, such as lung, testis or ependyma, suggesting a role for the protein in ciliary functions (22–25). This was confirmed in the EFHC1-deficient mice as the ciliary beating frequency of ependymal cells was affected in null mutants (26). In the latter suggestion, JME is considered as a ‘ciliopathy’. However, it is difficult to reconcile seizures, much less the establishment of epileptogenesis, resulting from a ‘ciliopathy’. In complement to the above-hypothesized functions of EFHC1, our research group has demonstrated that EFHC1 is a MAP that plays a role in cell division and radial migration during neocortical development (12,13). In a recent report, our results were confirmed when the Drosophila homolog of EFHC1 (Defhc1.1) was also shown to be a MAP (27). Furthermore, Defhc1.1 knockout flies displayed overgrowth of dendritic arbor, whereas overexpressing Defhc1.1 shows reduced dendrite elaboration (27). Taken together, these in vitro and in vivo experiments demonstrate that EFHC1 probably controls brain development by finely tuning microtubule cytoskeleton dynamics.
In our present study, we analyzed the impact of four pathological mutations (D210N, R221H, F229L and D253Y) and two coding polymorphisms (R159W and I619L) on the properties of EFHC1 at cellular level and during the development of the cerebral cortex. In HEK293 and stably transfected HEK Flp-In T-Rex-293 cells, we showed that all mutated and polymorphic forms of EFHC1 presented the same subcellular localization of the wild-type protein, with an association with the centrosome and the mitotic spindle. These results are not surprising since mutations and polymorphisms are located outside the microtubule-binding domain of EFHC1, i.e. the first 45 amino acids of the N-terminus (13). However, we observed that mutant proteins increased the number of abnormal mitotic spindles such as monopolar spindles and congression abnormalities. Similar results have been obtained by overexpression of EGFP-N45, a dominant-negative construct containing only the microtubule-binding domain of EFHC1 (13). These data suggest that mutant proteins seem to perturb functions of endogenous EFHC1 through dominant-negative effects.
In the developing NCx, we demonstrated that acute expression of mutants EFHC1 disrupts both radial and tangential migration in a dominant-negative manner by affecting the different steps of corticogenesis: (i) the extension of radial glia processes, (ii) the transition from the multipolar to bipolar stage within the IZ and (iii) the formation of leading processes of radially and tangentially migrating cells. These distinct phases of migrations imply cell shape remodeling, which largely depend on dynamic rearrangements of microtubules (16,28–30). Several MAPs and proteins localized at the centrosome, such as Lis1 and Dcx, are direct regulators of the microtubule dynamics underlying these morphological changes and are essential for cortical development (19,31,32). EFHC1, as a MAP, is implicated in the same cellular pathways that regulate morphological changes necessary for different steps of neuronal migration. These new results presented here strengthen the hypothesis that EFHC1 plays a role in microtubule dynamics as previously suggested (13,27). The range of defects that we observed in neuronal migration is consistent with multiple important roles for EFHC1 in cortical histogenesis. Obviously, the precise mechanisms how EFHC1 is involved in radial and tangential migrations remain to be determined.
In a previous study, we have shown that RNAi or dominant-negative loss of EFHC1 function leads to an alteration of radial migration with an accumulation of progenitor cells in the VZ/SVZ following mitosis and cell cycle exit defects (13). Our present results are slightly different as we did not observe any default in proliferation and cell cycle exit of cortical progenitors in the presence of mutants EFHC1. Cells were capable to exit the VZ/SVZ and to migrate into the IZ and CP, whereas some of them accumulated in the IZ. We observed the same discrepancy in HEK293 where effects on cell division were lower with mutations when compared with RNAi or dominant-negative construct. This paradox could be explained by a dose-dependent effect on EFHC1 impairment where a more severe reduction in EFHC1 function was induced by RNAi or dominant-negative construct compared with mutated proteins. Therefore, mutants EFHC1 seem to have mild dominant-negative effects, which would be fully consistent with our present data. The same observation has been made with DISC1, a MAP implicated in schizophrenia, where DISC1 RNAi induced a more pronounced failure of radial migration than did the addition of a carboxy-terminal-truncated mutant of DISC1 that function in a dominant-negative manner (33). Moreover, one has to consider differences in methodologies (ex vivo electroporation versus in utero electroporation) (34) and species (rat versus mouse) (35) used in both the studies.
To date, several quantitative neuroimaging studies have revealed multiple subtle structural abnormalities in JME brains such as an increase in cortical grey matter in mesial frontal lobes and an atrophy of the hippocampus, the thalamus and the corpus callosum (36–39). Moreover, post-mortem analysis of brains from JME patients has identified the presence of microscopic malformations called ‘microdysgenesis’, characterized by the presence of cortical and subcortical dystopic neurons (11). These may be indicative of areas of subtle cortical folding abnormality related to early disruption of cortical development. Appropriate neuronal positioning is essential for normal neocortical function. Several studies have shown that there is a connection between altered neuronal positioning and establishment of epileptogenesis (32,40,41). Moreover, subtle alterations in neuron position and/or function have been reported to contribute to disorders such as dyslexia, schizophrenia or mental retardation (33,42). This suggests that JME could be a neurodevelopmental disorder that may result from defects in neuronal integration.
All results obtained in our present work suggest that mutations of EFHC1 act as dominant-negative inducing the loss of protein functions during development with dual effects on the clinical phenotype depending on the mode of transmission. This functional dominant-negative effect seems to be relatively mild when JME patients carry one copy of an EFHC1 mutation, e.g. heterozygous transmitted as an autosomal dominant trait. The levels of a heterozygous mutant EFHC1 in JME patients are unknown, but may well be relatively low, producing myoclonias grand mal convulsions and absences easily controlled by antiepileptic drugs like valproate or levitiracetam. So, it is eminently reasonable to suggest that partial impairment of EFHC1 function may be responsible for the subtle ‘microdysgenesis’ in JME brains. Moreover, one has to consider the presence of EFHC2, the paralog of EFHC1, which could functionally compensate the absence of EFHC1 resulting in JME patients whose cognitive functions are normal (43). Therefore, we hypothesize that heterozygous mutations of EFHC1 might lead to subtle deficits in the number, composition or positioning of glutamatergic and GABAergic neurons in the cortex, which could explain the observed microdysgenesis, normal cognitive functions and hyperexcitability in JME patients. A more severe drug-intractable epilepsy with delay in psychomotor development, brain pathology and death in infancy could also be explained when two copies of EFHC1 mutations, e.g. homozygous mutations transmitted as an autosomal recessive trait, are present in EFHC1. Our results on the consequences of EFHC1 mutations linked to the adolescent JME phenotype and the severe intractable epilepsy of infancy emphasizes the important and vital functions of EFHC1 in brain development, improves our understanding of disease mechanisms in epilepsy and hopefully leads to improved diagnosis, safer drugs and curative therapies.
MATERIALS AND METHODS
Expression constructs
Point mutations were introduced in hEFHC1 by using the QuickChange site-directed mutagenesis method on the parent vectors pcDNA5/FRT/TO-EGFP-hEFHC1 or pCAGGS-EGFP-hEFHC1 (13). All constructs were verified by sequencing (Genotranscriptomics Platform, GIGA, University of Liege). The sequences of the mutagenic primers are available upon request. Plasmid solutions were prepared using the EndoFree plasmid purification kit (Qiagen).
Cell culture and transfections
HEK293 cells were maintained in Dulbecco's Modified Eagle's Medium (Gibco) supplemented with 10% fetal bovine serum (Greiner bio-one). Cells were grown at 37°C in a humidified incubator under 5% CO2 atmosphere. Stably transfected HEK Flp-In T-Rex-293 cells were generated and cultured as previously described (12). Cells were transfected using FuGENE 6 Transfection Reagent (Roche) according to the manufacturer's protocol.
Animals
Time-pregnant Wistar rats (E17) and NMRI mice (E14.5) were provided by the animal facility of the University of Liege. They were treated according to the guidelines of Belgian Ministry of Agriculture in agreement with European community laboratory Animal Care and Use regulations.
In utero electroporation
In utero electroporation was performed as described previously (44). Briefly, E14.5 timed-pregnant mice were deeply anesthetized with isoflurane in oxygen carrier (Abbot Laboratories Ltd). Uterine horns were exposed through a 1.5cm incision in the ventral peritoneum and 1–3 µl of plasmid solution (2.5 μg/μl) with 0.05% Fast Green (Sigma) were injected through the uterus into the lateral ventricles of embryos using a pulled glass micropipette and a microinjector (Femtojet, Eppendorf). Electroporation was accomplished by discharging five electrical pulses at 35 V (50 ms duration) across the uterine wall at 1 s intervals using 5 mm platinum tweezers electrodes (CUY650P5, Sonidel) and a square wave electroporator (ECM-830, BTX). Uterine horns were replaced in the abdominal cavity and the abdomen wall and skin were sutured using the surgical needle and thread. Buprenorphine was injected (Temgesic®, Schering-Plough) and the animals were warmed on heating pads until they woke up. At different times following the surgery, pregnant mice were sacrificed and embryos were processed for tissue analyses.
Focal electroporation of the MGE on organotypic brain slices
E17 time-pregnant rat were sacrificed by decapitation and embryos were removed from the uterus. Embryonic brains were dissected in 0.1 m phosphate-buffered saline (PBS, pH 7.4) containing 0.25 mm glucose (PBS/glucose). Brains were embedded in 3% low-melting agarose (37°C, Bio-Rad) in PBS and incubated on ice for 1 h. Brains were cut coronally into 300 µm slice with a vibratome (VT1000S, Leica) and transferred onto Nucleopore track-etched membrane (0.08 μm pore size, Whatman). Slices with their supporting membranes were placed onto a 1% low-melting agarose gel in PBS covering a Petridish square platinum plate electrode (NEPA GENE). One microliter of plasmid solution (2.5 μg/μl) with 0.05% Fast Green (Sigma) was injected into the MGE region of the brain slice using a pulled glass micropipette and a microinjector (Femtojet, Eppendorf). An agarose column was punched with a glass Pasteur pipette from a 1% (w/v) low-melting agarose gel in PBS and attached to a cover square platinum plate electrode (NEPA GENE). After adding one drop of PBS to the injected MGE region, the column was placed onto this region and square electric pulses (100 V, 10 ms) were passed five times with an interval of 1 s using an electroporator (ECM 830, BTX). The electroporated slices on their membranes were transferred into wells containing Neurobasal medium (Invitrogen) supplemented with 2% B27 (Gibco), 1% N2 (Gibco), 1% penicillin/streptomycin (Invitrogen) and 1% glutamine (Invitrogen). Slices were cultured for 3 days under semidry conditions in an incubator at 37°C and with 5% CO2.
Tissue processing
Embryonic brains were dissected in PBS/glucose and fixed in 4% paraformaldehyde for 1 h at 4°C. Cultured brain slices were fixed in 4% paraformaldehyde for 30 min at 4°C. Fixed samples were cryoprotected in 20% sucrose in PBS overnight at 4°C, embedded in Neg 50 frozen section medium (Thermo Scientific) and cut coronally at 20 µm onto slides (Superfrost Plus, Thermo Scientific) with a cryostat (HM 560 m, Microm).
Immunostaining procedures
For immunostaining, we used the following primary antibodies: mouse monoclonal antibody to α-tubulin (clone B-5-2-1, Sigma, 1:2000), mouse monoclonal antibody to γ-tubulin (clone GTU-88, Sigma, 1:500), rabbit polyclonal antibody GFP (Molecular probe, 1:2000), mouse monoclonal antibody to βIII-tubulin (Tuj-1, Covance, 1:1000), rabbit polyclonal antibody to BLBP (Chemicon, 1:500), rabbit monoclonal antibody to phospho-Histone H3 (PHH3, clone JY325, Upstate, 1:500), rat monoclonal antibody to BrdU (clone BU1/75, AbD serotec, 1:200) or mouse monoclonal antibody to Ki67 (clone B56, BD Pharmigen, 1:50). For secondary antibodies, we used RRX-, FITC- or Cy5-conjugated antibodies to rabbit, mouse or rat IgG (Jackson ImmunoResearch, 1:500).
For immunocytochemistry, cells were grown on glass coverslips coated with polyornithine (0.1 mg/ml, Sigma) for 1 h at room temperature and laminin (5 µg/ml, MP Biomedicals) for 2 h at 37°C. Cells were fixed in methanol for 6 min at −20°C or in 4% paraformaldehyde for 20 min at 4°C and rinsed three times in PBS. Cells were subsequently incubated in blocking solution (0.1% Triton X-100, 1.5% nonfat dried milk in PBS) for 1h at 37°C. They were incubated overnight at 4°C with primary antibodies diluted in blocking solution, washed three times with PBS before incubation with secondary antibodies in blocking solution for 1 h at room temperature. After three washes in PBS, coverslips were mounted in Vectashield Hard Set Mounting Medium with 4′,6′-diamidino-2-phenylindole (DAPI, Vector laboratories). Images were captured with an Olympus Fluoview FV1000 confocal system equipped with an Olympus IX81 inverted microscope (Olympus) (Imaging and Flow Cytometry Platform, GIGA, University of Liège). Images were processed using Photoshop software (Adobe Systems).
For immunohistochemistry, cryostat sections were washed one time with PBS and incubated 1 h at room temperature in blocking solution [0.1% Triton X-100, 10% normal goat serum (Jackson ImmunoResearch) in PBS]. For BrdU immunostaining, sections were pretreated with 2 N HCl for 30 min at 37°C followed by neutralization in Na2B4O7 for 15 min at 37°C before blocking. For Ki67 immunostaining, slices were pretreated with Target Retrieval Solution (Dako) for 20 min at 95°C before blocking. Slides were incubated overnight at 4°C with primary antibodies in blocking solution. After three washes in PBS, sections were incubated with secondary antibodies at room temperature in blocking solution. Slides were washed again with PBS and mounted in Vectashield Hard Set Mounting Medium with DAPI. Images were captured using an Olympus Fluoview FV1000 confocal system equipped with an Olympus IX81 inverted microscope (Olympus) (Imaging and Flow Cytometry Platform, GIGA, University of Liège).
Quantitative analysis
For quantification of abnormal spindles on HEK293 and HEK Flp-In T-Rex-293, cells were counted 48 h after transfection or doxycycline induction, respectively, in three independent experiments (500 cells were randomly counted in each experiment).
For quantification of neuronal migration 3 days after in utero electroporation, different subregions (VZ/SVZ, IZ and CP) of the cerebral cortex were identified on the basis of cell density visualized with DAPI nuclear staining and neuronal marker (β-III-tubulin) expression staining. The number of EGFP-positive cells in different cortical subregions was counted in two adjacent sections from six to eight independent brains.
For quantification of the mitotic index, cryosections were stained with anti-PHH3 antibody 48 h after in utero electroporation. The number of EGFP-positive and EGFP- and PHH3-double-positive cells in the VZ/SVZ was counted in two adjacent sections from six to eight independent brains. The mitotic index of EGFP-transfected cells was determined as the ratio of EGFP-positive mitotic cells (EGFP+ and PHH3+) to the total EGFP-positive population in the VZ/SVZ.
For cell cycle exit studies, mother mice were injected with BrdU (50 mg/g body weight) 24 h after in utero electroporation. Brains were fixed and stained with antibodies to GFP, Ki67 and BrdU 24 h after the BrdU pulse. EGFP and BrdU double-positive and EGFP, BrdU and Ki67 triple-positive cells in the VZ/SVZ cells were counted in two adjacent slices from six to eight independent embryos. The cell cycle exit index corresponds to the percentage of EGFP-positive cells that exited the cell cycle (EGFP+, BrdU+ and Ki67−) to total EGFP-labeled cells that incorporated BrdU (EGFP+ and BrdU+).
For quantification of tangential migration 3 days after focal electroporation, each brain slice was divided into two regions, i.e. the NCx and the striatum/MGE by drawing a line at the CSJ. The number of EGFP-positive cells in the two regions was counted in two slices from six to eight independent brains.
Statistical analyses
All experiments and quantifications were performed blind. Statistical analysis was carried out using GraphPad Prism software. All values are presented as mean ± SEM. Statistical analyses were performed using one-way ANOVA followed by Dunnett's post-hoc test for multiple comparisons. Differences between the groups were considered significant for P < 0.05. Asterisks indicate the level of significance: *P < 0.05, **P < 0.01 and ***P < 0.001.
SUPPLEMENTARY MATERIAL
FUNDING
This work was supported by the Fonds de la Recherche Scientifique-FNRS (FRSM 3.4531.09 to T.G. and B.L.); the Léon Fredericq Foundation (to L.d.N.) and the Fonds spéciaux of the University of Liège (C-10/31 and C-11/94 to T.G and B.L.). L.d.N. and B.L. are, respectively, postdoctoral researcher and research associates at the Fonds de la Recherche Scientifique-FNRS. A.V.D.-E. is supported by NIH grant 5R01NS055057 and VA Merit Review grant.
Supplementary Material
ACKNOWLEDGEMENTS
We thank S. Ormenese and G. Moraes from the GIGA-Imaging and Flow Cytometry platform of the University of Liège for support with confocal microscopy and V. Dhennin from the GIGA-Genotranscriptomics Platform for the sequencing of constructs.
Conflict of Interest statement. None declared.
References
- 1.Delgado-Escueta A.V., Enrile-Bacsal F. Juvenile myoclonic epilepsy of Janz. Neurology. 1984;34:285–294. doi: 10.1212/wnl.34.3.285. [DOI] [PubMed] [Google Scholar]
- 2.Delgado-Escueta A.V. Advances in genetics of juvenile myoclonic epilepsies. Epilepsy Curr. 2007;7:61–67. doi: 10.1111/j.1535-7511.2007.00171.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Noebels J.L., Avoli M., Rogawski M.A., Olsen R.W., Delgado-Escueta A.V. The next decade of research in the basic mechanisms of the epilepsies. In: Noebels J.L., Avoli M., Rogawski M.A., Olsen R.W., Delgado-Escueta A.V., editors. Jasper's Basic Mechanisms of the Epilepsies. Oxford University Press; 2012. pp. 3–11. [Google Scholar]
- 4.Suzuki T., Delgado-Escueta A.V., Aguan K., Alonso M.E., Shi J., Hara Y., Nishida M., Numata T., Medina M.T., Takeuchi T., et al. Mutations in EFHC1 cause juvenile myoclonic epilepsy. Nat. Genet. 2004;36:842–849. doi: 10.1038/ng1393. [DOI] [PubMed] [Google Scholar]
- 5.Ma S., Blair M.A., Abou-Khalil B., Lagrange A.H., Gurnett C.A., Hedera P. Mutations in the GABRA1 and EFHC1 genes are rare in familial juvenile myoclonic epilepsy. Epilepsy Res. 2006;71:129–134. doi: 10.1016/j.eplepsyres.2006.06.001. [DOI] [PubMed] [Google Scholar]
- 6.Stogmann E., Lichtner P., Baumgartner C., Bonelli S., Assem-Hilger E., Leutmezer F., Schmied M., Hotzy C., Strom T.M., Meitinger T., et al. Idiopathic generalized epilepsy phenotypes associated with different EFHC1 mutations. Neurology. 2006;67:2029–2031. doi: 10.1212/01.wnl.0000250254.67042.1b. [DOI] [PubMed] [Google Scholar]
- 7.Annesi F., Gambardella A., Michelucci R., Bianchi A., Marini C., Canevini M.P., Capovilla G., Elia M., Buti D., Chifari R., et al. Mutational analysis of EFHC1 gene in Italian families with juvenile myoclonic epilepsy. Epilepsia. 2007;48:1686–1690. doi: 10.1111/j.1528-1167.2007.01173.x. [DOI] [PubMed] [Google Scholar]
- 8.Medina M.T., Suzuki T., Alonso M.E., Duron R.M., Martinez-Juarez I.E., Bailey J.N., Bai D., Inoue Y., Yoshimura I., Kaneko S., et al. Novel mutations in myoclonin1/EFHC1 in sporadic and familial juvenile myoclonic epilepsy. Neurology. 2008;70:2137–2144. doi: 10.1212/01.wnl.0000313149.73035.99. [DOI] [PubMed] [Google Scholar]
- 9.Jara-Prado A., Martinez-Juarez I.E., Ochoa A., Gonzalez V.M., Fernandez-Gonzalez-Aragon Mdel C., Lopez-Ruiz M., Medina M.T., Bailey J.N., Delgado-Escueta A.V., Alonso M.E. Novel Myoclonin1/EFHC1 mutations in Mexican patients with juvenile myoclonic epilepsy. Seizure. 2012;21:550–554. doi: 10.1016/j.seizure.2012.05.016. [DOI] [PubMed] [Google Scholar]
- 10.Berger I., Dor T., Halvardson J., Edvardson S., Shaag A., Feuk L., Elpeleg O. Intractable epilepsy of infancy due to homozygous mutation in the EFHC1 gene. Epilepsia. 2012;58:1436–1440. doi: 10.1111/j.1528-1167.2012.03536.x. [DOI] [PubMed] [Google Scholar]
- 11.Meencke H.J., Janz D. Neuropathological findings in primary generalized epilepsy: a study of eight cases. Epilepsia. 1984;25:8–21. doi: 10.1111/j.1528-1157.1984.tb04149.x. [DOI] [PubMed] [Google Scholar]
- 12.de Nijs L., Lakaye B., Coumans B., Leon C., Ikeda T., Delgado-Escueta A.V., Grisar T., Chanas G. EFHC1, a protein mutated in juvenile myoclonic epilepsy, associates with the mitotic spindle through its N-terminus. Exp. Cell Res. 2006;312:2872–2879. doi: 10.1016/j.yexcr.2006.05.011. [DOI] [PubMed] [Google Scholar]
- 13.de Nijs L., Leon C., Nguyen L., Loturco J.J., Delgado-Escueta A.V., Grisar T., Lakaye B. EFHC1 interacts with microtubules to regulate cell division and cortical development. Nat. Neurosci. 2009;12:1266–1274. doi: 10.1038/nn.2390. [DOI] [PubMed] [Google Scholar]
- 14.Nadarajah B., Parnavelas J.G. Modes of neuronal migration in the developing cerebral cortex. Nat. Rev. Neurosci. 2002;3:423–432. doi: 10.1038/nrn845. [DOI] [PubMed] [Google Scholar]
- 15.Marin O., Rubenstein J.L. Cell migration in the forebrain. Annu. Rev. Neurosci. 2003;26:441–483. doi: 10.1146/annurev.neuro.26.041002.131058. [DOI] [PubMed] [Google Scholar]
- 16.Kriegstein A.R., Noctor S.C. Patterns of neuronal migration in the embryonic cortex. Trends Neurosci. 2004;27:392–399. doi: 10.1016/j.tins.2004.05.001. [DOI] [PubMed] [Google Scholar]
- 17.Bellion A., Metin C. Early regionalisation of the neocortex and the medial ganglionic eminence. Brain Res. Bull. 2005;66:402–409. doi: 10.1016/j.brainresbull.2005.07.010. [DOI] [PubMed] [Google Scholar]
- 18.Metin C., Vallee R.B., Rakic P., Bhide P.G. Modes and mishaps of neuronal migration in the mammalian brain. J. Neurosci. 2008;28:11746–11752. doi: 10.1523/JNEUROSCI.3860-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tsai L.H., Gleeson J.G. Nucleokinesis in neuronal migration. Neuron. 2005;46:383–388. doi: 10.1016/j.neuron.2005.04.013. [DOI] [PubMed] [Google Scholar]
- 20.Bellion A., Baudoin J.P., Alvarez C., Bornens M., Metin C. Nucleokinesis in tangentially migrating neurons comprises two alternating phases: forward migration of the Golgi/centrosome associated with centrosome splitting and myosin contraction at the rear. J. Neurosci. 2005;25:5691–5699. doi: 10.1523/JNEUROSCI.1030-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Katano M., Numata T., Aguan K., Hara Y., Kiyonaka S., Yamamoto S., Miki T., Sawamura S., Suzuki T., Yamakawa K., et al. The juvenile myoclonic epilepsy-related protein EFHC1 interacts with the redox-sensitive TRPM2 channel linked to cell death. Cell Calcium. 2012;51:179–185. doi: 10.1016/j.ceca.2011.12.011. [DOI] [PubMed] [Google Scholar]
- 22.Ikeda T., Ikeda K., Enomoto M., Park M.K., Hirono M., Kamiya R. The mouse ortholog of EFHC1 implicated in juvenile myoclonic epilepsy is an axonemal protein widely conserved among organisms with motile cilia and flagella. FEBS Lett. 2005;579:819–822. doi: 10.1016/j.febslet.2004.12.070. [DOI] [PubMed] [Google Scholar]
- 23.Suzuki T., Inoue I., Yamagata T., Morita N., Furuichi T., Yamakawa K. Sequential expression of Efhc1/myoclonin1 in choroid plexus and ependymal cell cilia. Biochem. Biophys. Res. Commun. 2008;367:226–233. doi: 10.1016/j.bbrc.2007.12.126. [DOI] [PubMed] [Google Scholar]
- 24.Léon C., de Nijs L., Chanas G., Delgado-Escueta A.V., Grisar T., Lakaye B. Distribution of EFHC1 or Myoclonin 1 in mouse neural structures. Epilepsy Res. 2010;88:196–207. doi: 10.1016/j.eplepsyres.2009.11.009. [DOI] [PubMed] [Google Scholar]
- 25.King S.M. Axonemal protofilament ribbons, DM10 domains, and the link to juvenile myoclonic epilepsy. Cell Motil. Cytoskeleton. 2006;63:245–253. doi: 10.1002/cm.20129. [DOI] [PubMed] [Google Scholar]
- 26.Suzuki T., Miyamoto H., Nakahari T., Inoue I., Suemoto T., Jiang B., Hirota Y., Itohara S., Saido T.C., Tsumoto T., et al. Efhc1 deficiency causes spontaneous myoclonus and increased seizure susceptibility. Hum. Mol. Genet. 2009;18:1099–1109. doi: 10.1093/hmg/ddp006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rossetto M.G., Zanarella E., Orso G., Scorzeto M., Megighian A., Kumar V., Delgado-Escueta A.V., Daga A. Defhc1.1, a homologue of the juvenile myoclonic gene EFHC1, modulates architecture and basal activity of the neuromuscular junction in Drosophila. Hum. Mol. Genet. 2012;20:4248–4257. doi: 10.1093/hmg/ddr352. [DOI] [PubMed] [Google Scholar]
- 28.Jaglin X.H., Chelly J. Tubulin-related cortical dysgeneses: microtubule dysfunction underlying neuronal migration defects. Trends Genet. 2009;25:555–566. doi: 10.1016/j.tig.2009.10.003. [DOI] [PubMed] [Google Scholar]
- 29.Ayala R., Shu T., Tsai L.H. Trekking across the brain: the journey of neuronal migration. Cell. 2007;128:29–43. doi: 10.1016/j.cell.2006.12.021. [DOI] [PubMed] [Google Scholar]
- 30.Schaar B.T., McConnell S.K. Cytoskeletal coordination during neuronal migration. Proc. Natl Acad. Sci. USA. 2005;102:13652–13657. doi: 10.1073/pnas.0506008102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Valiente M., Marin O. Neuronal migration mechanisms in development and disease. Curr. Opin. Neurobiol. 2010;20:68–78. doi: 10.1016/j.conb.2009.12.003. [DOI] [PubMed] [Google Scholar]
- 32.LoTurco J.J., Bai J. The multipolar stage and disruptions in neuronal migration. Trends Neurosci. 2006;29:407–413. doi: 10.1016/j.tins.2006.05.006. [DOI] [PubMed] [Google Scholar]
- 33.Kamiya A., Kubo K., Tomoda T., Takaki M., Youn R., Ozeki Y., Sawamura N., Park U., Kudo C., Okawa M., et al. A schizophrenia-associated mutation of DISC1 perturbs cerebral cortex development. Nat. Cell Biol. 2005;7:1167–1178. doi: 10.1038/ncb1328. [DOI] [PubMed] [Google Scholar]
- 34.Higginbotham H., Yokota Y., Anton E.S. Strategies for analyzing neuronal progenitor development and neuronal migration in the developing cerebral cortex. Cereb. Cortex. 2011;21:1465–1474. doi: 10.1093/cercor/bhq197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ramos R.L., Bai J., LoTurco J.J. Heterotopia formation in rat but not mouse neocortex after RNA interference knockdown of DCX. Cereb. Cortex. 2006;16:1323–1331. doi: 10.1093/cercor/bhj074. [DOI] [PubMed] [Google Scholar]
- 36.Kim J.H., Lee J.K., Koh S.B., Lee S.A., Lee J.M., Kim S.I., Kang J.K. Regional grey matter abnormalities in juvenile myoclonic epilepsy: a voxel-based morphometry study. Neuroimage. 2007;37:1132–1137. doi: 10.1016/j.neuroimage.2007.06.025. [DOI] [PubMed] [Google Scholar]
- 37.Woermann F.G., Free S.L., Koepp M.J., Sisodiya S.M., Duncan J.S. Abnormal cerebral structure in juvenile myoclonic epilepsy demonstrated with voxel-based analysis of MRI. Brain. 1999;122:2101–2108. doi: 10.1093/brain/122.11.2101. [DOI] [PubMed] [Google Scholar]
- 38.Tae W.S., Hong S.B., Joo E.Y., Han S.J., Cho J.W., Seo D.W., Lee J.M., Kim I.Y., Byun H.S., Kim S.I. Structural brain abnormalities in juvenile myoclonic epilepsy patients: volumetry and voxel-based morphometry. Korean J. Radiol. 2006;7:162–172. doi: 10.3348/kjr.2006.7.3.162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Betting L.E., Mory S.B., Li L.M., Lopes-Cendes I., Guerreiro M.M., Guerreiro C.A., Cendes F. Voxel-based morphometry in patients with idiopathic generalized epilepsies. Neuroimage. 2006;32:498–502. doi: 10.1016/j.neuroimage.2006.04.174. [DOI] [PubMed] [Google Scholar]
- 40.Kriegstein A.R. Constructing circuits: neurogenesis and migration in the developing neocortex. Epilepsia. 2005;46:15–21. doi: 10.1111/j.1528-1167.2005.00304.x. [DOI] [PubMed] [Google Scholar]
- 41.Marsh E., Fulp C., Gomez E., Nasrallah I., Minarcik J., Sudi J., Christian S.L., Mancini G., Labosky P., Dobyns W., et al. Targeted loss of Arx results in a developmental epilepsy mouse model and recapitulates the human phenotype in heterozygous females. Brain. 2009;132:1563–1576. doi: 10.1093/brain/awp107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Galaburda A.M., LoTurco J., Ramus F., Fitch R.H., Rosen G.D. From genes to behavior in developmental dyslexia. Nat. Neurosci. 2006;9:1213–1217. doi: 10.1038/nn1772. [DOI] [PubMed] [Google Scholar]
- 43.Young-Pearse T.L., Bai J., Chang R., Zheng J.B., LoTurco J.J., Selkoe D.J. A critical function for beta-amyloid precursor protein in neuronal migration revealed by in utero RNA interference. J. Neurosci. 2007;27:14459–14469. doi: 10.1523/JNEUROSCI.4701-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Creppe C., Malinouskaya L., Volvert M.L., Gillard M., Close P., Malaise O., Laguesse S., Cornez I., Rahmouni S., Ormenese S., et al. Elongator controls the migration and differentiation of cortical neurons through acetylation of alpha-tubulin. Cell. 2009;136:551–564. doi: 10.1016/j.cell.2008.11.043. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.