

Abstract
Our work and others’ over the past few years have led to the identification of new roles of PAK1 in cardiac physiology, such as the regulation of cardiac ion channel and actomyosin function. More recent studies have revealed that PAK1-deficient mice were vulnerable to cardiac hypertrophy and readily progress to failure under sustained pressure overload and susceptible to ischemia/reperfusion injury. Our further study indicated that the PAK1 activator FTY720 was able to prevent this pressure overload-induced hypertrophy in wild-type mice without compromising their cardiac functions. A cardiac protective effect against ischemia/reperfusion injury by FTY720 was also observed in both rat and mouse models by us and others. Thus, these studies suggest that PAK1 is more important in the heart than previously thought, in particular a therapeutic potential of PAK1 activators. In the future, in-depth investigations are required to further substantiate our hypotheses on mechanisms for PAK1 function in the heart and to explore a therapeutic potential of FTY720 and other PAK1 activators in heart disease conditions.
Keywords: PAK1, cardiac contractility, hypertrophic growth, ischemic injury
Expression and Physiological Roles of PAK1 in the Heart
Myocardial function of the heart relies on rhythmic and effective cardiac contraction and relaxation, which depends on the appropriately timed generation and spread of cardiac electrical activity itself. At the cellular level, excitation-contraction (E-C) coupling is initiated by action potential depolarization resulting, via a cascade of events, in an increase in intracellular calcium concentration, which ultimately leads to muscle contraction; subsequent removal of intracellular calcium via a number of mechanisms results in relaxation. Thus, this process involves multiple trans-membrane (e.g., ion channels) and intracellular proteins (e.g., Ca2+ handling and sarcomeric proteins) and is highly regulated by multiple extra- and intra-cellular signaling pathways. Recent work by us and others1-5 has laid the groundwork for elucidating key functional roles for p21 activated kinase 1 (PAK1) in this process including following aspects:
Expression and activation of PAK1 in the heart
The expression of PAK1 as well as its isoforms PAK2 and PAK3 in cardiac tissues has been investigated in several species.2,3,6 The protein level of PAK1 is high in all regions of the heart such as the sino-atrial node (SAN), atrial and ventricular tissue,2,3 whereas PAK2 is expressed at low or modest levels in cardiac tissues, and there is little expression of PAK3 in the heart. At the cellular level, it is localized to Z-disc, cell and nuclear membrane and intercalated disc of ventricular myocytes.2 Distinct subcellular distribution patterns have also revealed, for example, in guinea pig SAN cells, we found PAK1 was evenly expressed in central nodal cells, whereas it demonstrated a clear striation patterns in peripheral nodal cells (Fig. 1).
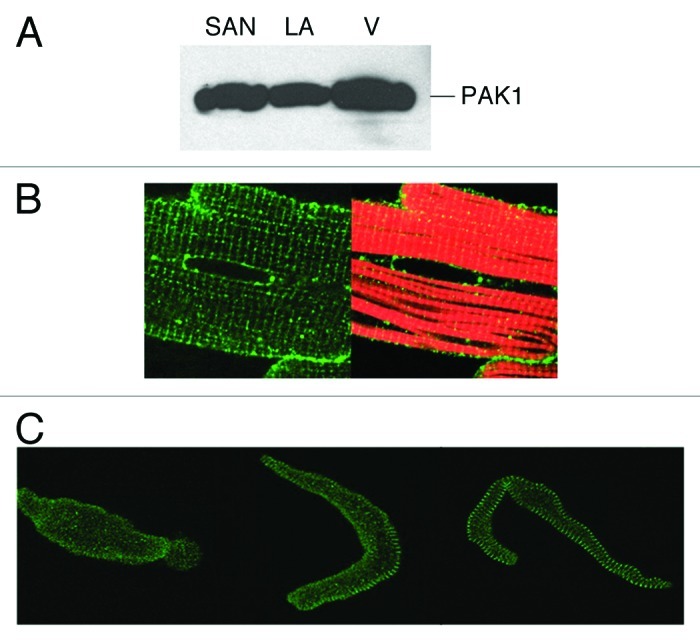
Figure 1. PAK1 expression in the heart. (A) Western blot detection of PAK1 expression in SAN, left atrial (LA) and ventricular tissues (V). PAK1 expression pattern was examined by immunocytochemical staining in (B) adult rat cardiomyocyte (green, anti-PAK1; red, rhodamine-conjugated phalloidin), in (C) guinea pig senatorial node (SAN) cells (adapted from refs. 3 and 4).
In cells PAK1 activation requires not only the GTPases (CDC42 and RAC), but also the Tyr-kinase ETK and the SH3 adaptor proteins such as PAK-interacting exchange factor (PIX) and non-catalytic region of tyrosine kinase adaptor protein (NCK), which directly interact with PAK1 (for detail, see Parrini in this issue). Furthermore, PAK1 is directly activated by a group of small molecules including C2/C6 ceramide and FTY720.7,8 These small molecules bear substantial structural similarity with sphingosine. Sphingosine also stimulates auto-phosphorylation of PAK2 at the same sites as CDC42 and RAC do.9 However, it is still unclear whether sphingosine activates PAK1 from the GTPase-binding domain or not. Early studies indicate that CDC42/RAC and sphingosine share the same molecular mechanism in the activation of PAK1.10 However, more recent studies indicate that these GTPases and sphingosine activate PAK1 in a synergetic way.11 PAK1 can also be activated by some other signal transducers that have prominent functional effects in the heart. For example, it was found that receptors coupled to the inhibitory G proteins Gi, such as bradykinin and acetylcholine, were able to activate PAK1 indirectly.12-14 Excellent evidence to support this comes from the observation that constitutively active PAK1 and ligands stimulating these receptors produce the same cytoskeletal re-organization in mammalian cells.12,13,15 Interestingly, recent studies have indicated that PAK1 is activated by a variety of growth factors such as EGF and other extracellular factors such as angiotensin II16 and isoproterenol (ISO).6,17 These observations reflect the complexity of PAK1 upstream signaling pathways that are still not well understood.
Regulation of cardiac ion channel activity
The potential role of PAK1 in the regulation of ion channel activities was first demonstrated by us in isolated guinea pig SAN preparations and single SAN pacemaker cells.3 In SAN expressing the constitutively active (CA) PAK1, responses to ISO stimulation manifested by the frequency of electrical impulse firing were significantly lower compared with those of controls. We also characterized the beating rate of cultured SAN cells infected with AdPak1 or the control AdLacZ.3 In the control SAN cells infected with AdLacZ, 5 nM ISO increases the beating rate by 40-50%. However, this ISO-induced increase in beating rate is diminished, when they express the CA-PAK1, clearly indicating that PAK1 antagonizes ISO. Whole cell patch clamping demonstrated that both L-type Ca2+ current and the delayed rectifier K+ current had a repressed response to ISO in cells expressing the CA-PAK1 (Fig. 2). In the SAN cells, CA-PAK1 redirects intracellular localization of protein phosphatase 2A (PP2A).3 A recent study further demonstrated PAK1 regulation of connexion (Cx) 43 phosphorylation in both isolated rabbit ventricular myocytes and HEK cells expressing Cx43 is likely through the PP2A activity.18

Figure 2. PAK1 regulation of cardiac ion channel activity. (A and B) Representative L-type Ca2+ current recordings from cultured SAN cells infected with Ad-LacZ or Ad-PAK1 in the absence and presence of 100 nmol/L ISO for 5 min. Currents were recorded during 200-ms step depolarizations from a holding potential of -50 mV to a range of potentials between -40 and +50 mV. (C and D) Current-voltage relationship of ICaL in cells infected with Ad-LacZ or Ad-PAK1, in the absence and presence of ISO (adapted from ref. 4).
Based on these results, we suggested that there is a dynamic balance between the kinase and the phosphatase activity in controlling L-type Ca2+ channel as well as delayed rectifier K+ channel activity in heart cells. The balance between these kinase and phosphatase actions may be of importance in controlling cardiac pacemaker activity in response to autonomic and humoral stimulation. The importance of this balance is highlighted by a recent report by Vinogradova et al. that a high basal PKA-dependent phosphorylation in SAN pacemaker cells drives rhythmic internal Ca2+ store oscillations and spontaneous beating of these cells.19 Alterations of PAK1 activity may lead to the alteration of the dynamic regulatory processes and balance between kinase and phosphatase activity in cardiac cells and thus result in change in various ion channel activity. The latter may clearly be associated with arrhythmogenesis. It will be important to further investigate the regulatory role of PAK1 on ion channels in the heart under physiological and pathological conditions.
Regulation of Ca2+ handling
A recent study by us also presented a novel evidence for the regulatory role of PAK1 on Ca2+ handling in cardiac myocytes.20 Specifically, we examined the effects of constitutively active (CA)-PAK1 on [Ca2+]i transients and shortening of electrically stimulated ventricular myocytes under basal and β-adrenergic-stimulated conditions (Fig. 3). Under basal conditions, a change in the [Ca2+]i characteristics consisting of a significantly slower rate of Ca2+ decline in single adult rat ventricular myocytes expressing CA-PAK1 than in control cells, concurrent with longer time for cellular re-lengthening. There was no change in AdPAK1 cell sarcoplasmic reticulum (SR) Ca2+ content as assessed by caffeine, consistent with the lack of change in phospholamban (PLB) phosphorylation. This indicates CA-PAK1 slows the intracellular Ca2+ decay rate independently of PLB, although the exact mechanism remains to be determined. There was also a significant reduction in the amplitude of spontaneous Ca2+ sparks in CA-PAK1 compared with the control cells but no change in the frequency of occurrence. They suggested that lower amplitude may be in part due to the slight decrease in SR.

Figure 3. PAK1 regulation of Ca2+ handling. Line-scan images and whole cell-width line plots of fluo-4 fluorescence recorded parallel to the longitudinal axis of the cell from representative field-stimulated adult rat ventricular myocytes expressing LacZ [blue trace, (A)] or CA-PAK1 [red trace, (B)] under control conditions and during steady-state stimulation with 100 nM ISO. (C) Average intracellular Ca2+ concentration ([Ca2+]i) transient amplitudes in LacZ (875 ± 153 and 1,889 ± 286 nM without and with ISO, respectively) and CA-PAK1 (845 ± 99 and1,299 ± 138 nM without and with ISO, respectively) cells. (D) Average time constant for [Ca2+]i transient decay (τdecay) for LacZ (276 ± 8 and 149 ± 6 ms without and with ISO, respectively) and CA-PAK1 (308 ± 10 and 180 ± 10 ms without and with ISO, respectively) cells. Values are means ± SE *, statistically significant difference between LacZ and CA-PAK1. +, statistically significant difference between ISO and +ISO (adapted from ref. 20).
Under β-adrenergic stimulation by treatment of the cells with ISO, a significant blunting of ISO induced increases in [Ca2+]i transient amplitude, SR Ca2+ content and cell shortening was observed in CA-PAK1 myocytes. Propagation of spontaneous Ca2+ waves resulting from SR Ca2+ overload was significantly slower in CA-PAK1 myocytes. Thus PAK1 activity may alter ryanodine receptor (RyR) 2 gating via dephosphorylation by PP2A, but a full characterization is required. The level of PLB phosphorylation at Ser16 and Thr17 was not significantly different between CA-PAK1 and control myocytes, nor was cardiac troponin I (cTnI) phosphorylation at Ser23/24 significantly different.
Alternatively, the lower spark amplitude in CA-PAK1 myocytes may suggest that the RyR channel Po may be lower, so that fewer RyRs are gating within a cluster. The spatial spread of the sparks was reduced, but the temporal duration was unchanged, suggesting that fewer channels opened but the kinetics of opening and closing were the same. The slower propagation velocity of spontaneous Ca2+ waves indicates a resistance to activation of adjacent RyR clusters by cytosolic Ca2+ in CA-PAK1 cells.
Regulation of cardiac contractility
As well as a regulator of electrical activity and Ca2+ handling in myocytes, PAK1 is emerging as a potent modulator of cardiac contractility. Activation of PAK1 appears likely to alter the sarcomeric response to Ca2+ by modification of phosphorylation of contractile proteins. The phosphorylation state of myofilament regulatory proteins such as cTnI, cardiac TnT (cTnT), myosin binding protein C (MyBP-C) and tropomyosin are critical in regulation of contractility and are major determinants of myofilament Ca2+ sensitivity and cross-bridge cycling kinetics.21
In our previous study,2 we compared the phosphorylation levels of both cTnI and MyBP-C between the control and CA-PAK1 myocytes. Basal phosphorylation levels of both proteins were relatively high in the control myocytes. However, their phosphorylation levels in CA-PAK1 cells were significantly reduced. Measurement of Ca2+ tension relations in single myocytes demonstrated that this reduction in their phosphorylation was associated with the predicted increase in sensitivity to Ca2+. PAK1 is emerging as a potent regulator of cardiac contractility not only through PP2A-mediated dephosphorylation of cTnI and MyBP-C but may also through modification of the cellular proteins that regulate Ca2+ fluxes. As previously discussed, the potential targets for the PAK1-PP2A signaling module in intracellular Ca2+ regulation include L type-Ca2+ channels, RyR and PLB in control of sarcoplasmic reticulum Ca2+-ATPase (SERCA) activity.
Cardiac Protective Effects of PAK1
Anti-hypertrophy
Hypertrophy is defined as proliferation-independent cardiomyocyte growth, which bears some similarity to tumor growth. In response to acute or chronic insults the heart initially undergoes hypertrophic growth as an adaptive response to normalize ventricular wall stress; however, the capacity of this compensation is limited, prolonged application of deleterious stimuli eventually causes decompensated remodeling, such as chamber dilation, reduced contractile function, interstitial fibrosis and myocyte loss. These maladaptive effects subsequently lead to heart failure, a devastating condition currently affecting millions people world-wide. Since hypertrophy is generally regarded as a determinant of heart failure; studies to discover molecular and cellular mechanisms underlying hypertrophic remodeling and to identify potential therapeutic approaches for treating heart failure are of paramount importance.
To date, many oncogenes have been demonstrated to positively regulate cardiac hypertrophy. For example, mutations in the GTPase RAS can ultimately lead to oncogenesis.22 Cardiac overexpression of constitutively active RAS (H-v12-RAS) manifested ventricular hypertrophy.23 Similarly, aberrant activation of RAF is a step in the development of many types of cancers.24 Moreover, transgenic mice with cardiac-specific expression of a dominant-negative (DN) form of RAF-1 or cardiac-specific RAF-1 knockout mice demonstrate resistance to stresses inducing cardiac hypertrophy.25,26 In line with these observations, a recent study by Oceandy et al. revealed that the tumor suppressor RASSF1A (RAS-association domain family 1 isoform A) inhibits pressure overload-induced hypertrophy in mice.27 RASSF1 is known to block signal transmission from RAS to RAF. Of note, patients with Noonan or Costello syndrome in which a number of mutations occur in genes of the RAS signaling pathway develop both tumors and hypertrophic cardiomyopathy.28,29 Thus, above evidence indicates that the oncogenic signaling programs may be closely related to the programs that control the growth of post-mitotic adult cardiomyocytes.
Thus, we predicted that PAK1, which is activated by RAS and in turn activates RAF and is responsible for the growth of more than 70% of human cancers, may exert a similar function to that of the RAS-RAF pathway in facilitating cardiac hypertrophy. However, to our surprise, our recent study using primary cardiomyocytes and cardiomyocyte-specific PAK1 knockouts (PAK1cko) revealed an anti-hypertrophic function of PAK1.6 We found in cultured neonatal rat cardiomyocytes (NRCMs) with overexpression of constitutively active PAK1 attenuated phenylephrine-induced hypertrophic responses, whereas knockdown of PAK1 in NRCM caused a greater hypertrophy after phenylephrine stimulation. This anti-hypertrophic property of PAK1 was further substantiated by the study of PAK1cko mice. The PAK1cko mice manifested cardiac hypertrophy greater than controls with evident apoptosis following two weeks of pressure overload, and a rapid progression to heart failure after five weeks of load stress (Fig. 4). The PAK1cko mice also presented enhanced hypertrophy in response to angiotensin II infusion. We found that PAK1 activates another kinase called JNK (c-Jun N-terminal kinase) which in turn phosphorylates and inactivates a transcription factor called NFAT, which is essential for activation of the hypertrophic genes such as atrial natriuretic peptide (ANP), brain natriuretic peptide (BNP).30,31 Thus, the loss of PAK1 would lead to inactivation of this JNK signaling cascade and an enhancement of NFAT (Fig. 5). Furthermore, application of FTY720 (a synthetic analog structurally similar to sphingosine) induced PAK1 activation and restrained the development of cardiac hypertrophy in pressure overload stressed-wild type mice, but not in PAK1cko mice, suggesting the anti-hypertrophic effect of FTY720 was likely due to its function on activation of PAK1. Furthermore, a recent examination of PAK1 global knockout mice revealed a similar phenotype as that in PAK1cko mice.32 Absence of PAK1 in the whole heart rendered mice more capable of hypertrophic growth under a condition of ISO stimulation. Overall, these data demonstrate that PAK1 plays differing roles in promoting the malignant cell growth and in cardiac hypertrophy and PAK1 activators, which could be oncogenic, may thus be potential therapeutics for the cardiac hypertrophy.

Figure 4. Exacerbated cardiac hypertrophy in PAK1cko mice after 5 weeks transverse aortic constriction (TAC). (A) Heart weight/Tibia length (HW/TL) ratios of PAK1f/f and PAK1cko mice (upper panel). Morphometry demonstrates greater hypertrophy in PAK1cko-TAC mice (lower panel, scale bar: 2 mm). (B) Measurements of mean cross-sectional areas (upper panel) and hematoxylin and eosin staining of heart cross-sections (lower panel, scale bar: 20 µm) (adapted from ref. 6).

Figure 5. Regulation of cardiac excitation/contraction and hypertrophy by PAK1. In cardiomyocytes, PAK1 regulates activities of ion channels and myofilaments through PP2A activation. Cardiac hypertrophy induced under pathological conditions is suppressed by PAK1, which inactivates the transcription factor NFAT through JNK.
Protective effect in cardiac ischemic/reperfusion injury
Acute myocardial ischemia-reperfusion injury is the major pathophysiological manifestation of ischemic heart disease (i.e., coronary heart disease). Interestingly, the heart has ability to render itself resistant to ischemia/reperfusion injury, such phenomenon was first discovered by Murry et al. to describe as ischemic preconditioning.33 “Conditioning” the heart to tolerate the effects of acute ischemia-reperfusion injury can be initiated through inducing brief non-lethal episodes of ischemia and reperfusion to the heart either prior to, during, or even after an episode of sustained lethal myocardial ischemia-a phenomenon termed ischemic preconditioning (IPC), preconditioning or post-conditioning, respectively. To date, at least three different cardio-protective protein kinase programs have been proposed to be initiated by different G-protein coupled receptors including the adenosine, bradykinin, opioid, protease activated receptor 2 (PAR2) and sphingosine-1-phosphate (S1P) receptors, etc. The activation of these cell-surface receptors activates several intracellular signaling cascades including AKT and ERK1/2 components of the reperfusion injury salvage kinase (RISK) pathways, which then activate downstream targets that terminate at the level of the mitochondria.
S1P has been shown to be an important mediator (through S1P receptor signaling) of cardiac ischemic pre- and post-conditioning in both pharmacological and knockout animal studies.34,35 FTY720 demonstrated a protective effect on preventing organ ischemia/reperfusion (I/R) injury in animal models.8,36 In our recent study, we demonstrated that prevention of arrhythmias by FTY720 in I/R model was attributed to the activation of PAK1/AKT. FTY720 is able to stimulate the auto-phosphorylation of both PAK1 and AKT and their kinase activities in cardiomyocytes. AKT is a well-established regulator of myocardial growth and survival, contractile function and coronary angiogenesis.37 Using both gain- and loss-of-function approaches in vitro and in vivo, Mao et al. demonstrated that PAK1 alone is sufficient to activate AKT.5 The functional significance of this PAK1-AKT signaling is underscored by the observation that the pro-survival effect of PAK1 is diminished by AKT inhibition.5 The PAK1-conferred protection was blocked by the AKT inhibitor X (a selective Akt phosphorylation inhibitor), suggesting that the protective effect of PAK1 is mediated, at least in part, by AKT signaling. Our more recent study also demonstrated that PAK1 improves cardiac contractile function in the I/R model of PAK1 global KO mice through regulation of troponin-T and myosin light chain 2 phosphorylation.38
These findings demonstrate an important role for PAK1-AKT signaling in cardiomyocyte survival. Our further experiments on a mouse model of cardiac-specific deletion of PAK1 demonstrate a crucial role of PAK1 in cardiac protection against I/R injury. The recognition of the functional significance of PAK1 in preventing arrhythmogenesis associated with I/R injury may lead to the development of more potent therapeutics for treating I/R injury induced ventricular arrhythmias.
Acknowledgments
We would like to thank our co-authors who contributed to the original papers that produce Figures 1-4. We thank the Wellcome Trust (UK), Medical Research Council (UK), British Heart Foundation (UK) and National Institute of Health (USA) for funding our present research.
Glossary
Abbreviations:
- SAN
sino-atrial node
- ISO
isoproterenol
- PP2A
protein phosphatase 2A
- SR
sarcoplasmic reticulum
- RyR
ryanodine receptor
- PLB
phospholamban
- cTnI
cardiac troponin I
- cTnT
cardiac TnT
- MyBP-C
myosin binding protein C
- SERCA
sarcoplasmic reticulum Ca2+-ATPase
- GPCR
G protein-coupled receptor
- JNK
c-Jun N-terminal kinase
- NFAT
nuclear factor of activated T cell
- ANP
atrial natriuretic peptide
- BNP
brain natriuretic peptide
Footnotes
Previously published online: www.landesbioscience.com/journals/cellularlogistics/article/21497
References
- 1.Clerk A, Sugden PH. Activation of p21-activated protein kinase α (α PAK) by hyperosmotic shock in neonatal ventricular myocytes. FEBS Lett. 1997;403:23–5. doi: 10.1016/S0014-5793(97)00020-3. [DOI] [PubMed] [Google Scholar]
- 2.Ke Y, Wang L, Pyle WG, de Tombe PP, Solaro RJ. Intracellular localization and functional effects of P21-activated kinase-1 (Pak1) in cardiac myocytes. Circ Res. 2004;94:194–200. doi: 10.1161/01.RES.0000111522.02730.56. [DOI] [PubMed] [Google Scholar]
- 3.Ke Y, Lei M, Collins TP, Rakovic S, Mattick PA, Yamasaki M, et al. Regulation of L-type calcium channel and delayed rectifier potassium channel activity by p21-activated kinase-1 in guinea pig sinoatrial node pacemaker cells. Circ Res. 2007;100:1317–27. doi: 10.1161/01.RES.0000266742.51389.a4. [DOI] [PubMed] [Google Scholar]
- 4.Frazier DP, Wilson A, Dougherty CJ, Li H, Bishopric NH, Webster KA. PKC-alpha and TAK-1 are intermediates in the activation of c-Jun NH2-terminal kinase by hypoxia-reoxygenation. Am J Physiol Heart Circ Physiol. 2007;292:H1675–84. doi: 10.1152/ajpheart.01132.2006. [DOI] [PubMed] [Google Scholar]
- 5.Mao K, Kobayashi S, Jaffer ZM, Huang Y, Volden P, Chernoff J, et al. Regulation of Akt/PKB activity by P21-activated kinase in cardiomyocytes. J Mol Cell Cardiol. 2008;44:429–34. doi: 10.1016/j.yjmcc.2007.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Liu W, Zi M, Naumann R, Ulm S, Jin J, Taglieri DM, et al. Pak1 as a novel therapeutic target for antihypertrophic treatment in the heart. Circulation. 2011;124:2702–15. doi: 10.1161/CIRCULATIONAHA.111.048785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ke Y, Solaro RJ. Use of a decoy peptide to purify p21 activated kinase-1 in cardiac muscle and identification of ceramide-related activation. Biologics. 2008;2:903–9. doi: 10.2147/BTT.S3870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Egom EE, Ke Y, Musa H, Mohamed TM, Wang T, Cartwright E, et al. FTY720 prevents ischemia/reperfusion injury-associated arrhythmias in an ex vivo rat heart model via activation of Pak1/Akt signaling. J Mol Cell Cardiol. 2010;48:406–14. doi: 10.1016/j.yjmcc.2009.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Roig J, Tuazon PT, Traugh JA. Cdc42-independent activation and translocation of the cytostatic p21-activated protein kinase gamma-PAK by sphingosine. FEBS Lett. 2001;507:195–9. doi: 10.1016/S0014-5793(01)02965-9. [DOI] [PubMed] [Google Scholar]
- 10.Zenke FT, King CC, Bohl BP, Bokoch GM. Identification of a central phosphorylation site in p21-activated kinase regulating autoinhibition and kinase activity. J Biol Chem. 1999;274:32565–73. doi: 10.1074/jbc.274.46.32565. [DOI] [PubMed] [Google Scholar]
- 11.Chong C, Tan L, Lim L, Manser E. The mechanism of PAK activation. Autophosphorylation events in both regulatory and kinase domains control activity. J Biol Chem. 2001;276:17347–53. doi: 10.1074/jbc.M009316200. [DOI] [PubMed] [Google Scholar]
- 12.Kozma R, Ahmed S, Best A, Lim L. The Ras-related protein Cdc42Hs and bradykinin promote formation of peripheral actin microspikes and filopodia in Swiss 3T3 fibroblasts. Mol Cell Biol. 1995;15:1942–52. doi: 10.1128/mcb.15.4.1942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kozma R, Sarner S, Ahmed S, Lim L. Rho family GTPases and neuronal growth cone remodelling: relationship between increased complexity induced by Cdc42Hs, Rac1, and acetylcholine and collapse induced by RhoA and lysophosphatidic acid. Mol Cell Biol. 1997;17:1201–11. doi: 10.1128/mcb.17.3.1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ke Y, Sheehan KA, Egom EE, Lei M, Solaro RJ. Novel bradykinin signaling in adult rat cardiac myocytes through activation of p21-activated kinase. Am J Physiol Heart Circ Physiol. 2010;298:H1283–9. doi: 10.1152/ajpheart.01070.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Manser E, Huang HY, Loo TH, Chen XQ, Dong JM, Leung T, et al. Expression of constitutively active alpha-PAK reveals effects of the kinase on actin and focal complexes. Mol Cell Biol. 1997;17:1129–43. doi: 10.1128/mcb.17.3.1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Woolfolk EA, Eguchi S, Ohtsu H, Nakashima H, Ueno H, Gerthoffer WT, et al. Angiotensin II-induced activation of p21-activated kinase 1 requires Ca2+ and protein kinase Cdelta in vascular smooth muscle cells. Am J Physiol Cell Physiol. 2005;289:C1286–94. doi: 10.1152/ajpcell.00448.2004. [DOI] [PubMed] [Google Scholar]
- 17.Cheng G, Kasiganesan H, Baicu CF, Wallenborn JG, Kuppuswamy D, Cooper G., 4th Cytoskeletal role in protection of the failing heart by β-adrenergic blockade. Am J Physiol Heart Circ Physiol. 2012;302:H675–87. doi: 10.1152/ajpheart.00867.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ai X, Jiang A, Ke Y, Solaro RJ, Pogwizd SM. Enhanced activation of p21-activated kinase 1 in heart failure contributes to dephosphorylation of connexin 43. Cardiovasc Res. 2011;92:106–14. doi: 10.1093/cvr/cvr163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Vinogradova TM, Maltsev VA, Bogdanov KY, Lyashkov AE, Lakatta EG. Rhythmic Ca2+ oscillations drive sinoatrial nodal cell pacemaker function to make the heart tick. Ann N Y Acad Sci. 2005;1047:138–56. doi: 10.1196/annals.1341.013. [DOI] [PubMed] [Google Scholar]
- 20.Sheehan KA, Ke Y, Wolska BM, Solaro RJ. Expression of active p21-activated kinase-1 induces Ca2+ flux modification with altered regulatory protein phosphorylation in cardiac myocytes. Am J Physiol Cell Physiol. 2009;296:C47–58. doi: 10.1152/ajpcell.00012.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Solaro RJ. Multiplex kinase signaling modifies cardiac function at the level of sarcomeric proteins. J Biol Chem. 2008;283:26829–33. doi: 10.1074/jbc.R800037200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bar-Sagi D, Hall A. Ras and Rho GTPases: a family reunion. Cell. 2000;103:227–38. doi: 10.1016/S0092-8674(00)00115-X. [DOI] [PubMed] [Google Scholar]
- 23.Hunter JJ, Tanaka N, Rockman HA, Ross J, Jr., Chien KR. Ventricular expression of a MLC-2v-ras fusion gene induces cardiac hypertrophy and selective diastolic dysfunction in transgenic mice. J Biol Chem. 1995;270:23173–8. doi: 10.1074/jbc.270.39.23173. [DOI] [PubMed] [Google Scholar]
- 24.Davies H, Bignell GR, Cox C, Stephens P, Edkins S, Clegg S, et al. Mutations of the BRAF gene in human cancer. Nature. 2002;417:949–54. doi: 10.1038/nature00766. [DOI] [PubMed] [Google Scholar]
- 25.Harris IS, Zhang S, Treskov I, Kovacs A, Weinheimer C, Muslin AJ. Raf-1 kinase is required for cardiac hypertrophy and cardiomyocyte survival in response to pressure overload. Circulation. 2004;110:718–23. doi: 10.1161/01.CIR.0000138190.50127.6A. [DOI] [PubMed] [Google Scholar]
- 26.Yamaguchi O, Watanabe T, Nishida K, Kashiwase K, Higuchi Y, Takeda T, et al. Cardiac-specific disruption of the c-raf-1 gene induces cardiac dysfunction and apoptosis. J Clin Invest. 2004;114:937–43. doi: 10.1172/JCI20317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Oceandy D, Pickard A, Prehar S, Zi M, Mohamed TM, Stanley PJ, et al. Tumor suppressor Ras-association domain family 1 isoform A is a novel regulator of cardiac hypertrophy. Circulation. 2009;120:607–16. doi: 10.1161/CIRCULATIONAHA.109.868554. [DOI] [PubMed] [Google Scholar]
- 28.Tartaglia M, Mehler EL, Goldberg R, Zampino G, Brunner HG, Kremer H, et al. Mutations in PTPN11, encoding the protein tyrosine phosphatase SHP-2, cause Noonan syndrome. Nat Genet. 2001;29:465–8. doi: 10.1038/ng772. [DOI] [PubMed] [Google Scholar]
- 29.Aoki Y, Niihori T, Kawame H, Kurosawa K, Ohashi H, Tanaka Y, et al. Germline mutations in HRAS proto-oncogene cause Costello syndrome. Nat Genet. 2005;37:1038–40. doi: 10.1038/ng1641. [DOI] [PubMed] [Google Scholar]
- 30.Liang Q, Bueno OF, Wilkins BJ, Kuan CY, Xia Y, Molkentin JD. c-Jun N-terminal kinases (JNK) antagonize cardiac growth through cross-talk with calcineurin-NFAT signaling. EMBO J. 2003;22:5079–89. doi: 10.1093/emboj/cdg474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Molkentin JD. Calcineurin-NFAT signaling regulates the cardiac hypertrophic response in coordination with the MAPKs. Cardiovasc Res. 2004;63:467–75. doi: 10.1016/j.cardiores.2004.01.021. [DOI] [PubMed] [Google Scholar]
- 32.Taglieri DM, Monasky MM, Chernoff J, Wang X, Lei M, Ke Y, et al. Ablation of p21-activated kinase-1 in mice promotes isoproterenol-induced cardiac hypertrophy in association with activation of Erk1/2 and inhibition of protein phosphatase 2A. J Mol Cell Cardiol. 2011;51:988–96. doi: 10.1016/j.yjmcc.2011.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Murry CE, Jennings RB, Reimer KA. Preconditioning with ischemia: a delay of lethal cell injury in ischemic myocardium. Circulation. 1986;74:1124–36. doi: 10.1161/01.CIR.74.5.1124. [DOI] [PubMed] [Google Scholar]
- 34.Jin ZQ, Zhang J, Huang Y, Hoover HE, Vessey DA, Karliner JS. A sphingosine kinase 1 mutation sensitizes the myocardium to ischemia/reperfusion injury. Cardiovasc Res. 2007;76:41–50. doi: 10.1016/j.cardiores.2007.05.029. [DOI] [PubMed] [Google Scholar]
- 35.Lecour S, Smith RM, Woodward B, Opie LH, Rochette L, Sack MN. Identification of a novel role for sphingolipid signaling in TNF alpha and ischemic preconditioning mediated cardioprotection. J Mol Cell Cardiol. 2002;34:509–18. doi: 10.1006/jmcc.2002.1533. [DOI] [PubMed] [Google Scholar]
- 36.Hofmann U, Burkard N, Vogt C, Thoma A, Frantz S, Ertl G, et al. Protective effects of sphingosine-1-phosphate receptor agonist treatment after myocardial ischaemia-reperfusion. Cardiovasc Res. 2009;83:285–93. doi: 10.1093/cvr/cvp137. [DOI] [PubMed] [Google Scholar]
- 37.Shiojima I, Walsh K. Regulation of cardiac growth and coronary angiogenesis by the Akt/PKB signaling pathway. Genes Dev. 2006;20:3347–65. doi: 10.1101/gad.1492806. [DOI] [PubMed] [Google Scholar]
- 38.Monasky MM, Taglieri DM, Patel BG, Chernoff J, Wolska BM, Ke Y, et al. p21-activated kinase improves cardiac contractility during ischemia-reperfusion concomitant with changes in troponin-T and myosin light chain 2 phosphorylation. Am J Physiol Heart Circ Physiol. 2012;302:H224–30. doi: 10.1152/ajpheart.00612.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]