

Abstract
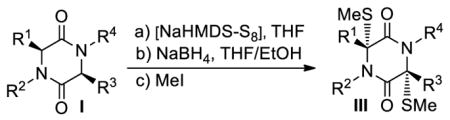
An improved sulfenylation method for the preparation of epidithio-, epitetrathio- and bis-(methylthio)diketopiperazines from diketopiperazines has been developed. Employing NaHMDS and related bases and elemental sulfur or bis[bis(trimethylsilyl)amino]trisulfide (23) in THF, the developed method was applied to the synthesis of a series of natural and designed molecules, including epicoccin G (1), 8,8′-epi-ent-rostratin B (2), gliotoxin (3), gliotoxin G (4), emethallicin E (5) and haematocin (6). Biological screening of selected synthesized compounds led to the discovery of a number of nanomolar anti poliovirus agents (i.e. 46, 2,2′-epi-46 and 61, Table 5) and several low micromolar anti Plasmodium falciparum lead compounds (i.e. 46, 2,2′-epi-46, 58, 61 and 1, Table 5).
INTRODUCTION
2,5-Diketopiperazines are a ubiquitous class of compounds of diverse molecular architectures and biological activities.1 Numerous have been discovered from natural sources while many more have been synthesized in the laboratory for biological investigations and drug discovery purposes.1 The 2,5-diketopiperazine structural motif constitutes a unique scaffold upon which three dimensional molecules, including chiral ones, may be constructed,1,2 thereby providing a useful alternative to the planar structural motifs commonly found in drugs and drug candidates, the latter being often far from ideal in terms of pharmacological properties.3
Of particular interest are the naturally occurring epidithiodiketopiperazines and bis-(methylthio)diketopiperazines, whose biological activities include antiviral, antibacterial, antiallergic, antimalarial and cytotoxic properties.1,4 Despite their promising biological profiles, however, these compounds remain largely unexplored, primarily due to their natural scarcity and the synthetic laboratory challenge they pose.5,6
In order to alleviate some of these deficiencies and facilitate biological investigations in this area, we recently initiated a research program directed toward the development of improved methods of sulfenylation of 2,5-diketopiperazines and applied them to the total synthesis of natural and designed epidithio-, epitetrathio- and bis-(methylthio)diketopiperazines. In preliminary communications we already reported an improved method for the sulfenylation of 2,5-diketopiperazines7 and the total synthesis8 of epicoccin G9 (1, Figure 1) and 8,8′-epi-ent-rostratin B10 (2, Figure 1). In this article we describe further studies in this area that include enantioselective total syntheses of gliotoxin11 (3, Figure 1), gliotoxin G12 (4, Figure 1), emethallicin E13 (5, Figure 1) and haematocin14 (6, Figure 1), as well as the monomeric unit (7, Figure 1) of aranotin15,16 (8, Figure 1). We also report our biological evaluation of a number of selected synthesized compounds that led to the discovery of potent anti poliovirus and anti Plasmodium falciparum agents.
Figure 1.

Selected naturally occurring epidithio-, epitetrathio- and bis-(methylthio)diketopiperazines.
RESULTS AND DISCUSSION
Methodology Development
Recognizing the deficiencies of the then available sulfenylation methods of 2,5-diketopiperazines, we set as part of our goals the development of improved sulfenylation methods to construct epidithiodiketopiperazines and bis-(methylthio)diketopiperazines. Figure 2 depicts a number of selected sulfenylation methods of 2,5-diketopiperazines known at the outset of our investigations. Thus, as early as 1968, Trown,17 and subsequently Hashimoto18 (1987), pioneered the use of 3,6-dibromodiketopiperazines (9) as substrates and KSAc as a sulfur source to prepare epidithiodiketopiperazines (18). In 1971, Poisel and U. Schmidt19 introduced the use of sodium tetrasulfide (Na2S4) as a source of sulfur to produce epidithiodiketopiperazines (10→18, Figure 2), and in 1972 the classical U. Schmidt method20 for the synthesis of these compounds from 2,5-diketopiperazines employing sulfur (S8) and NaNH2 in liq. NH3 (11→18, Figure 2) was reported. In 1973, Kishi21 reported a method of masking 3,6-dithiodiketopiperazines with anisaldehyde and then generating the desired epidithiodiketopiperazines at a later stage (12→18, Figure 2), a tactic that he elegantly applied to synthesize gliotoxin (3).6h,6i In 1975, Matsunari23 utilized 3,6-dimethoxydiketopiperazines as substrates in conjunction with H2S as a source of sulfur to prepare epidithiodiketopiperazines (13→18, Figure 2), where-as in 2002 Overman and Sato24 employed the corresponding bis-acetates and H2S in their quest of similar epidithiodiketopiperazines (14→18, Figure 2). In 2009, Movassaghi6e and Sodeoka6f applied the use of 3,6-dihydroxydiketopiperazines (15 and 16, respectively, Figure 2) and H2S to construct the epidithiodiketopiperazine structural motifs (18, Figure 2) of their synthetic targets, 11,11′-dideoxyverticillin A and chaetocin, respectively. In 2010, Kim and Movassaghi described the use of potassium trithiocarbonate (K2CS3) to generate an epidithiodiketopiperazine moiety from monosilylated 3,6-dihydroxydiketopiperazine intermediate (17→18, Figure 2) in their elegant synthesis of chaetocins A and C and 12,12′-dideoxychetracin A.6g
Figure 2.
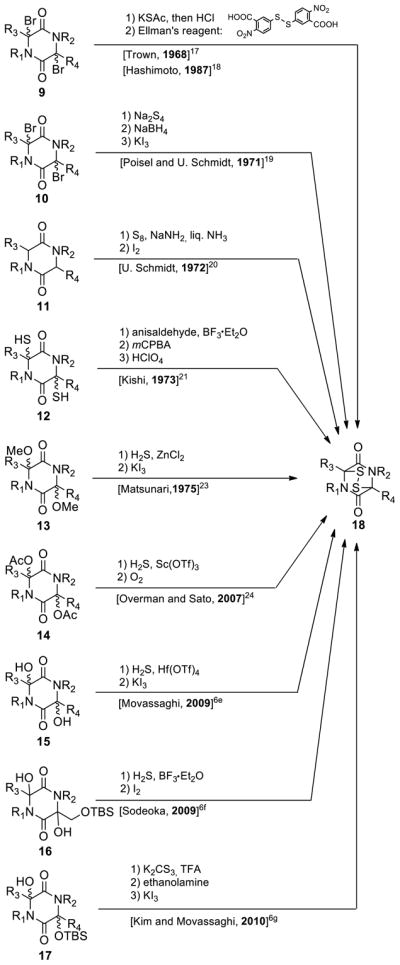
Selected sulfenylation methods of 2,5-diketopiperazines.
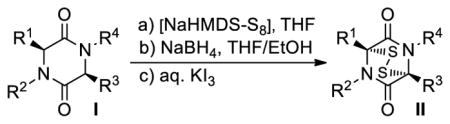
Inspired by the U. Schmidt method20 of introducing sulfur atoms into 2,5-diketopiperazines directly using S8 and NaNH2 in liq. NH3, we opted to employ S8 and NaHMDS or LiHMDS as the base in THF. Our expectations included not only the convenience of carrying out the sulfenylation reaction in an organic solvent rather than liq. NH3, but also the possibility of generating more well-defined sulfenylating species to effect the desired reaction more efficiently and with stereocontrol. In retrospect, we realized that the reaction of S8 with NaHMDS had already been studied by M. Schmidt24a–c in the 1960s, a study24a that we inadvertently missed in our preliminary communications.7,8 Our investigations with this reaction are summarized in Scheme 1. Thus, from the reaction of S8 (19) and NaHMDS, we were able to isolate, chromatographically, and characterize three reactive species: tetrasulfide 21 [40% yield, 1H NMR (CDCl3, 600 MHz): δ = 0.26 ppm; 13C NMR (CDCl3, 150 MHz): δ = 2.4 ppm; HRMS [M+H]+: calcd for C12H36N2O2S4+H: 449.0911; found: 449.0908], pentasulfide 22 [5% yield, HRMS [M+H]+: calcd for C12H36N2O2S5+H: 512.0280; found: 512.0241], and trisulfide 23 [8% yield, HRMS [M+H]+: calcd for C12H36N2O2S3+H: 417.1190; found: 417.1186]. Their formation, presumably through intermediate 20, may be explained as shown in Scheme 1 and it is consistent with the observations of M. Schmidt et al.24 The predominance of the tetrasulfide 21 is most likely due to steric shielding and charge repulsion during the second nucleophilic attack by the (TMS)2N− species on the sulfur chain (see 20, Scheme 1). The reaction of the resulting mixture with 2,5-diketopiperazines as exemplified with substrate 24 in the presence of excess base (NaHMDS) as shown in Scheme 2 is consistent with the presence of these species, although only epidi- and epitetrasulfides were isolated. Reduction of the mixture (presumably containing additional sulfenylated species, such as epitri- and epipentasulfides as well as open-chain oligosulfides) with NaBH4 followed by oxidation of the resulting dithiolate 26 (aq. NH4Cl; then KI3) led to a good yield of the epidithiodiketopiperazine 27 (69%). The same result was obtained from the pure tetrasulfide 25 (obtained in 22% yield from 24, see Scheme 2) upon reduction/oxidation (94%). Reaction of dithiolate 26 with MeI furnished bis-(methylthio)diketopiperazine 28 in 72% overall yield from 24. In support of the proposed mechanism (Scheme 2), we found that only mono-deuteration occurs upon quenching the initially formed species from substrate 24 and NaHMDS (2.2 equiv). Also in support of the intramolecular nature of the second C-S bond formation are the good yields of the epidithio- and epitetrathiodiketopiperazine products observed.
Scheme 1.

Reaction of Sulfur (S8) with NaHMDS [NaN(TMS)2]a
aReagents and conditions. NaHMDS (0.6 M in PhMe, 3.0 equiv), S8 (1.0 equiv), THF, 25 °C, 5 min, 21: 40%, 22: 5%, 23: 8%.
Scheme 2.

Sulfenylation of 2,5-Diketopiperazines with [NaHMDS-S8]. Preparation of Epitetrathiodiketopiperazine 25, Epidithiodiketopiperazine 27 and bis-(Methylthio)diketopiperazine 28a
aReagents and conditions. a) NaHMDS (0.6 M in PhMe, 3.0 equiv), S8 (1.0 equiv), THF, 25 °C, 1 min; then 24 (1 M in THF, 1.0 equiv), 1 min; then NaHMDS (0.6 M in PhMe, 2.0 equiv), 25 °C, 30 min; b) NaBH4 (25 equiv), THF/MeOH (1:1), 0→25 °C, 45 min; c) NH4Cl aq. (1.0 M), 25 °C; d) KI3 aq. (1.4 M), 25 °C, 10 min, 69% over the four steps from 24; e) MeI (50 equiv), 25 °C, 15 h, 72% over the three steps from 24.
The generality and scope of the sulfenylation reaction was explored with a variety of substrates. These explorations led to a series of epidithiodiketopiperazines (Table 1) and bis-(methylthio)diketopiperazines (Table 2). Thus, under the reaction conditions shown in Table 1, 3,6-unsubstituted diketopiperazines such as 29 (entry 1) reacted to form epidithiodiketopiperazines (i.e. 30, entry 1), albeit in modest yield (40%), the latter observation being attributed to possible unhindered intermolecular reactions of the intermediate sulfur species. This speculation is supported by the higher yields observed with 3,6-mono- and 3,6-disubstituted substrates (e.g. entries 2–5). The relatively low yield of epidithiodiketopiperazine 38 (entry 6) is most likely due to the steric congestion at the sites of sulfenylation (i.e. positions 3 and 6). It is notable that both syn (entries 4, 8, 10–12) and anti (entries 3 and 9) 3,6-disubstituted diketopiperazine systems enter the reaction equally well. These include monocyclic (entries 3–7) and polycyclic (entries 8–12) systems. The fact that sulfenylation occurs from the same side of the molecule in both the syn and the anti series provides support for the intramolecular nature of the second C-S bond formation (see 24d→25, Scheme 2). All epidithiodiketopiperazine products shown in Table 1 are racemic as a consequence of the enolate intermediacy in these reactions. Enantiopure compound 45 (entry 10) gave a mixture of enantiopure diastereoisomers (ca. 1.4:1 dr) due to the additional chiral centers within the structure.
Table 1.
Preparation of Epidithiodiketopiperazinesa
| |||
|---|---|---|---|
| Entry | Substrate | Productb | Overall Yield [%]c |
| 1 |
 29 |
 30 |
40 |
| 2 |
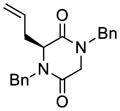 31 |
 32 |
63 |
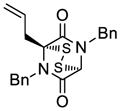
| 3 |
 33 |
 34 |
70 |
| 4 |
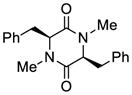 24 |
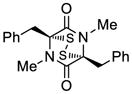 27 |
69 |
| 5 |
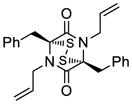
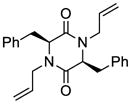 35 |
 36 |
65 |
| 6 |
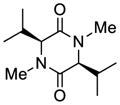 37 |
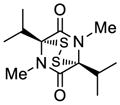 38 |
45 |
| 7 |
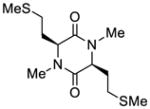 39 |
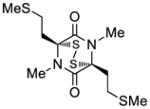 40 |
43 |
| 8 |
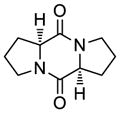 41 |
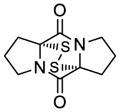 42 |
65 |
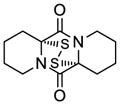
| 9 |
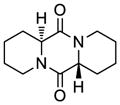 43 |
 44 |
68 |
| 10 |
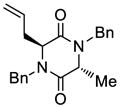
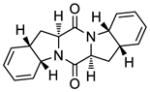 45 |
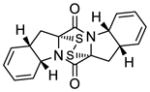 46 |
55d |
| 11 |
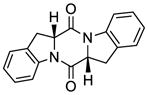 47 |
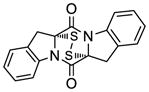 48 |
68 |
| 12 |
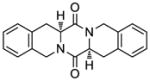 49 |
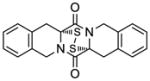 50 |
70 |
Reactions were performed on 100 mg scale at 25 °C.
Racemic mixture unless otherwise stated.
Yield of isolated products after chromatography.
ca. 1.4:1 dr.
Table 2.
Preparation of bis-(Methylthio)diketopiperazinesa
| |||
|---|---|---|---|
| Entry | Substrate | Productb | Overall Yield [%]c |
| 1 |
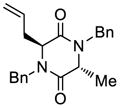 33 |
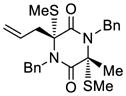 51 |
70 |
| 2 |
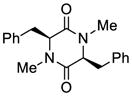 24 |
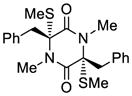 28 |
72 |
| 3 |
 35 |
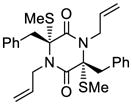 52 |
63 |
| 4 |
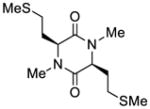 39 |
 53 |
51 |
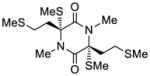
| 5 |
 41 |
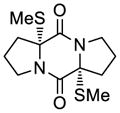 54 |
64 |
| 6 |
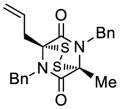
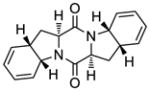 45 |
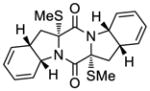 55 |
58d |
| 7 |
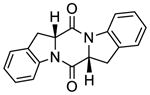 47 |
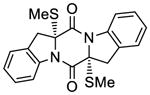 56 |
61 |
| 8 |
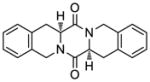 49 |
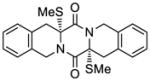 57 |
67 |
Reactions were performed on 100 mg scale at 25 °C.
Racemic mixture unless otherwise stated.
Yield of isolated products after chromatography.
ca. 1.4:1 dr.
Table 3.
Influence of the Base in the Sulfenylation of Selected Epidithiodiketopiperazinesa
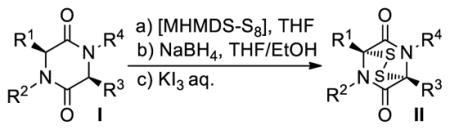
| |||||
|---|---|---|---|---|---|
| Entry | Substrate | Productb | LiHMDS | NaHMDS | KHMDS |
| Yield [%]c | |||||
| 1 |
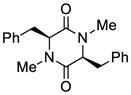 24 |
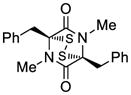 27 |
45 | 69 | 50 |
| 2 |
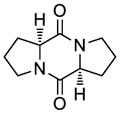 41 |
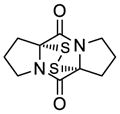 42 |
50 | 65 | 53 |
| 3 |
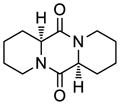 43 |
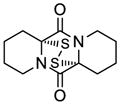 44 |
53 | 69 | 43 |
Reactions were performed on 50 mg scale at 25 °C.
Racemic mixtures were obtained.
Yield of isolated products after chromatography.
Employment of the reaction conditions shown in Table 2 on the indicated substrates led to the corresponding bis-(methylthio)diketopiperazines. All products were isolated as single racemic syn compounds with the exception of 55 (entry 6), which was formed as a mixture of enantiopure diastereoisomers (ca. 1.4:1 dr) due to the additional stereocenters within the substrate. Again, the observation of only the syn product provides support for the intramolecularity of the second sulfenylation step. The excellent stereoselectivity and good yields obtained in this sulfenylation reaction and its epidithiodiketopiperazine-forming counterpart (see Table 1) demonstrate the superiority of this method in comparison to the traditional U. Schmidt process that often leads to mixtures of the syn and anti products in lower yields.
The effect of the alkali metal in the base on the efficiency of the reaction was then examined. Thus, KHMDS, NaHMDS and LiHMDS were used in the sulfenylation protocol shown in Table 1 using diketopiperazine substrates 24, 41 and 2-epi-43 to generate epidithiodiketopiperazines 27, 42 and 44, respectively. As shown in Table 3, the results consistently point to NaHMDS as the preferred base for this reaction, although all three bases gave good yields of the epidithiodiketopiperazine products. As we shall see below, however, this is not always the case, especially with more sensitive substrates (see Table 4).
Table 4.
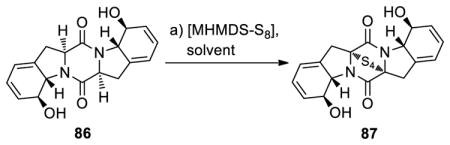
Optimization Study of the Sulfenylation of Diketopiperazine 86a
| ||||
|---|---|---|---|---|
| Entry | Solvent | LiHMDS | NaHMDS | KHMDS |
| Yield of 87 (%) [rsm]b | ||||
| 1 | THF | 20 [70] | 10 [40] | <5 [35] |
| 2 | CH2Cl2 | <5 [<5] | <5 [<5] | <5 [<5] |
| 3 | PhMe | 15 [70] | <5 [45] | <5 [30] |
| 4 | Et2O | 25 [60] | 15 [35] | <5 [35] |
| 5 | THFc | 46 [43] | 28 [30] | <5 [40] |
Reactions were performed on 5 mg scale at 25 °C.
Yields of product and recovered starting material (rsm) are isolated yields after chromatography, <5% yield refers to no detectable product or starting material as determined by 1H NMR spectroscopic analysis of the crude reaction mixture.
Reverse addition of preformed sulfenylation reagent to substrate and base.
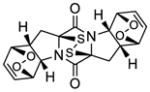
Previously known24a bis[bis(trimethylsilyl)amino]trisulfide (23, Scheme 3) was prepared and investigated for its suitability as a sulfenylating agent of 2,5-diketopiperazines in the presence of base. Thus, pure 23 reacted with diketopiperazine 24 in the presence of NaHMDS in THF at ambient temperature to produce a mixture of epidithiodiketopiperazine 27 (43%) and epitetrathiodiketopiperazine 25 (22%). A speculative mechanism for the formation of these products is shown in Scheme 3.25 Thus, the initially formed enolate 24b may react with trisulfide 23 through path a (attack at terminal S) to afford trisulfide intermediate 24e, which may then suffer in-tramolecular attack by the second enolate (24f) to afford epidithiodiketopiperazine 27 and (TMS)2NSNa (23b). The same product (27) could be formed from enolate 24b and trisulfide 23 through path b (attack at the central S) via the intermediacy of species 24g and 24h by intramolecular attack as shown in the scheme. Alternatively, trisulfide intermediate 24f may undergo different intramolecular collapse to generate, through path c, epitrithiodiketopiperazine 24i,26 whose opening with (TMS)2N− as shown may form epitetrathiodiketopiperazine 25 via intermediate species 24j.
Scheme 3.

Reaction of Diketopiperazine 24 with bis[bis(trimethylsilyl)amino]trisulfide [(TMS)2SSS(TMS)2] and NaHMDS and Mechanistic Considerations. Direct Formation of Epidithio- and Epitetrathiodiketopiperazinesa
aReagents and conditions. a) (TMS)2NSSSN(TMS)2 (4.0 equiv), NaHMDS (0.6 M in PhMe, 4.0 equiv), THF, 25 °C, 30 min, 25: 22%, 27: 43%.
Total Syntheses of Epicoccin G (1), 8,8′-epi-ent-Rostratin B (2), Gliotoxin (3), Gliotoxin G (4), Emethallicin E (5) and Haematocin (6). Empowered with the improved sulfenylation method7,8 we were able to synthesize a number of biologically active sulfenylated diketopiperazine natural products8 (Figure 1), including the antiviral agent epicoccin G9 (1), the 8,8′-epi-ent-isomer (2) of the cytotoxic agent rostratin B,10 the antiviral and antibiotic gliotoxin11 (3), and its epitetrathio counterpart gliotoxin G12 (4), the immunosuppressant emethallicin E13 (5), and the antifungal agent haematocin14 (6). The designed synthetic strategies employed to construct these molecules are exemplified with those depicted for epicoccin G [1, a bis-(methylthio)diketopiperazine] and 8,8′-epi-ent-rostratin B (2, an epidithiodiketopiperazine), in retrosynthetic format, in Scheme 4. Thus, epicoccin G (1) was disconnected retrosynthetically to its bis-unsaturated precursor 58 through a bis-hydrogenation step. The latter intermediate was then traced to bis-endoperoxide 60 through the rarely used Kornblum–DeLaMare rearrangement,27 anticipating a regioselective rupture of the endoperoxide moieties under basic conditions. Steric control in the latter process was envisioned to furnish the desired regioisomer (58). Through a bis-photooxygenation/bis-sulfenylation sequence, bis-endoperoxide 60 was traced back to bis-diene diketopiperazine 45 through the intermediacy of bis-diene 55. Similar retrosynthetic analysis of 8,8′-epi-ent-rostratin B (2) led to the same precursor (45) as shown in Scheme 4. The latter was envisioned to arise from L-N-Boc-tyrosine (62) via bicyclic intermediate 6328 (see Scheme 4) through appropriate elaboration and dimerization procedures.
Scheme 4.

Retrosynthetic Analysis of Epicoccin G (1) and 8,8′-epi-ent-Rostratin B (2)
The synthesis of the bis-diene 45 from the known tyrosine-derived hydroxy enone 6328 is shown in Scheme 5. Thus, acetylation of 63 followed by treatment with Zn and AcOH in MeOH at 65 °C and exposure to DBU led to the deoxygenation product bicyclic enone 64 possessing the desired syn ring junction (51% yield for the three steps). Luche reduction29 of the latter (NaBH4, CeCl3) gave allylic alcohol 65 (possessing the α configuration as expected on steric grounds; inconsequential) in 92% yield. In preparation for the pending cyclodimerization, key intermediate 65 was separately processed with LiOH and TFA to afford coupling partners 66 (99% yield, TFA salt) and 67 (99% yield), respectively. N-Boc carboxylic acid 67 and amine methyl ester TFA salt 66 were coupled in the presence of BOP-Cl and Et3N to afford amide 68 in 86% yield. Treatment of the latter with TFA followed by exposure to Et3N led to the formation of pentacyclic diketopiperazine 69 in 77% yield for the two steps. The desired bis-dehydration of bis-allylic alcohol 69 was achieved through the intermediacy of bis-allylic trifluoroacetate 70 formed by treatment of the former with (CF3CO)2O in the presence of Et3N and 4-DMAP (69% yield). The latter intermediate (70) was smoothly converted to the targeted bis-diene 45 upon exposure to catalytic amounts of Pd(PPh3)4 in the presence of K2CO3 (90% yield).30
Scheme 5.

Synthesis of bis-Diene Diketopiperazine 45a
aReagents and conditions. a) Ac2O (2.0 equiv), Et3N (3.0 equiv), 4-DMAP (0.2 equiv), CH2Cl2, 0→25 °C, 4 h; b) Zn (8.0 equiv), AcOH (2.0 equiv), MeOH, 65 °C, 30 min; c) DBU (5.0 equiv), PhMe, 65 °C, 3 h, 51% for the three steps; d) NaBH4 (1.1 equiv), CeCl3·7H2O (1.0 equiv), MeOH, −78→0 °C, 1 h, 92%; e) TFA/CH2Cl2 (1:1), 0→25 °C, 30 min, 99%; f) aq. LiOH (1.0 M)/THF (4:1), 0→25 °C, 3 h, 99%; g) 66, 67 (1.0 equiv each), BOP-Cl (1.1 equiv), Et3N (3.0 equiv), CH2Cl2, 0→25 °C, 15 h, 86%; h) TFA (32 equiv), CH2Cl2, 0→25 °C, 1.5 h; then Et3N (5.0 equiv), CH2Cl2, 0→25 °C, 15 h, 77% for the two steps; i) (CF3CO)2O (4.0 equiv), Et3N (6.0 equiv), 4-DMAP (0.3 equiv), MeCN, −40→25 °C, 1 h, 69%; j) Pd(PPh3)4 (0.1 equiv), K2CO3 (2.1 equiv), dioxane, 65 °C, 30 min, 90%.
The advancement of bis-diene 45 to the desired sulfenylated intermediates epidithiodiketopiperazine 46 and bis-(methylthio)diketopiperazine 55 and their diastereoisomers is summarized in Scheme 6. Thus, sulfenylation of 45 according to the developed procedure [NaHMDS-S8] furnished a mixture of oligosulfides (71) from which emerged epidithiodiketopiperazines 46 and 2,2′-epi-46 and bis-(methylthio)diketopiperazines 55 and 2,2′-epi-55 upon reduction/oxidation (NaBH4; KI3; 55% combined yield for 46 and 2,2′-epi-46, ca. 1.4:1 dr) and reduction/methylation (NaBH4; MeI; 58% overall yield for 55 and 2,2′-epi-55, ca. 1.4:1 dr). The stereochemical configurations of these chromatographically separated products were deciphered by NOESY correlations as indicated in Scheme 6 (bottom).
Scheme 6.

Synthesis of Dithiodiketopiperazines 46 and 2,2′-epi-46, and bis-(Methylthio)diketopiperazines 55 and 2,2′-epi-55a and Stereochemical Assignments of 55 and 2,2′-epi-55 by NOESY Studies (Arrows Designate NOESY Correlations)
aReagents and conditions. a) NaHMDS (0.6 M in PhMe, 3.0 equiv), S8 (1.0 equiv), THF, 25 °C, 1 min; then 45 (1 M in THF, 1.0 equiv), 1 min; then NaHMDS (0.6 M in PhMe, 2.0 equiv), 25 °C, 30 min; b) NaBH4 (25 equiv), THF/MeOH (1:1), 0→25 °C, 45 min; then MeI (50 equiv), 25 °C, 15 h, 58% over the three steps from 45 (55:2,2′-epi-55 ca. 1.4:1 dr); c) NaBH4 (25 equiv), THF/MeOH (1:1), 0→25 °C, 0.75 h; then KI3 aq. (1.4 M), 25 °C, 10 min, 55% over the three steps from 45 (46: 2,2′-epi-46 ca. 1.4:1 dr); d) NaBH4 (25 equiv), THF/MeOH (1:1), 0→25 °C, 45 min; then MeI (50 equiv), 25 °C, 15 h, 65% from 46(55:2,2′-epi-55 ca. 1.4:1 dr).
The correct diastereoisomers 55 and 2,2′-epi-46 were elaborated to the target molecules epicoccin G (1) and 8,8′-epi-ent-rostratin B (2) through similar pathways as shown in Schemes 7 and 8, respectively. Thus, reaction of bis-(methylthio)diketopiperazine bis-diene 55 with singlet oxygen (generated from triplet oxygen and UV light in the presence of tetraphenylporphyrin sensitizer)31 in CH2Cl2 at −45 °C furnished bis-endoperoxide 60, which was treated with DBU (−45→0 °C) without isolation to afford bis-hydroxy enone 58 as the major product (52% overall yield, Scheme 7). The latter compound was subjected to catalytic hydrogenation [H2, 20% Pd(OH)2/C] to give smoothly epicoccin G in 86% yield. Processing epidithio bis-diene 2,2′-epi-46 with singlet oxygen (0 °C) followed by treatment of the resulting bis-endoperoxide (61) with Et3N (0→25 °C) furnished epidithio bis-hydroxy enone 59 in 55% overall yield (Scheme 8). The sensitivity of the epidithiodiketopiperazine structural motif within 59 dictated the use of Stryker’s reagent32 [CuH(PPh3)]6 (as opposed to the hydrogenation conditions employed for the conversion of 58 to epicoccin G, Scheme 7) for the required reduction of the olefinic bonds, followed by re-oxidation with KI3 to regenerate the partially cleaved epidithio moiety, thereby furnishing 8,8′-epi-ent-rostratin B (2) in 82% overall yield.
Scheme 7.

Completion of the Total Synthesis of Epicoccin G (1)a
aReagents and conditions. a) O2, TPP (0.02 equiv), CH2Cl2, −45 °C, 45 min; then DBU (10.0 equiv), −45→0 °C; 1 h, 52% from 55; b) H2, Pd(OH)2/C (20% w/w, 0.4 equiv), MeOH, 25 °C, 1 h, 86%.
Scheme 8.

Completion of the Total Synthesis of 8,8′-epi-ent-Rostratin B (2)a
aReagents and conditions. a) O2, TPP (0.02 equiv), CH2Cl2, 0 °C, 2 h; then Et3N (5.0 equiv), 0→25 °C, 3 h, 55% for the two steps; b) [CuH(PPh3)]6 (10.0 equiv), benzene, 25 °C, 30 min; then KI3 aq. (1.4 M), 25 °C, 10 min, 82%.
As further demonstrations of the applicability of the present improved sulfenylation method, we pursued the enantioselective total synthesis of gliotoxin (3) and gliotoxin G (4), as well as emethallicin E (5) and haematocin (6) (Figure 1). The devised synthetic strategy toward these target molecules envisioned bicyclic hydroxy diene 78 (see Scheme 9) as a common intermediate. This key building block was obtained in multi-gram quantities from the tyrosin-derived hydroxy enone N-Boc methyl ester 63 as shown in Scheme 9.33 Thus, Luche reduction (NaBH4, CeCl3) of 6328 gave diol 72 stereoselectively (99% yield), which was smoothly acetylated to afford hydroxy acetate 73 in 91% yield. The latter was converted to hydroxy diene 74 through palladium-catalyzed elimination [Pd(OAc)2 (cat.), PPh3 (cat.), Et3N, 86%]. Photooxygenation of this diene (O2, TPP, hν, 73%) generated hydroxy endoperoxide 75, whose reduction with thiourea afforded triol 76 in 84% yield. Selective monosilylation of the latter (TIPSOTf, 96% yield) followed by engagement of the 1,2-diol system into a thionocarbonate moiety [(im)2C=S, 90% yield] furnished intermediate 77. The latter was deoxygenated [P(OMe)3, 82% yield] and desilylated (aq. HCl, 98% yield) to afford the desired building block hydroxy diene 78.
Scheme 9.

Synthesis of Common Key Building Block 78a
aReagents and conditions. a) NaBH4 (2.0 equiv), CeCl3·7H2O (1.3 equiv), MeOH, −20→0 °C, 3 h, 99%; b) Ac2O (2.0 equiv), Et3N (3.0 equiv), 4-DMAP (0.2 equiv), CH2Cl2, 0 °C, 1 h, 91%; c) Pd(OAc)2 (0.02 equiv), PPh3 (0.1 equiv), Et3N (1.2 equiv), PhMe, 25→110 °C, 3 h, 86%; d) O2, TPP (0.0036 equiv), CH2Cl2, 25°C, 24 h, 73%; e) thiourea (2.0 equiv), MeOH, 25 °C, 2 h, 84%; f) TIPSOTf (1.1 equiv), Et3N (2.0 equiv), CH2Cl2, 0 °C, 30 min, 96%; g) (im)2C=S (1.2 equiv), PhMe, 110 °C, 3 h, 90%; h) P(OMe)3, 111 °C, 12 h, 82%; i) HCl aq. (1.0 M), CH2Cl2/Et2O (1:1), 0 °C, 10 min, 98%.
The enantioselective total synthesis of gliotoxin (3) and gliotoxin G (4) from the common building block 78 is summarized in Scheme 10. Thus, hydrolysis of the methyl ester within 78 (LiOH) led to carboxylic acid 79 (99% yield), which was coupled with L-serine derivative 8034 (HATU, HOAt, DIPEA) to afford amide 81 in 88% yield. Removal of the Boc group from the latter and exposure of the resulting amino ester to Et3N furnished tricyclic diketopiperazine 82 (63% overall yield), whose structure was proven beyond doubt through X-ray crystallographic analysis (see ORTEP, Scheme 10). Sulfenylation of the latter required the use of S8 and LiHMDS, conditions that furnished directly gliotoxin (3, 23% yield) and gliotoxin G (4, 33% yield) (plus 6% recovered starting material 82). Interestingly, attempts to effect the sulfenylation of 82 with [NaHMDS-S8] failed to produce gliotoxin or gliotoxin G, leading instead to aromatization of the cyclohexadiene ring and decomposition. These results underscore the subtle differences in reactivity of the various alkali metal HMDS bases and point to the importance of thorough experimentation in attempting to achieve certain transformations, including the present sulfenylation.
Scheme 10.

Completion of the Enantioselective Total Syntheses of Gliotoxin (3) and Gliotoxin G (4)a
aReagents and conditions. a) aq. LiOH (1.0 M)/THF (6:1), 0→25 °C, 5 h, 99%; b) 79 (1.0 equiv), 80 (2.0 equiv), HOAt (1.1 equiv), HATU (1.1 equiv), DIPEA (3.0 equiv), CH2Cl2, 0→25 °C, 15 h, 88%; c) TFA/CH2Cl2 (1:1), 0→25 °C, 3 h; d) Et3N (5.0 equiv), CH2Cl2, 0→25 °C, 15 h, 63% for the two steps; e) LiHMDS (1.0 M in THF, 4.0 equiv), S8 (8.0 equiv), THF, 25 °C, 5 min; then 82 (0.06 M in THF, 1.0 equiv) 5 min; then LiHMDS (1.0 M in THF, 4.0 equiv), 25 °C, 1.5 h, 3: 23%, 4: 33%, plus 6% recovered starting material 82.
Scheme 11 summarizes the enantioselective total syntheses of emethallicin E (5) and haematocin (6) from common intermediate 78. Thus, a three-step sequence involving replacement of the Boc protective group with Alloc (TFA, 95% yield; then AllocCl, 88% yield) followed by saponification of the methyl ester group (LiOH) furnished hydroxy carboxylic acid 83 in high yield. Coupling of building blocks 83 and 84 (obtained in the first step of the above sequence 78→83) under the influence of BOP-Cl and DIPEA led to amide 85 in 83% yield over the two steps. Pentacyclic bis-hydroxy diketopiperazine 86 was generated from amide 85 in 84% overall yield upon cleavage of the Alloc protecting group [Pd2(dba)3 (cat.)] in the presence of Et2NH. The structure of intermediate 86 was unambiguously confirmed by X-ray crystallographic analysis (see ORTEP, Scheme 11). Sulfenylation of bis-hydroxy diketopiperazine 86 with [LiHMDS-S8] led to tetrasulfide 87 as the major product (46% yield, plus 43% recovered starting material 86).
Scheme 11.

Completion of the Enantioselective Total Syntheses of Emethallicin E (5) and Haematocin (6)a
aReagents and conditions. a) TFA/CH2Cl2 (1:2.5), 25 °C, 4 h, 95%; b) AllocCl (1.7 equiv), NaHCO3 (10.0 equiv), dioxane/H2O (1:1), 0→25 °C, 3 h, 88%; c) LiOH aq. (1.0 M)/THF (1:1), 0→25 °C, 5 h; d) 83, 84 (1.0 equiv each), BOP-Cl (1.1 equiv), DIPEA (3.0 equiv), CH2Cl2, 0→25 °C, 15 h, 83% for the two steps; e) Pd2(dba)3 (0.02 equiv), dbbp (0.05 equiv), THF/Et2NH (2:1), 25 °C, 2 h, 84%; f) LiHMDS (1.0 M in THF, 20 equiv), S8 (37 equiv), THF, 25 °C, 5 min; then 86 (0.06 M in THF/Et2O (9:1), 1.0 equiv), 5 min; then LiHMDS (1.0 M in THF, 20 equiv), 25 °C, 5 h, 46%, plus 43% recovered starting material 86; g) PhCH2COOH (30 equiv), DCC (30 equiv), 4-DMAP (3.0 equiv), 0→25 °C, 15 h, 71%, plus 26% recovered starting material 87; h) AcOH (30 equiv), DCC (30 equiv), 4-DMAP (3.0 equiv), 0→25 °C, 15 h, 71%, plus 24% recovered starting material 87; i) 1,3-propane dithiol (90 equiv), Et3N (0.32 equiv), MeCN/CH2Cl2 (25:1), 25 °C; then concentrate; then O2, MeOH, 2 h, 25 °C, 54% overall; j) NaBH4 (80 equiv), MeOH/py (1:1), 0 °C; then MeI (485 equiv) 0→25 °C, 4 h; 97% overall.
As in the case of the gliotoxins discussed above (Scheme 10), the standard [NaHMDS-S8] conditions failed to produce the sulfenylated product from substrate 86 in satisfactory yield, leading only to 10% yield of epitetrasulfide 87 (Scheme 11). This observation prompted a systematic investigation to optimize the yield of this sulfenylation reaction varying the HMDS base and the solvent. The results of this study, shown in Table 4, revealed LiHMDS in THF as the optimum conditions (entry 5/LiHMDS). The formation of the epitetrasulfide as the predominant product in this case is also of interest. This example underscores once again the importance of careful optimization of conditions to achieve the best results in diketopiperazine sulfenylation reactions. It is also noteworthy that the use of bis[bis(trimethylsilyl)amino]trisulfide (23, Scheme 3) as a sulfenylating reagent in the presence of LiHMDS as a base proved less reactive than the corresponding in situ generated species [LiHMDS-S8], leading to recovery of 80% of starting material (86) and no epidisulfide or epitetrasulfide products. The use of NaHMDS or KHMDS and trisulfide 23 led primarily to aromatization under the same sulfenylation conditions.
The stereochemical configuration of the epitetrasulfide 87 was based on NMR spectroscopic studies and was confirmed by the successful synthesis of the natural products 5 and 6. Indeed, intermediate 87 served as a common precursor to emethallicin E (5) and haematocin (6) as shown in Scheme 11. Thus, bis-esterification of 87 with phenylacetic acid (PhCH2COOH) in the presence of DCC and 4-DMAP gave bis-phenylacetate 88 (71% yield, plus 26% recovered starting material 87), whose reduction/oxidation (1,3-propane dithiol; then O2) furnished the desired product emethallicin E (5) in 54% overall yield. Alternatively, bis-acetylation of 87 (AcOH, DCC, 4-DMAP, 71% yield, plus 24% recovered starting material 87) followed by reduction/methylation (NaBH4; then MeI) of the resulting bis-acetate afforded haematocin (6) in 97% overall yield. The use of the DCC/4-DMAP esterification protocol instead of the more conventional acid anhydride or chloride methods was dictated by the sensitivity of the substrate (87) and products (88, 89) under the reaction conditions, especially toward aromatization.
As part of a program directed toward the total synthesis of aranotin (8) we attempted to construct its monomeric unit (7, see Scheme 13) through diazo epoxide precursor 95 as shown in Scheme 12.35 Thus, bicyclic diene system 90 (for its synthesis see Scheme 9, 77→78, step h) was reacted with bis-trichloroethylazodicarboxylate (TrocN=NTroc) to afford Diels–Alder adduct 91 stereoselectively (steric control) and in 93% yield. Desilylation of the latter with TBAF gave hydroxy derivative 92 (92% yield), whose treatment with methyl(trifluoromethyl) dioxirane followed by sequential exposure to Zn and CuCl2 in the presence of NH4OH afforded diazo epoxide 93 as a single diastereoisomer (76% overall yield). The stereochemical configuration of 93 was established through X-ray crystallographic analysis (see ORTEP, Scheme 12). As expected, this epoxide did not enter the obligatory rearrangement with loss of N2 by virtue of the syn arrangement of the diazo and epoxide moieties that does not allow for the proper orbital orientations.35a
Scheme 13.

Synthesis of Oxepin (7) by Ring Expansion of Nitroso Epoxide 98a
aReagents and conditions. a) TrocNHOH (2.5 equiv), NaIO4 (1.0 equiv), TBAI (1.0 equiv), CH2Cl2/H2O (4:1), 0→25 °C, 10 min, 88%; b) 1,1,1-trifluoro acetone (7.0 equiv), Na2EDTA, Na-HCO3 aq. (60 equiv), oxone(R) (20 equiv), MeCN/H2O (1:1), 0 °C, 15 h, 40%.
Scheme 12.

Attempted Synthesis of Oxepin 7 by Ring Expansion of a Diazo Epoxidea
aReagents and conditions. a) TrocN=NTroc (1.2 equiv), CH2Cl2, 41 °C, 4 h, 93%; b) TBAF (1.0 M in THF, 2.0 equiv), THF, 0 °C, 30 min, 92%; c) 1,1,1-trifluoro acetone (62 equiv), Na2EDTA, NaHCO3 aq. (15 equiv), oxone(R) (38 equiv), MeCN, 0→25 °C, 15 h; d) Zn (21 equiv), MeOH/NH4Cl aq. (1.0 M) (4:1), 25 °C, 2 h; then NH4OH aq. (15 M), CuCl2 aq. (1.0 M), 25 °C, 5 min, 76% for the two steps.
Our inability to reach the anti diazo epoxide 95 (Scheme 12), whose rearrangement to oxepin 94 was anticipated to be facile, prompted us to pursue the alternative pathway (Scheme 13) involving trichloroethyl nitrosoformate compound 96 (generated from TrocNHOH and NaIO4) as the dienophile. The latter reacted with bicyclic diene 90 to give Diels–Alder adduct 97 (88% yield) diastereo- and regioselectively (presumably due to steric control).36 The structure of 97 was assigned based on NMR spectroscopic analysis (COSY, NOESY, HMBC, HSQC). This intermediate was epoxidized with methyl(trifluoromethyl) dioxirane to afford directly oxepin system 7 (40% yield), presumably via the fleeting epoxide 98 through a retro-Diels–Alder/epoxide opening. Epoxide 98 apparently must be of the anti configuration with respect to the N–O bridge, which allows for the facile rearrangement/extrusion of 96 (which undergoes disproportionation with expulsion of oxygen to form TrocN=NTroc).36
Biological Evaluation
Having synthesized various types of epidithio- and bis-(methylthio)diketopiperazines we selected a number of them for biological evaluation. Specifically, selected compounds were tested against poliovirus and Plasmodium falciparum.37 Table 5 summarizes the results of these biological assays. Thus, in the anti poliovirus assays (carried out in the laboratory of D. F. S. under the auspices of the National Institute of Allergy and Infectious Diseases, NIAID), epidithiodiketopiperazines 46 (code number KCN-19), 2,2′-epi-46 (code number KCN-2,2′-epi-19) and epidithio-bis-endoperoxide-diketopiperazine 61 (code number KCN-21) proved to be the most potent, exhibiting EC50 = 101–115 nM, 107–123 nM, and EC50 = 21.4 nM values, respectively, depending on the assay (see Table 5, entries 2, 3 and 7). Table 5 also displays selectivity indices (SI = CC50/EC50 or EC90, with CC50 = 50% cell-inhibitory, cytotoxic concentration deter-mined in stationary cells and EC50/90 = 50%/90% poliovirus-inhibitory, effective concentration) for some of these compounds. Compounds 46 (SI = 41–70), 2,2′-epi-46 (SI = 27–75) and 61 (SI = 23–59) were the most impressive in this regard (see Table 5). The anti poliovirus drug Pirodavir(R) (99, Table 5, entry 9), used as a control in this poliovirus assay, exhibited EC50 = 1.58 μM, underscoring the significant activities of KCN-19 (46), KCN-2,2′-epi-19 (2,2′-epi-46), and KCN-21 (61).
Table 5.
Biological Evaluation of Selected Compounds in Poliovirus and Plasmodium falciparum Assaysa
| Entry | Structure | Code Number | Poliovirus EC50 [visual;b neutral redc] | Poliovirus EC90 [virus yieldd] | Selectivity Index (Poliovirus) [visual;b neutral red;c virus yieldd] | Plasmodium falciparum IC50 |
|---|---|---|---|---|---|---|
| 1 |
 45 |
KCN-7 | >50 μM;b n.d.c,e |
n.d.d,e | n.d.e | >50 μM |
| 2 |
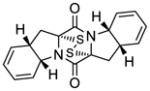 46 |
KCN-19 | 101±59 nM;b 115±59 nMc |
149±65 nMd | 70±48;b 56±45;c 41±23d |
3.6 μM |
| 3 |
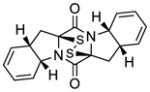 2,2′-epi-46 |
KCN-2,2′-epi-19 | 107±73 nM;b 123±90 nMc |
177±45 nMd | 59±59;b 75±92;c 27±11d |
2.7 μM |
| 4 |
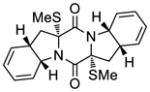 55 |
KCN-18 | 14.5 μM;b 25.6 μMc |
n.d.d,e | 5.7;b 3.4;c n.d.d,e |
>50 μM |
| 5 |
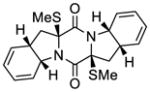 2,2′-epi-55 |
KCN-2,2′-epi-18 | 56.9 μM;b >50 μMc |
n.d.d,e | 1.5;b n.d.;c,e n.d.d,e |
>50 μHM |
| 6 |
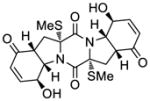 58 |
KCN-20 | >50 μM;b n.d.c,e |
n.d.d,e | n.d.e | 1.2 μM |
| 7 |
 61 |
KCN-21 | 21.4±11.9 nM;b 21.4±2.4 nMc |
38.1±7.1 nMd | 59±33;b 41±17;c 23±8d |
2.5 μM |
| 8 |
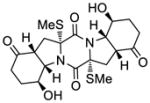 1: epicoccin G |
KCN-1 | >50 μM;b n.d.c,e |
n.d.d,e | n.d.e | 2.5 μM |
|
| ||||||
| 9 |
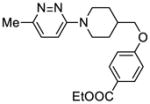 99:Pirodavir®f |
— | n.d.;b,e 1.58 μMc |
1.55 μMd | >18c | n.d.e |
Assays for entries 2, 3 and 7 were carried out as triplicates; mean and standard deviation are given. For experimental details of all assays, see Supporting Information.
Visual assay.
Neutral red assay.
Virus yield reduction assay.
Not determined.
Standard anti poliovirus drug used as a control.
In the anti Plasmodium falciparum assays (carried out in the laboratories of E. A. W. at TSRI), epidithiodiketopiperazines 46 (IC50 = 3.6 μM, Table 5, entry 2), 2,2′-epi-46 (IC50 = 2.7 μM, entry 3), 59 (IC50 = 4.5 μM; not included in the table, for structure see Scheme 4), 61 (IC50 = 2.5 μM, entry 7), and bis-(methylthio)diketopiperazines 58 (IC50 = 1.2 μM, entry 6), 2,2′-epi-58 (IC50 = 4.4 μM; not included in the table, for structure see Scheme 4), and epicoccin G (1, IC50 = 2.5 μM) proved to be the most potent.
CONCLUSION
An improved method for the sulfenylation of 2,5-diketopiperazines based on the use of alkali metal hexame-thyldisilazide bases (i.e. NaHMDS, LiHMDS and KHMDS) and sulfur (S8) in THF at 25 °C as a means to prepare epidithio-, epitetrathio- and bis-(methylthio)diketopiperazines has been developed. A second method involving the use of bis[bis(trimethylsilyl)amino]trisulfide [(TMS)2NSSSN(TMS)2] and NaHMDS for the direct preparation of epidithio- and epitetrathiodiketopiperazines has also been developed.
Application of these methods led to the synthesis of an array of sulfenylated diketopiperazine systems, including the natural products epiccocin G (1), gliotoxin (3), gliotoxin G (4), emethallicin E (5), haematocin (6) and the 8,8′-epi-ent-isomer (2) of rostratin B. With the exception of gliotoxin (3),6h these accomplishments represent the first enantioselective total syntheses of these natural products and their analogs and feature a number of novel synthetic strategies and reactions, including the [2+2] photooxygenation and the rarely used Kornblum–DeLaMare rearrangement.
Biological investigations of selected members of the synthesized compound libraries led to the discovery of a number of potent anti poliovirus agents (i.e. 46, 2,2′-epi-46 and 61) and a series of anti Plasmodium falciparum lead compounds (i.e. 46, 2,2′-epi-46, 58, 61 and 1) that may facilitate biological investigations and drug discovery efforts in the antiviral and anti-malarial areas, respectively.
By blending total synthesis of natural products of biological and medical interest with method development endeavors and chemical biology studies, the work described herein exemplifies the modern paradigm of natural product synthesis and underscores its relevance and importance to chemistry, biology and medicine.
Supplementary Material
Acknowledgments
Funding Sources
K.C.N. acknowledges funding from the National Institute of Health (USA) (grant nos. AI055475 and CA100101), as well as fellowships from Fonds de Recherche Québec (to D. G.), Bristol-Myers Squibb (to D. S.) and The Skaggs Research Institute. This project was funded in part for D.F.S. from the Division of Microbiology and Infectious Diseases, National Institute of Allergy and Infectious Diseases, National Institute of Health (USA), Department of Health and Human Services, under Contract No. HHSN272201100019I/HHSN27200001/B01., and for E.A.W. by the National Institute of Health (USA) (grant no. R01 AI090141).
We thank Drs. D. H. Huang and L. Pasternack for NMR spectroscopic, Dr. G. Siuzdak for mass spectrometric, and Dr. R. K. Chadha for X-ray crystallographic assistance.
Footnotes
Supporting Information. Experimental procedures and characterization data for key compounds (pdf cif files). This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.(a) Bull SD, Davies SG, Parkin RM, Sancho FS. J Chem Soc, Perkin Trans 1. 1998:2313–2319. [Google Scholar]; (b) Ding G, Jiang L, Guo L, Chen X, Zhang H, Che Y. J Nat Prod. 2008;71:1861–1865. doi: 10.1021/np800357g. [DOI] [PubMed] [Google Scholar]; (c) Kamei H, Oka M, Hamagishi Y, Tomita K, Komishi M, Oki T. J Antibiot. 1990;43:1018–1020. doi: 10.7164/antibiotics.43.1018. [DOI] [PubMed] [Google Scholar]; (d) Huang R, Zhou X, Xu T, Yang X, Liu Y. Chem Biodiv. 2010;7:2809–2829. doi: 10.1002/cbdv.200900211. [DOI] [PubMed] [Google Scholar]; (e) Cornacchia C, Cacciatore I, Baldassarre L, Mollica A, Feliciani F, Pinnen F. Mini-Rev Med Chem. 2012;12:2–12. doi: 10.2174/138955712798868959. [DOI] [PubMed] [Google Scholar]; (f) Borthwick AD. Chem Rev. 2012;112:3641–3716. doi: 10.1021/cr200398y. [DOI] [PubMed] [Google Scholar]
- 2.Ressurreicao ASM, Delatouche R, Gennari C, Piarulli U. Eur J Org Chem. 2011:217–228. [Google Scholar]
- 3.Stepan AF, et al. J Med Chem. 2012;55:3414–3424. doi: 10.1021/jm300094u. and references cited therein. [DOI] [PubMed] [Google Scholar]
- 4.(a) Gardiner MD, Waring P, Howlett BJ. Microbiol. 2005;151:1021–1032. doi: 10.1099/mic.0.27847-0. [DOI] [PubMed] [Google Scholar]; (b) Rezanka T, Sobotka M, Spizek J, Sigler K. Anti-infect Agents Med Chem. 2006;5:187–224. [Google Scholar]; (c) Greiner D, Bonaldi T, Eskeland R, Roemer R, Imhof A. Nat Chem Biol. 2005;1:143–145. doi: 10.1038/nchembio721. [DOI] [PubMed] [Google Scholar]; (d) Isham CR, Tibodeau JD, Jin W, Xu R, Timm MM, Bibblel KC. Blood. 2007;109:2579–2588. doi: 10.1182/blood-2006-07-027326. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Jiang CS, Müller WEG, Schröder HC, Guo YW. Chem Rev. 2012;112:2179–2207. doi: 10.1021/cr200173z. [DOI] [PubMed] [Google Scholar]; (f) Waring P, Eichner RD, Müllbacher A. Med Res Rev. 1988;8:499–524. doi: 10.1002/med.2610080404. [DOI] [PubMed] [Google Scholar]; (g) Waring P, Beaver J. Gen Pharmacol. 1996;27:1311–1316. doi: 10.1016/s0306-3623(96)00083-3. [DOI] [PubMed] [Google Scholar]
- 5.Iwasa E, Hamashima Y, Sodeoka M. Isr J Chem. 2011;51:420–433. [Google Scholar]
- 6.For selected epidithiodiketopiperazine total syntheses, see: Williams RM, Rastetter WH. J Org Chem. 1980;45:2625–2631.Wu Z, Williams LJ, Danishefsky SJ. Angew Chem, Int Ed. 2000;39:3866–3868. doi: 10.1002/1521-3773(20001103)39:21<3866::AID-ANIE3866>3.0.CO;2-E.DeLorbe JE, Salman YJ, Mennen SM, Overman LE, Zhang F. J Am Chem Soc. 2011;133:6549–6552. doi: 10.1021/ja201789v.Boyer N, Movassaghi M. Chem Sci. 2012;3:1798–1803. doi: 10.1039/C2SC20270K.Kim J, Ashenhurst JA, Movassaghi M. Science. 2009;324:238–241. doi: 10.1126/science.1170777.Iwasa E, Hamashima Y, Fujishiro S, Higuchi E, Ito A, Yoshida M, Sodeoka M. J Am Chem Soc. 2010;132:4078–4079. doi: 10.1021/ja101280p.Kim J, Movassaghi M. J Am Chem Soc. 2010;132:14376–14378. doi: 10.1021/ja106869s.Fukuyama T, Nakatsuka S, Kishi Y. Tetrahedron. 1981;37:2045–2078.Fukuyama T, Kishi Y. J Am Chem Soc. 1976;98:6723–6724. doi: 10.1021/ja00437a063.
- 7.Nicolaou KC, Giguère D, Totokotsopoulos S, Sun Y. Angew Chem, Int Ed. 2012;51:728–732. doi: 10.1002/anie.201107623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nicolaou KC, Totokotsopoulos S, Giguère D, Sun Y, Sarlah D. J Am Chem Soc. 2011;133:8150–8153. doi: 10.1021/ja2032635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.(a) Guo H, Sun B, Gao H, Chen X, Liu S, Yao X, Liu X, Che Y. J Nat Prod. 2009;72:2115–2119. doi: 10.1021/np900654a. [DOI] [PubMed] [Google Scholar]; (b) Wang JM, Ding GZ, Fang L, Dai JG, Yu SS, Wang YH, Chen XG, Ma SG, Qu J, Xu S, Du D. J Nat Prod. 2010;73:1240–1249. doi: 10.1021/np1000895. [DOI] [PubMed] [Google Scholar]
- 10.Tan RX, Jensen PR, Williams PG, Fenical W. J Nat Prod. 2004;67:1374–1382. doi: 10.1021/np049920b. [DOI] [PubMed] [Google Scholar]
- 11.(a) Weindling R, Emerson OH. Phytopathol. 1936;26:1068–1070. [Google Scholar]; (b) Johnson JR, Bruce WF, Dutcher JD. J Am Chem Soc. 1943;65:2005–2009. [Google Scholar]; (c) Beecham AF, Fridrichsons J, Mathieson AMcL. Tetrahedron Lett. 1966;27:3131–3138. doi: 10.1016/s0040-4039(01)99927-7. [DOI] [PubMed] [Google Scholar]
- 12.Waring P, Eichner RD, Palni UT, Müllbacher A. Tetrahedron Lett. 1986;27:735–738. [Google Scholar]
- 13.Kawahara N, Nozawa K, Yamazaki M, Nakajima S, Kawai K. Heterocycles. 1990;30:507–515. [Google Scholar]
- 14.Suzuki Y, Takahashi H, Esumi Y, Arie T, Morita T, Koshino H, Uzawa J, Uramoto M, Yamaguchi I. J Antibiot. 2000;53:45–49. doi: 10.7164/antibiotics.53.45. [DOI] [PubMed] [Google Scholar]
- 15.Nagarajan R, Huckstep LL, Lively DH, DeLong DC, Marsh MM, Neuss N. J Am Chem Soc. 1968;90:2980–2982. [Google Scholar]
- 16.For a total synthesis of acetylaranotin, see: Codelli JA, Puchlopek ALA, Reisman SE. J Am Chem Soc. 2012;134:1930–1933. doi: 10.1021/ja209354e.
- 17.Trown PW. Biochem Biophys Res Commun. 1968;33:402–407. doi: 10.1016/0006-291x(68)90585-8. [DOI] [PubMed] [Google Scholar]
- 18.Shimazaki N, Shima I, Hemmi K, Tsurumi Y, Hashimoto M. Chem Pharm Bull. 1986;35:3527–3530. doi: 10.1248/cpb.35.3527. [DOI] [PubMed] [Google Scholar]
- 19.(a) Poisel H, Schmidt U. Angew Chem, Int Ed Engl. 1971;10:130–131. [Google Scholar]; (b) Poisel H, Schmidt U. Chem Ber. 1971;104:1714–1721. [Google Scholar]
- 20.Öhler E, Poisel H, Tataruch F, Schmidt U. Chem Ber. 1972;105:635–641. doi: 10.1002/cber.19721050229. [DOI] [PubMed] [Google Scholar]
- 21.Kishi Y, Fukuyama T, Nakatsuka S. J Am Chem Soc. 1973;95:6490–6492. doi: 10.1021/ja00800a078. [DOI] [PubMed] [Google Scholar]
- 22.Yoshimura J, Nakamura H, Matsunari K. Bull Chem Soc. 1975;48:605–609. [Google Scholar]
- 23.Overman LE, Sato T. Org Lett. 2007;9:5267–5270. doi: 10.1021/ol702518t. [DOI] [PubMed] [Google Scholar]
- 24.Scherer O, Schmidt M. Naturwissenschaften. 1963;50:304.Schmidt M, Scherer O. Naturwissenschaften. 1963;50:302–304.Scherer O, Schmidt M. Z Naturforsch Pt B. 1963;18:415. See also: Siivari J, Maaninen A, Haapaniemi E, Laitinen RS, Chivers T. Z Naturforsch Pt B. 1995;50:1575–1582.
- 25.For a review on the reactivity of organic polysulfides, see: Steudel R. Chem Rev. 2002;102:3905–3945. doi: 10.1021/cr010127m.
- 26.For the isolation of epitrithiodiketopiperazines, see: Waring P, Eichner RD, Tiwari-Palni U, Müllbacher A. Aust J Chem. 1987;40:991–997.Dong JY, He HP, Shen YM, Zhang KQ. J Nat Prod. 2005;68:1510–1513. doi: 10.1021/np0502241.
- 27.(a) Kornblum N, DeLaMare HE. J Am Chem Soc. 1951;73:880–881. [Google Scholar]; (b) Staben ST, Linghu X, Toste FD. J Am Chem Soc. 2006;128:12658–12659. doi: 10.1021/ja065464x. [DOI] [PubMed] [Google Scholar]
- 28.(a) Wipf P, Kim Y. Tetrahedron Lett. 1992;33:5477–5480. [Google Scholar]; (b) Pierce JG, Kasi D, Fushimi M, Cuzzupe A, Wipf P. J Org Chem. 2008;73:7807–7810. doi: 10.1021/jo801552j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Luche JL. J Am Chem Soc. 1978;100:2226–2227. [Google Scholar]
- 30.Barrero AF, Arseniyadis S, Quilez del Moral J, Mar Herrador M, Valdivia M, Jeminez D. J Org Chem. 2002;67:2501–2508. doi: 10.1021/jo0161882. [DOI] [PubMed] [Google Scholar]
- 31.For a review on synthetic applications of bicyclic endoperoxides, see: Balci M. Chem Rev. 1981;81:91–108.
- 32.Mahoney WS, Brestensky DM, Stryker JM. J Am Chem Soc. 1988;110:291–293. [Google Scholar]
- 33.Henninger TC, Sabat M, Sundberg RJ. Tetrahedron. 1996;52:14403–14418. [Google Scholar]
- 34.Turos E, Audia JE, Danishefsky SJ. J Am Chem Soc. 1989;111:8231–8236. [Google Scholar]
- 35.(a) Rastetter WH. J Am Chem Soc. 1976;98:6350–6353. doi: 10.1021/ja00436a046. [DOI] [PubMed] [Google Scholar]; (b) Haas DD, Rastetter WH. J Am Chem Soc. 1976;98:6353–6359. doi: 10.1021/ja00436a047. [DOI] [PubMed] [Google Scholar]; (c) Rastetter WH, Richard TJ. J Am Chem Soc. 1979;101:3893–3897. [Google Scholar]
- 36.Kirby GW, McGuigan H, Mackinnon JWM, McLean D, Sharma RP. J Chem Soc, Perkin Trans I. 1985:1437–1442. [Google Scholar]
- 37.For current efforts in the global eradication of the poliovirus, see: Callaway E. Nature. 2012;485:563. doi: 10.1038/485563a.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.