

Abstract
Background
Experimental autoimmune encephalomyelitis (EAE), the best available model of multiple sclerosis, can be induced in different animal strains using immunization with central nervous system antigens. EAE is associated with inflammation and demyelination of the nervous system. Micro-array can be used to investigate gene expression and biological pathways that are altered during disease. There are few studies of the changes in gene expression in EAE, and these have mostly been done in a chronic mouse EAE model. EAE induced in the Lewis with myelin basic protein (MBP-EAE) is well characterised, making it an ideal candidate for the analysis of gene expression in this disease model.
Methodology/Principal Findings
MBP-EAE was induced in female Lewis rats by inoculation with MBP and adjuvants. Total RNA was extracted from the spinal cords and used for micro-array analysis using AffimetrixGeneChip Rat Exon 1.0 ST Arrays. Gene expression in the spinal cords was compared between healthy female rats and female rats with MBP-EAE. Gene expression in the spinal cord of rats with MBP-EAE differed from that in the spinal cord of normal rats, and there was regulation of pathways involved with immune function and nervous system function. For selected genes the change in expression was confirmed with real-time PCR.
Conclusions/Significance
EAE leads to modulation of gene expression in the spinal cord. We have identified the genes that are most significantly regulated in MBP-EAE in the Lewis rat and produced a profile of gene expression in the spinal cord at the peak of disease.
Introduction
On the basis of pathological findings and genetic studies that show that the majority of genes that predispose to multiple sclerosis (MS) are associated with T cell activation [1], MS is thought to be primarily a T-cell mediated autoimmune disease. B cells and antibodies are also thought to play a role in pathogenesis [2]. The symptoms of MS are due to demyelination and axonal loss, and possibly to temporary loss of nerve conduction due to circulating factors [3]. Experimental autoimmune encephalomyelitis (EAE) is the best available animal model of MS, and can be induced by inoculation of susceptible animals with a range of central nervous system (CNS) antigens.
EAE was first induced by inoculation of whole brain tissue [4]; [5]. Myelin basic protein (MBP) was the first encephalitogenic protein to be studied in detail. Later studies have used myelin proteolipid protein (PLP) [6]; [7] and myelin oligodendrocyte glycoprotein (MOG) [8] to induce EAE. The susceptibility to EAE varies among animal strains and with the antigen used to induce the disease. There have been studies of the genetic loci that contribute to this susceptibility. The most important genes are the MHC class II, but other genes have been implicated, including other immune related genes [9]–[11]. Studies have also identified genetic loci that contribute to different EAE traits such as the severity of disease, maximum clinical score and the presence of demyelination [9]; [10].
In this study we have investigated MBP-EAE, which is characterized by weight loss and ascending paralysis [12] followed by spontaneous recovery. It has been clearly shown that recovery from weakness is associated with restoration of electrical conduction [13]. The pathology of MBP-EAE involves infiltration of the spinal cord and nerve roots with inflammatory cells [14], followed by demyelination of axons. In MBP-EAE there is apoptosis within the spinal cord [15]; [16] and there is also axonal damage [17]. The metabolic processes that cause damage in EAE include glutamate toxicity [18] and dysregulation of ion channels including calcium channels [19]. Because of the detailed knowledge of the pathogenesis of this EAE model, it is of interest to determine how gene expression correlates with the known features of this disease. The advantages of using EAE for studies of gene expression in the CNS are that tissue can be easily obtained at the peak of disease, without a significant post-mortem delay, allowing good preservation of RNA for expression studies.
Because of the extensive characterization of MPB-EAE in the Lewis rat, it is an excellent candidate for studies aimed at defining the gene expression profile of the disease. There have been no previous gene array studies of MBP-EAE in the Lewis rat; however, the expression profile of selected cytokines, chemokines, adhesion molecules, cell markers, matrix metalloproteinases and other genes relevant to disease etiology have been examined in this model by real-time PCR (RT-PCR) [20]. There have been micro-array studies of gene expression in MBP-EAE in SJL/J mice [21] and of the effects of estrogen treatment on gene expression in MBP-EAE in BV8S2/AV4 double transgenic mice [22]. There have also been microarray studies of EAE induced with myelin-oligodendrocyte glycoprotein (MOG) in the spinal cords of mice [23] and rats [24]. The present study was carried out to investigate changes in gene expression in MBP-EAE compared to control Lewis rats, and found that there were significant differences from controls.
Results
a) Differential Gene Expression in MBP Induced EAE in the Lewis Rat
Rats were inoculated with MBP to induce EAE. The typical clinical course of this disease is shown in Figure 1A, which demonstrates that disease is generally monophasic with spontaneous recovery. Rats were sacrificed on day 13 post injection which corresponds approximately with the peak of clinical disease in our model. The course of disease of the rats that were used in the micro-array study is shown in Figure 1B.
Figure 1. Clinical course of EAE. A.

The figure shows the results of a clinical course experiment (n = 10) in which female Lewis rats (10–12 wks old) were injected with 50 µg Guinea Pig MBP emulsified in incomplete Freund’s Adjuvant containing 4 mg/ml heat inactivated M.Butyricium. The peak of disease generally occurs between 12 and 14 days post injection and resolves, usually within 7–10 days later. Mild relapses occur in a few animals between 28 and 40 dpi. B. Spinal cords used to prepare RNA for microarray analysis were collected on day 13 when the disease course was either at its peak or in the phase of very early resolution. Clinical course scores were recorded using a 12 point scale [43].
RNA quality analysis was carried out on the BioRad Experion automated electrophoresis system. All preparations used in both assays had values of values of >9.5 for RNA quality indicator (RQI) [25]. This is illustrated in Figures S1 and S2. After array hybridization with the Affimetrix Rat Exon 1.0 ST array, which contains probe sets for 27,324 genes, data was uploaded for analysis with the Partek genomics suite. Transcripts from 8,793 genes were detected. Of these, we identified 2,350 genes that were significantly regulated between the healthy rats and rats with MBP-EAE (t test, (p-value <0.05). The overall gene expression changes in MBP induced EAE are illustrated in the volcano plot in Figure 2, which shows a large number of genes that were highly regulated compared to healthy controls.
Figure 2. Volcano plot.

This shows the significance (y axis) versus the fold change (x axis) for genes in rats with EAE compared to healthy controls.
Of the 2,350 differentially regulated genes, 2,265 mapped to known locations in the Partek data-base and the remainder were unmapped. Of these mapped genes, 1,665 were mapped to named genes. Of the 1,665 differentially regulated genes, 998 were found to be down-regulated and 667 were up-regulated. These are listed in Tables S1 and S2.
The 2,530 genes that were differentially regulated were up-loaded into the Ingenuity Pathway analysis software package (IPA) (Ingenuity ® Systems, www.ingenuity.com ) for functional and pathway analysis. After filtering to exclude genes that were not of relevance to the nervous system, we produced an annotated set of 1,190 differentially regulated genes. These genes were used for our further analysis of functions, pathways and upstream regulators.
To produce a list of the genes that are most significantly and highly regulated in MBP-EAE, we used a False Discovery Rate (FDR) [26] of 5% to further refine the data-set. This produced a list of the most significantly regulated genes (p values ≤0.0045). These are shown in Table 1, which lists the most significantly up-regulated genes (fold-change limit + or –4.0) and Table 2 which contains the most significantly down-regulated (fold change limit + or –2.0) transcripts in the data-set. A number of these genes mapped to loci that predispose to EAE in the rat (http://rgd.mcw.edu), or have been associated with one or more of the commonly used EAE models, as shown in Tables 1 and 2.
Table 1. Genes most significantly up-regulated at the peak of Clinical disease in MBP induced EAE in the Lewis Rat.
| Entrez gene symbol | Entrez Gene Name | Fold change | p-value | QTL | Function | Previous association with EAE or Inflammation in the CNS | Ref |
| Cd74 | CD74 molecule, major histocompatibility complex, class II invariant chain | 139.3 | 3.35E–07 | EAEy EAEz | Antigen processing and presentation | Cd74 maps to a gene regions associated with EAE in the DA rat. | [45] |
| Psmb9 | proteasome (prosome, macropain) subunit, beta type, 9 (large multifunctional peptidase 2) | 8.2 | 8.80E–10 | EAE1 | Antigen processing and presentation | Psmb9 maps to a gene region associated with EAE in the DA rat | [45] |
| Rt1-a1 | rat major histocompatibility complex, class I | 6.5 | 2.06E–05 | – | Antigen processing and presentation | – | – |
| Rt1-dmb | rat major histocompatibility complex, class II, RTI-DM β chain | 9.2 | 2.37E–05 | – | Antigen processing and presentation | – | – |
| Rt1-Ba | rat major histocompatibility complex, class II, RT1-B α chain | 32.7 | 1.33E–06 | – | Antigen processing and presentation | Rt1-Ba involved in antigen presentation in MBP induced EAE in the Lewis rat. | [46]; [47] |
| Rt1-Da | rat major histocompatibility complex, class II, RT1-D α chain | 116.7 | 4.04E–06 | – | Antigen processing and presentation | Rt1-Da involved in antigen presentation in MBP induced EAE in the Lewis rat. | [46]; [47] |
| Rt1-Db1 | rat major histocompatibility complex, class II, RT1-Dβ 1 chain | 8.3 | 7.54E–05 | – | Antigen processing and presentation | Rt1-Db1 involved in antigen presentation in MBP induced EAE in the Lewis rat. | [46]; [47] |
| Tap1 | transporter 1, ATP-binding cassette, sub-family B (MDR/TAP) | 4.3 | 1.08E–06 | EAE1 | Antigen processing and presentation | Tap1 maps to a gene region associated with EAE in the DA rat | [45] |
| Timp1 | TIMP metallopeptidase inhibitor 1 | 8.0 | 2.86E–05 | Antigen processing and presentation | Timp1 has been previously identified as up-regulated in mouse models of EAE. | [48] | |
| Casp1 | caspase 1, apoptosis-related cysteine peptidase (interleukin 1, beta, convertase) | 6.3 | 4.48E–06 | – | Apoptosis and cell death | Caspase 1 levels correlate with clinical disease course and the transcription rate of pro-inflammatory cytokines. | [32]; [33] |
| Casp4 | caspase 4, apoptosis-related cysteine peptidase | 7.1 | 2.52E–06 | – | Apoptosis and cell death | Oligodendrocyte cell death in MOG induced EAE in mice was mediated by a pathway that involves Casp4. | [49] |
| Casp8 | caspase 8, apoptosis-related cysteine peptidase | 4.3 | 1.93E–05 | – | Apoptosis and cell death | Casp8 involved in neuronal cell death by apoptosis in EAE in the Lewis rat. | [50] |
| Gda | guanine deaminase | 15.1 | 5.82E–06 | – | Apoptosis and cell death | – | – |
| Irf1 | interferon regulatory factor 1 | 8.6 | 5.89E–06 | EAE3 EAE18 | Apoptosis and cell death | Irf1 maps to a gene locus conferring resistance to MOG induced EAE in the rat. | [51] |
| Irgm | immunity-related GTPase family, M | 14.0 | 1.93E–05 | EAE3 EAE18 | Apoptosis and cell death | Irgm maps to a gene region conferring resistance to MOG induced EAE in the rat. | [51] |
| Lcn2 | lipocalin 2 | 75.2 | 5.72E–06 | – | Apoptosis and cell death | Lcn2 has previously been identified as upregulated in rat and mouse models of EAE. | [28]; [29]; [52] |
| Tlr2 | toll-like receptor 2 | 6.0 | 4.77E–05 | – | Apoptosis and cell death | Tlr2 is upregulated in active lesions of MS and MOG induced EAE in the DA rat. | [35] |
| Tlr4 | Tlr4 | 4.9 | 8.16E–04 | – | Apoptosis and cell death | Tlr4 is upregulated in active lesions of MS and MOG induced EAE in the DA rat. | [34]; [35] |
| Trpm2 | transient receptor potential cation channel, subfamily M, member 2 | 4.6 | 8.25E–05 | – | Apoptosis and cell death | – | – |
| Ubd | ubiquitin D | 4.3 | 7.31E–04 | – | Apoptosis and cell death | – | – |
| Anxa3 | annexin A3 | 8.1 | 5.01E–06 | – | Cellular movement | – | [52] |
| Ccl7 | chemokine (C-C motif) ligand 7 | 6.7 | 2.65E–04 | EAE18b | Cellular movement | Ccl7 maps to a gene region associated with MOG EAE in the rat | [53] |
| Ccr5 | chemokine (C-C motif) receptor 5 | 6.5 | 2.25E–05 | – | Cellular movement | Ccr5 was upregulated in the spinal cords of rats with MOG induced EAE | [54] |
| Cx3cr1 | chemokine (C-X3-C motif) receptor 1 | 5.6 | 1.19E–03 | – | Cellular movement | Cx3cr1 is involved in neuroinflammation and upregulated in phagocytic cells in the rat brain in MOG induced EAE. | [55]; [56] |
| Cxcl9 | chemokine (C-X-C motif) ligand 9 | 113.50 | 2.51E–06 | – | Cellular movement | Cxc19 is involved in neuroinflammation. | [55] |
| Cxcl16 | chemokine (C-X-C motif) ligand 16 | 20.1 | 5.28E–06 | EAE18 EAE18a | Cellular movement | Cxcl16 maps to gene region previously associated with a number of inflammatory diseases including MOG induced EAE in the DA rat. | [53]; |
| Fn1 | fibronectin 1 | 5.3 | 1.35E–03 | – | Cellular movement | – | – |
| Apobec1 | apolipoprotein B mRNA editing enzyme, catalytic polypeptide 1 | 46.8 | 5.61E–05 | EAE20, EAE22 | Cellular growth and proliferation | Apobec1 maps to gene regions previously associated with EAE in the mouse. | [48] |
| Gpnmb | glycoprotein (transmembrane) nmb | 16.1 | 2.89E–05 | – | Cellular growth and proliferation | – | – |
| Grn | Granulin | 6.0 | 1.61E–05 | – | Cellular growth and proliferation | Grn enhances the proliferation of mouse neural progenitor cells in culture | [58] |
| IL2ra | interleukin 2 receptor, alpha | 7.6 | 1.94E–04 | – | Cellular growth and proliferation | MOG Induced EAE susceptible DA rats had higher IL-2Rα, expression in naïve T cells compared to the resistant PVG rats. | [59] |
| IL2rg | interleukin 2 receptor, gamma | 34.7 | 3.46E–07 | – | Cellular growth and proliferation | Common cytokine receptor gamma-chain (gamma(c)) family cytokines have crucial roles in the development, proliferation, survival and differentiation of multiple cell lineages of both the innate and adaptive immune systems. | [60] |
| Klrk1 | killer cell lectin-like receptor subfamily K, member 1 | 13.5 | 2.66E–05 | – | Cellular growth and proliferation | – | – |
| Laptm5 | lysosomal protein transmembrane 5 | 9.2 | 5.08E–04 | – | Cellular growth and proliferation | – | – |
| Stat1 | signal transducer and activator of transcription 1, 91 kDa | 12.5 | 2.11E–05 | – | Cellular growth and proliferation | Expression of Stat1 increases to the recovery stage of MBP induced EAE in the Lewis rat. | [61] |
| Irf8 | interferon regulatory factor 8 | 4.8 | 1.37E–05 | – | Cellular development and differentiation | – | – |
| Arhgdib | rho GDP dissociation inhibitor (GDI) beta | 8.4 | 1.18E–04 | EAE22 | Cell- cell signalling and interaction | Arhgdib maps to gene region previously associated with a number of inflammatory diseases including MOG induced EAE in the DA rat. | [57] |
| Emr1 | egf-like module containing, mucin-like, hormone receptor-like 1 | 16.9 | 1.60E–06 | – | Cell- cell signalling and interaction | – | – |
| Prkcd | protein kinase C, delta | 4.1 | 2.49E–04 | – | Cell- cell signalling and interaction | – | – |
| Ptafr | platelet-activating factor receptor | 6.5 | 3.74E–05 | – | Cell- cell signalling and interaction | – | – |
| Rac2 | ras-related C3 botulinum toxin substrate 2 (rho family, small GTP binding protein Rac2) | 6.5 | 2.31E–06 | – | Cell- cell signalling and interaction | – | – |
| Tyrobp | TYRO protein tyrosine kinase binding protein | 7.3 | 2.02E–04 | – | Cell- cell signalling and interaction | Tyrobp maps to gene regions previously associated with EAE in the mouse. | [48] |
| C3 | complement component 3 | 26.1 | 4.58E–05 | – | Complement activation | C3 is required for full development of EAE in MOG induced EAE in mice | [62] |
| C1qa | complement component 1, q subcomponent, A chain | 5.8 | 1.27E–04 | – | Complement activation | The complement system has previously been implicated in the development of EAE, C1qa has been previously identified as up-regulated in mouse models of EAE. | [31]; [48] |
| C1qb | complement component 1, q subcomponent, B chain | 4.6 | 1.06E–03 | – | Complement activation | The complement system has previously been implicated in the development of EAE C1qb has been previously identified as up-regulated in mouse models of EAE | [31]; [48] |
| C1qc | complement component 1, q subcomponent, C chain | 10.6 | 8.76E–05 | – | Complement activation | The complement system has previously been implicated in the development of EAE C1qc has been previously identified as upregulated in mouse models of EAE | [31]; [48] |
| C3ar1 | complement component 3a receptor 1 | 4.1 | 1.43E–03 | EAE20 EAE22 | Complement activation | C3ar1 maps to gene region previously associated with a number of inflammatory diseases including MOG induced EAE in the DA rat. | [57] |
| Cfb | complement factor B | 4.7 | 2.89E–04 | – | Complement activation | – | |
| Serping1 | serpin peptidase inhibitor, clade G (C1 inhibitor), member 1 | 20.5 | 7.24E–05 | – | Complement activation | – | – |
| Soat1 | sterol O-acyltransferase 1 | 4.8 | 1.72E–05 | EAE17 | Complement activation | Soat1 maps to gene regions associated with MOG EAE in the rat, | [63] |
| Btk | Bruton agammaglobulinemia tyrosine kinase | 5.5 | 1.80E–05 | – | Inflammatory Response | – | – |
| Cd4 | CD4 molecule | 12.5 | 7.05E–07 | EAE20 EAE22 | Inflammatory Response | Cd4 maps to gene region previously associated with a number of inflammatory diseases including MOG induced EAE in the DA rat, CD4 expression on microglia correlates with resolution of disease in MBP induced EAE in the Lewis rat. | [57]; [64] |
| Cd14 | CD14 molecule | 4.4 | 4.44E–03 | EAEz | Inflammatory Response | Cd14 maps to a gene region associated with EAE in the DA rat. | [45] |
| Cd86 | CD86 molecule | 10.5 | 7.23E–06 | – | Inflammatory Response | B7/CD86 costimulation is critical for activation and expansion of MOG Fcgr2 areactive T cells in a mouse model of EAE. | [65] |
| Chi3l1 | chitinase 3-like 1 (cartilage glycoprotein-39) | 9.1 | 7.66E–04 | EAE14 EAE17 | Inflammatory Response | Chi3l1 maps to gene regions associated with MOG EAE in the rat, and has previously been identified as upregulated in mouse models of EAE. | [63]; [66] [48] |
| Fcgr2a | Fc fragment of IgG, low affinity IIa, receptor (CD32) | 9.5 | 2.69E–04 | EAE17 | Inflammatory Response | Fcgr2a maps to gene regions associated with MOG EAE in the rat. | [63] |
| Fcgr2b | Fc fragment of IgG, low affinity IIb, receptor (CD32) | 11.6 | 6.35E–06 | EAE17 | Inflammatory Response | Fcgr2b maps to gene regions associated with MOG EAE in the rat. | [63] |
| F10 | coagulation factor X | 5.4 | 4.64E–04 | – | Inflammatory Response | – | – |
| Il1b | interleukin 1, beta | 4.8 | 1.37E–03 | – | Inflammatory Response | Il1b is expressed by activated microglia in MOG induced EAE in the mouse. | [30] |
| IL1rn | interleukin 1 receptor antagonist | 7.5 | 7.42E–04 | – | Inflammatory Response | IL1rn modulates a variety of interleukin 1 related immune and inflammatory responses. | [67] |
| Lyn | v-yes-1 Yamaguchi sarcoma viral related oncogene homolog | 9.4 | 6.06E–08 | – | Inflammatory Response | Severity of EAE is increased in lyn−/− mice compared to WT controls. | [68] |
| Ptprc | protein tyrosine phosphatase, receptor type, C | 17.0 | 2.78E–07 | EAE14 EAE17 | Inflammatory Response | Ptprc maps to gene regions associated with MOG EAE in the rat. | [63]; [66] |
| Vav1 | vav 1 guanine nucleotide exchange factor | 4.0 | 4.36E–05 | – | Inflammatory Response | – | – |
| Pygl | phosphorylase, glycogen, liver | 8.0 | 4.28E–06 | – | Other metabolic processes | – | – |
| Cp | ceruloplasmin (ferroxidase) | 6.8 | 5.35E–05 | – | Other metabolic processes | CP Increases with oedema and has previously been identified as upregulated in mouse models of EAE | [48]; [69] |
Table 2. Genes most significantly down-regulated at the peak of Clinical disease in MBP induced EAE in the Lewis Rat.
| Gene Symbol | Entrez Gene name | Fold change | p-value | QTL | Function | Previous association with EAE or Inflammation in the CNS | Ref |
| Idi1 | isopentenyl-diphosphate delta isomerase 1 | −3.4 | 5.34E–04 | – | Cholesterol Biosynthesis | – | – |
| Hmgcs1 | 3-hydroxy-3-methylglutaryl-CoA synthase 1 | −3.2 | 1.28E–04 | – | Cholesterol Biosynthesis | – | – |
| Cyp51a1 | cytochrome P450, family 51, subfamily A, polypeptide 1 | −2.9 | 2.31E–03 | – | Cholesterol Biosynthesis | Cytochhrome P450 dependent enzyme activities in the brain were reduced in both LPS induced and EAE models of inflammation in astrocytes in LPS induced inflammation. | [70] |
| Msmo1 | methylsterol monooxygenase 1 | −2.4 | 4.67E–04 | – | Cholesterol Biosynthesis | – | – |
| Sc5dl | sterol-C5-desaturase | −2.2 | 1.07E–03 | – | Cholesterol Biosynthesis | – | – |
| Sqle | squalene epoxidase | −2.1 | 2.58E–03 | – | Cholesterol Biosynthesis | – | – |
| Npy | neuropeptide Y | −2.1 | 3.00E–04 | EAE25 | Cellular growth and proliferation | Npy maps to a gene regipon that regulates EAE and suppresses EAE in the rat. | [71]; [72] |
| Ptn | pleiotrophin | −2.1 | 7.33E–04 | – | Cellular growth and proliferation | Ptn found to be up-regulated in recovery from EAE in the rat. | [73] |
| Sept3 | septin 3 | −2.1 | 5.28E–04 | – | Cellular growth and proliferation | – | – |
| Gjb1 | gap junction protein, beta 1 | −2.0 | 1.08E–03 | – | Cell-cell signalling and Interaction | Gjb1 (Connexin32) null mice develop demyelinating peripheral neuropathy | [74] |
| Grb14 | growth factor receptor-bound protein 14 | −2.1 | 5.55E–04 | Neuinf1 | Cell-cell signalling and Interaction | Grb14 maps to a gene region regulating the inflammatory microglial response in the rat. | [75] |
| Hnmt | histamine N-methyltransferase | −2.8 | 6.78E–04 | – | Cell-cell signalling and Interaction | – | – |
| Il1rapl1 | interleukin 1 receptor accessory protein-like 1 | −2.5 | 2.29E–04 | – | Cell-cell signalling and Interaction | – | – |
| Nlgn1 | neuroligin 1 | −2.0 | 9.21E–04 | – | Cell-cell signalling and Interaction | – | |
| Ppp1r14c | protein phosphatase 1, regulatory (inhibitor) subunit 14C | −2.2 | 1.74E–03 | Neuinf 4 | Cell-cell signalling and Interaction | Ppp1r14c maps to a gene region regulating the inflammatory microglial response in the rat. | [75] |
| Trhr | thyrotropin-releasing hormone receptor | −2.5 | 1.82E–03 | – | Cell-cell signalling and Interaction | – | – |
| Ugt8 | UDP glycosyltransferase 8 | −2.2 | 3.64E–03 | – | Myelination | – | – |
| Gal | -galanin prepropeptide | −2.2 | 1.20E–04 | EAE6 | Inflammatory Response | Gal maps to a gene region in the rat associated with models of rheumatoid arthritis and EAE. | [45] |
| Galnt13 | -UDP-N-acetyl-alpha-D-galactosamine:polypeptide N-acetylgalactosaminyltransferase 13 (GalNAc-T13) | −2.8 | 1.66E–03 | – | Post translational protein modification | – | – |
b) Quantitative Real-Time PCR (rt PCR) Validation of Micro-array Data
To confirm our micro-array findings, expression levels of a the genes caspase 1 (Casp1), oligodendrocyte myelin glycoprotein (OMG), alpha subunit voltage-gated sodium channel type 1 (Scn1a), Fas (TNF receptor superfamily member 6) and superoxide dismutase 2, mitochondrial (Sod2) were validated using RT-PCR in 8 rats in each group. These genes were selected as being regulated in EAE and of biological interest. Gene transcripts were amplified from cDNA preparations. These were obtained from the original total RNA samples used in the micro-array analysis and an additional 4 normal rats with MBP-EAE and an additional 4 healthy rats. The results are shown in Figure 3. All transcripts amplified showed similar expression patterns in the RT-PCR assay compared to the micro-array analysis.
Figure 3. RT- PCR validation of 5 genes expressed differentially in the spinal cords of Lewis rats with MBP induced EAE.

Tissue samples were snap frozen in liquid nitrogen and stored at −80°C prior to total RNA preparation using the QIAGEN RNeasy Lipid tissue kit. RNA quality analysis was carried out on the BioRadExperion automated electrophoresis system. All preparations used in both assays had RNA quality indicator (RQI) values of >9.5. For RT-PCR, total RNA was reverse transcribed and amplified as described in the methods. Analysis of selected genes up or down regulated at the peak of disease in EAE. Bars represent the average fold change between expression in the spinal cord level at peak of disease compared to normal healthy animals (+/− SEMs, Microarray n = 4, RT-PCR n = 8). Dark columns represent fold change derived from the microarray data. Similar amplification patterns were obtained from RT-PCR amplification of the same total RNA samples and a second set of 4 animals samples at an identical time point.
c) Biological Functions Most Significantly Regulated in MBP-EAE
The functional predictions that are generated from IPA are based on the direction of expression of a number of downstream genes which have been previously shown to be associated with that function. The functions that are most significantly altered in MBP-EAE are shown in Tables 3 and 4. The functions that are predicted to be up-regulated include activation of central nervous system cells, cell death or apoptosis (of leukocytes, microglia and dopaminergic neurons), formation of amyloid fibrils, and proliferation of oligodendrocyte precursor cells. The functions that are predicted to be down-regulated include long term-potentiation, neurotransmission, synaptic transmission, quantity of vesicles and delay in the death of neurons.
Table 3. Cellular functions significantly up-regulated in MBP induced EAE in the Lewis rat.
| Function | Prediction | No of genes in direction of prediction | Z score | Overlap p-value | Genes for which expression direction predicts an increase in function* | Genes for which expression direction predicts a decrease in function | Genes which affect the function |
| Activation of central nervous system cells | Increased | 9 of 12 | 2.268 | 2.83E–03 | Il1b,Gfap, Ptgs2, Vim, Il1rn, Cebpb, Tlr2, Tlr4, Npc1 | Kcnj10,Ace | Gjd2 |
| Activation of astrocytes | Increased | 9 of 10 | 2.595 | 1.55E–03 | Il1b,Gfap, Ptgs2, Vim, Il1rn, Cebpb, Tlr2, Tlr4, Npc1 | Kcnj10,Ace | |
| Apoptosis of leukocytes | Increased | 82 of 136 | 2.328 | 2.06–06 | Tnfrsf1a, Fas, Irf1, Myd88, Lcn2, Tlr2, Tlr4, Stat1 | Irgm | Casp3, Casp4 |
| Apoptosis of microglia | Increased | 7 of 9 | 2.570 | 1.08–06 | Fas, Irf1, Myd88, Lcn2, Tlr2, Tlr4, Stat1 | Casp3, Casp4 | |
| Cell death of dopaminergic neurons | Increased | 10 of 13 | 2.071 | 4.94E–04 | Park2, Ppargc1a, TRB@, Cd4, Casp7, Casp8 , Lep , Casp3, Shh, Mapk8ip1, | Snca , Cybb , | Fas |
| Formation of amyloid fibrils | Increased | 5 of 6 | 2.213 | 4.55E–03 | Tnfrsf1a , Mme, Apoe, Apba2, Sod2 | Psen1 | |
| Proliferation of oligodendrocyte precursor cells | Increased | 5 of 5 | 2.236 | 7.83E–03 | Cxcl2, Rps6kb2 , Thrb, Igf1 , Plp1 |
Table 4. Cellular Functions significantly down-regulated in MBP induced EAE in the Lewis rat.
| Function | Prediction | No of genes in direction of prediction | Z score | Overlap p value | Genes for which expression direction predicts a decrease in function* | Genes for which expression direction predicts an increase in function | Genes which affect the function |
| Long term-potentiation | Decreased | 28 of 43 | −2.397 | 2.49E–06 | Chrnb2, Rhob, Gnaq, Il1b, Ptk2b, Grin1, Itpr1 , Kcna4, Reln, Vldlr, ApoE, Grin2a, Grik1, Snca, Gucy1a3, Ppp1r1a, Rab3a, B2m , Psen1, Grik2, Rims1, Chrna7, Tap1 , Ppp1r1b, Cplx2, Pcdh8, Jph4, Adcy8 | Ppp3r1, Gria2, Nrg1, Pcp4, Mgll, Cybb , Il1rn , Erbb4, Gal, Dlg4, Gria3, Casp1, Omg, Ncf1 | Tlr4 |
| Neurotransmission | Decreased | 13 of 26 | −2.043 | 4.79E–03 | Park2 , Il1b, Grin1,Grik1,Grm5, Psen1, Grik2, Htr1a,Gria3, Snap29 , Cacna1b, Slc8a3, Nlgn1 | Gria2, Gal, | Chrna7, Chrm3, Nqo1, Rab3a, Slc5a7, Snca, Apo1, Chrna3, Chrnb2 |
| Synaptic transmission | Decreased | 10 of 12 | −2.578 | 2.45E–02 | Grin1,Grik1, Grm5, Psen1, Grik2, Htr1a, Gria3, Snap29 , Slc8a3, Nlgn1 | Gria2, Gal, | Chrna7, Chrm3, Nqo1, Rab3a, Slc5a7, Scna, Apoe, Chrna3, Chrnb2 |
| Quantity of vesicles | Decreased | 7 of 8 | −2.137 | 7.33E –03 | Grn, Snca, Bm2, Ptprn, Rims1, Tap1, Syn1, | Abc1a | |
| Quantity of dendritic spines | Decreased | 7 of 9 | score -2.044 | 3.08E–03 | Rhob, Grin1, Gda, Pfn2, Il1rapl1, Lcn2, Akap5, | Mark2 | Arhgef7 |
| Size of axons | Decreased | 4 of 4 | −2.000 | 1.24E–04 | Nefm, Sod1, Nefh, Nefl | ||
| Delay in the death of neurons | Decreased | 4 of 4 | −2.000 | 4.56E–02 | Tp53 , Casp2, Casp3 , Bak1 |
d) Canonical Pathways Regulated in MBP-EAE
A number of Canonical pathways were identified in IPA as being significantly regulated in MBP-EAE, based on the expression profile of genes in MBP treated animals compared to healthy controls. These are shown in Figure 4. These include immune related pathways and neural pathways. The details of some of the pathways that were regulated are shown in Figures S3, S4, S5, S6, S7, S8, S9, S10, S11, S12, S13, S14, S15, S16, S17, S18, S19, S20.
Figure 4. Summary of Canonical pathways that are regulated in MBP-EAE.

The most regulated canonical pathways are listed. The percentage of genes of each specific pathway that are up-regulated are shown in red and the percentage of genes that are down-regulated are shown in green. The genes involved in each pathway are shown in individual supplementary figures.
e) Upstream Factors Predicted to be Regulated in MBP-EAE
From IPA we have identified the upstream regulators that are predicted to be activated or inactivated in MBP-EAE. These are shown in Table 5. These include Sirtuin 2, stat1 and presenilin 2.
Table 5. Upstream Regulators.
| Gene ID | Gene name | Molecular Type | Predicted Activation State | Fold Change | Activation z-score | p-value of overlap | Gene regulation consistent with activation (Number of genes regulated of all potential gene targets)* | Genes regulation consistent with inhibition (Number of genes regulated of all potential gene targets) |
| Sirt2 | Sirtuin2 | Transcription regulator | Inhibited | – | −3.317 | 3.85E–09 | Sqle, Sc5dl, Pmvk, Mvd, Lss,Idi1, Hmgcr, Fnta, Fdft1, Dhcr7,Aacs(11 of 11) | |
| IL1b | interleukin 1, beta | cytokine | Activated | 4.8 | 2.422 | 6.34E–06 | Vim, Sod2, Serpina3, Ptgs2, Psen1, Plau, Il1rn, Il1b, Igf1,Icam1, Fas, Cxcl2, Csf1, Casp4 (14 of 18) | Tac1, Snca, Slc1a3 , Lep (4 of 18) |
| Mapt | microtubule-associated protein tau | other | Activated | 2.0 | 9.66E–05 | Pclo, Grin1, Dlgap2, Dlg4 (4 of 46) | ||
| Crebbp | CREB binding protein | Transcription regulator | Inhibited | – | −2.828 | 1.14E–04 | Grm1, Grin2d, Grin2c, Grin2a, Grin1, Gria3, Gria2, Camk1g, (8 of 9) | |
| Stat1 | signal transducer and activator of transcription 1 | Transcription regulator | Activated | 12.5 | 2.197 | 2.27E–04 | Ubd, Stat2, Rsad2, Psmb9, Irf9, Irf8, Irf1, Icam1, Gbp2, Cxcl9, Ceacam1 (11 of 15) | Ubd, Stat2, Rsad2, Psmb9, Irf9, Irf8, Irf1, Icam1, Gbp2, Cxcl9, Ceacam1 (3 of 15) |
| Nfe2l2 | nuclear receptor subfamily 3, group C, member 1 (glucocorticoid receptor) | Transcription regulator | Inhibited | 3.8 | −2.125 | 4.04E–04 | Slc7a8, Ptpro, Nucb2, Nqo1 (4 of 21) | Vsnl1, Syt5, Syn2, Slc30a4, Scg2, Reln, Pkia, Pafah1b, Kifc2, Hpca, Fgf13, Chgb, Calm1, Calb1 (14 of 21) |
| Tnf | tumor necrosis factor | cytokine | Activated | – | 3.575 | 7.32E–04 | Vcam1, Stat1, Sp1, Sod2, Ptgs2, Plau, Icam1, Fas, Csf1, Cflar, Cd14, Ccl7, Casp4 (13 0f 13) | |
| Tgfbr2 | transforming growth factor, beta receptor II (70/80 kDa) | kinase | Activated | 3.1 | 2.137 | 1.61E–03 | Tgif1, Stat3 , Kcnn2, Gfap, Fn1 , Chrm3, Cd44, Cd14, Ccl7 (9 of 11) | Gja1 , Gal (2 of 11) |
| Nr3c1 | nuclear receptor subfamily 3, group C, member 1 (glucocorticoid receptor) | Ligand dependent nuclear receptor | Activated | – | 2.626 | 3.36E–03 | Grm7,Grm5, Grm1, Grin2c, Gria4, Gria3, Gria2 (7 of 11) | |
| Tnfrsf1a | tumor necrosis factor receptor superfamily, member 1A | Trans-membrane receptor | Activated | 1.8 | 2.000 | 3.78E–03 | Sod2, P2rx7, Dio2,Cflar (4 of 4) | |
| Parp1 | poly (ADP-ribose) polymerase 1 | enzyme | Activated | 1.6 | 2.429 | 9.30E–03 | Vcam1,Stat1, Sp1, Ptgs2, Il1b, Icam1 (6 of 6) | |
| Slc30a3 | solute carrier family 30 (zinc transporter), member 3 | transporter | Inhibited | −1.3 | −2.000 | 1.98E–02 | Snap25, Grin2a, Dlg4, Dcx (4 of 4) | |
| Psen2 | presenilin 2 | peptidase | Inhibited | – | −2.747 | 3.51E–02 | Gba2 (1 of 11) | Klra4 , Kcnab2, Gja1, Gfap, Ctss, CD74, Cap1, C3ar1, C1qb, C1qa (10 of 11) |
Discussion
In this study, using female Lewis rats, we have investigated the expression of genes in MBP-EAE compared to controls. We have studied rats at day 13, which is the peak of clinical disease, when processes that lead to recovery are under way. The pathology of MBP-EAE involves infiltration of the CNS with macrophages and activated T lymphocytes [27]. There are morphological changes in microglia and astrocytes and there is demyelination of axons and axonal transaction. Consistent with this, we have found that MBP-EAE is associated with changes in expression of genes and biological functions of the immune system and also of the nervous system. We found markedly increased expression of MHC class II molecules and other immune related genes. This is consistent with the pathological findings of upregulation of MHC class II molecules and infiltration with immune cells [14]. Micro-array analysis of C57/BL6 mice with EAE induced with myelin-oligodendrocyte protein also found upregulation of numerous genes [23] including MHC class II and immune related genes. We note increased expression of lipocalin 2, which is a protein that can be involved in the immune response and that has been found to be upregulated in MOG-EAE in DA rats [28] and in C57/BL mice [29].
Genes involved in the inflammatory response and in antigen processing and presentation were some of the most highly regulated genes identified. Cell-cell signalling and interaction and cell movement were also highly represented amongst the most differentially regulated gene transcripts identified. A number of chemokines and their receptors, annexin 3A and fibronectin were all up-regulated more than four-fold in the diseased spinal cords. Cellular growth factors, notably IL2-α and the common gamma chain were highly up-regulated as was the inflammatory cytokine IL1-β which is expressed in activated microglial cells [30]. IL18, the IL1 receptor agonist IL1rn, the interleukin receptors IL1r1, IL4r, IL6r, and the interferon gamma inducible cytokine IL18 were all moderately up-regulated (Table S1).
The regulation of key components of the classical (C1q, C4, C3b, Serping1) and alternative (C3, Cfb) complement pathways and the complement component 3a receptor 1 (3ar1), but not molecules in the terminal pathway, is in accordance with previous studies which indicate that the common terminal pathway (culminating in the membrane attack complex) is not important for the development of fulminant demyelinating EAE [31].
Genes associated with apoptosis which were highly regulated in our data set include the apoptosis related serine peptidases (Caspases) which have all been associated with disease in various EAE models [32]; [33] and the toll like receptors 2 and 4 (Tlr2, Tlr4) which have previously been found up-regulated in active lesions of MS and MOG induced EAE in the DA rat [34]; [35]. As mentioned above, the apoptotic inducer lipocalin 2 (Lcn2) [36] which has previously been identified as up-regulated in MOG induced EAE in DA rats [28] and C57BL mice [29] was also highly expressed in MBP-EAE in the present study.
Down-regulation of gene transcripts in MBP induced EAE in the Lewis rat was much more subtle than up-regulation however almost a thousand genes were found to be significantly down-regulated in the model (Table S2). Eighteen of these were down-regulated more than 2-fold, notably including key enzymes in the cholesterol biosynthesis pathway and molecules involved in cellular growth and proliferation, signalling and interaction.
Of the most highly regulated genes in the spinal cord (Tables 1 & 2), many are associated with processes associated with induction of disease, such as cell-cell signalling, chemotaxis antigen presentation, activation of both infiltrating cells such as T-lymphocytes and macrophages and resident astrocytes and microglia. There is also regulation of genes associated with resolution of disease, for example apoptosis of immune infiltrating cells and activated glial cells, and growth and proliferation of oligodendrocyte precursor cells which migrate to the sites of demyelination and differentiate into mature oligodendrocytes able to carry out the remyelination of axonal processes damaged during the inflammatory phase of the disease [37].
EAE can only be induced in susceptible animals. In studies of different mouse and rat strains, some genetic loci have been identified as being important in the predisposition to EAE. In our study, many of the genes identified as being differentially regulated found to map to those regions (http://rgd.mcw.edu/).
From IPA, the cellular functions that are regulated can be predicted. The upregulated functions in MBP-EAE included activation of CNS cells, cell death or apoptosis (of immune cells, microglia and dopaminergic neurons), formation of amyloid fibrils, and proliferation of oligodendrocyte precursor cells. Apoptosis is known to be an important function in EAE. Apoptosis of T cells is important in the regulation of MBP-EAE [15]; [16]. Apoptosis of oligodendrocytes could also contribute to pathology in EAE. Consistent with this, we have found upregulation of genes associated with apoptosis. Our finding of increased proliferation of oligodendrocyte precursor cells is consistent with the observation that recovery in MBP-EAE is due to restoration of conduction in demyelinated fibres.
The functions that are predicted to be downregulated include long term-potentiation, neurotransmission, synaptic transmission, quantity of vesicles and delay in the death of neurons. These downregulated functions presumably reflect damage to neural tissue.
From IPA we also obtained a list of the canonical pathways that were regulated in MBP-EAE. There were changes in immune pathways and neural pathways. The immune related pathways include antigen presentation, T cell differentiation and complement pathways, all of which would be expected to be activated in EAE. The neural related pathways include dopamine feedback of cAMP signalling, Creb signalling in neurones, biosynthesis of steroids and ALS signalling.
From IPA we have identified the upstream regulators that are predicted to be activated or inactivated in MBP-EAE. The greatest inactivation was predicted in sirtuin 2 (sirt2). Sirt 2 is is a member of the sirtuin family of histone deacetylases and has been shown to have a role in promoting longevity [38]. With proteomic studies, sirtuin 2 has also been found to be downregulated in EAE [39]. Resveratrol which is an agonist of sirt2 has been shown to reduce the severity of EAE [40]. Stat1 is a member of the JAK/STAT group of proteins [41]. Stat 1 regulates genes involved in interferon signalling which is shown in this study to be upregulated in MBP-EAE. Presenilin 2 is a member of the gamma-secretase family and is predominantly expressed in neurones where it plays a role in the processing of amyloid precursor protein [42] and was predicted in this study to be downregulated.
In conclusion, we have shown that MBP induced EAE alters gene expression in the spinal cord. We have produced a gene expression profile for this model in the Lewis rat which correlates well with the documented characteristics of the disease. Further examination of the pathways and functions regulated should pave the way for future studies into their regulation which may lead to the identification of potential drug targets for enhancement of the resolution of inflammatory disease in the CNS.
Materials and Methods
Ethics Statement
This research was approved by the Animal Ethics Committee of the University of Queensland. The approval number was UQCCR/770/08/MSRA.
Induction of EAE
EAE was induced in 11 wk old female Lewis rats by subcutaneous injection at the base of the tail of 50 µg recombinant MBP, using our standard methods (McCombe et al., 1996). Healthy age-matched unimmunized rats were used as controls. Weakness of the tail, hindlimbs and forelimbs was assessed by grading the degree of weakness of each region separately on a scale of 0 (no weakness) to 4 (total paralysis), as described previously [43]. On each day of examination, the scores from each region were added together to give a total clinical score per rat (maximum total clinical score = 12). Figure 1 shows the typical clinical course of rats with this form of EAE.
Tissue Collection
On day 13 after inoculation, which is the peak of clinical disease, rats were sacrificed and the spinal cord was excised from the spinal column by insufflation using a large bore blunt ended needle and a 60 ml syringe. Figure 1B shows the clinical course of these rats up to the day of sacrifice. Tissue was snap frozen in liquid nitrogen prior to being stored at −80°C. Four rats in each group were used for micro-array analysis and a total of 8 rats in each group were used for rt-PCR.
RNA Extraction
Total RNA was extracted from tissues using QIAGEN RNeasy® Lipid tissue Midi kits as per the manufactures instructions and treated with DNase1 (QIAGEN) to remove all traces of genomic DNA prior to storage at −80°C. The RNA quality analysis was carried out using the BioRadExperion automated gel electrophoresis system (BioRad Laboratories Inc.).
Array Hybridization
For each group, 4 biological replicates were used. cDNA synthesis and amplification was performed using the Applause WT-Amp Plus ST kit (NuGEN). Samples were enzymatically fragmented and biotinylated using the Encore Biotin Module labelling kit (NuGEN). Samples were hybridized to AffimetrixGeneChip Rat Exon 1.0 ST Arrays as per the manufacturer’s instructions. Briefly, 5 µg of fragmented biotinylated ssDNA was hybridised for 16 hrs at 45°C, 60 rpm to the array chip. After 16 hrs GeneChips were washed on a FS_450 Fluidics station using the washing script FS450_0001 with buffers and stains supplied with the GeneChip Hybridisation, Wash and Stain Kit from Affymetrix.
Data Acquisition and Analysis
Data was acquired on a 7G GeneChip Scanner 3000 and.CEL file generation performed using AGCC. Expression Console with Robust Multi-chip Average (RMA) was used initially to extract probe intensity data. This data was used to access Affimetrix supplied PolyA and Eucaryotic Hybridisation controls to confirm consistent processing of the RNA samples. Subsequent statistical analysis was performed in Partek Genomics Suite – Version 6.110801 (Partek Inc. St Louis, MO). The Affymetrix RaEx-1_0-st-v1.r2.dt.core.mps was used to filter probe sets. RMA background correction was applied including pre-background adjustment for GC content and quantile normalization across all chips in the experiment. Probe data was log2 transformed.
Gene Level Expression Analysis
For gene transcript expression analysis, core probe sets were averaged for each gene. The significance of differences in gene expression between the EAE and healthy control data sets was evaluated by analysis of variance (ANOVA) that compared the controls and normal rats and investigated the effect of different batches. There was no significant batch effect in this study.
Although we are aware that the data may not be distributed normally and that a reduced homogeneity of variance may exist, the alternative non-parametric tests such as the Kruskal Wallis analysis would not produce the ratio of means required to identify statistically significant functional categories and pathways and make predictions about the possible downstream functional effects of fold change for various groups of genes. As a result, the above violations to the assumptions made by this analysis were considered to be adequately compensated for by the robust nature of the analysis. We have produced a list of all the genes that are regulated in EAE, and also a smaller list of the genes that are most highly regulated in MBP-EAE.
Functional Association Analysis
IPA (Ingenuity® systems, www.ingenuity.com) was used to identify biological functions, gene networks and pathways which were significantly regulated in the spinal cord in EAE compared to healthy controls. In IPA, right-tailed Fisher’s exact test was used to calculate a p-value determining the probability that each function network or pathway assigned to that data set is due to chance alone. Molecules from the data set that met the p<0.05 cut off and were associated with biological functions in the Ingenuity knowledge base were considered for the analysis.
Downstream Effects Analysis in IPA
The Ingenuity downstream effects analytic were used to identify biological functions which were predicted to be increased or decreased based on the observed gene expression changes in the data-set. The software compared the direction of change for each gene to the causal effects information between genes and biological functions stored in the IPA Knowledge base. Statistical validation of the predictions made were carried out based on the calculation of the regulation z score “which is designed such that data sets composed of randomly chosen perturbed genes with random sign of fold change do not lead to significant results on average” (Ingenuity Downsteam Effects Analysis, whitepaper). Each biological function considered is associated with a number of upstream genes based on annotations derived from the literature and stored in the Ingenuity knowledgebase. The previously observed effects on function of each gene in this set are then used to predict an increase or decrease in the function under the conditions studied. Table 3 shows the biological functions found to be significantly up or down-regulated in MBP induced EAE in the Lewis rat.
Quantitative RT-PCR Validation of Micro-array Data
Validation of the results obtained from the micro-array experiments was carried out using real time PCR. Five genes (rtCasp1, rtOmg, rtScn1a, rtFas and rtSod2) found to be differentially regulated in EAE in the micro-array gene expression experiments, were selected for validation by RT-PCR. Rat primer pairs were purchased from SA BioSystems (RT2qPCR Primer Assay, QIAGEN). Real time PCR analysis was carried on the MyIQ™ real time PCR thermo-cycler and detection system (BioRad Laboratories Inc.) using the RT2 QPCR Primer assay according to the manufactures’ protocol. Briefly, 1 µg of total rat spinal cord RNA was treated with QIAGEN Genomic DNA Elimination mix for 5 min at 42?C prior to being reverse transcribed using the kit protocol. RT-PCR conditions were as follows: 95?C, 10 min (Hot Start Taq polymerisation activation step) followed by 40 cycles of 95?C, 15 sec, 60?C, 1 min. The reactions were monitored using SYBR Green containing fluorescein (RT2 SYBR Green Fluor qPCR Master mix, QIAGEN). At the end of each PCR run a melting curve analysis was carried out to verify that homogeneous products of equal size had been amplified for each primer pair. The same set of RNA samples that was used for the micro-array experiments was also analysed by RT-PCR and in addition we tested four more independent validation samples from each group, which had been collected and prepared in an identical manner, to test for sampling error. Expression was normalized to rat peptidylprolylisomerase H (rtPpih). Fold changes were calculated using the ΔΔCT method [44] and expressed as fold-change at peak of disease in EAE compared to the expression levels in healthy control Lewis rats (Figure 3).
Supporting Information
RNA Virtual Gel Image. Tissue samples were snap frozen in liquid nitrogen and stored at −80°C prior to total RNA preparation using the QIAGEN RNeasy Lipid tissue kit. RNA quality analysis was carried out on the BioRad Experion automated electrophoresis system. This shows an example of the virtual gel image, illustrate the high quality of samples used. All preparations had RQI values of >9.5.
(DOCX)
RNA Electropherogram Profile. Tissue samples were snap frozen in liquid nitrogen and stored at −80°C prior to total RNA preparation using the QIAGEN RNeasy Lipid tissue kit. RNA quality analysis was carried out on the BioRad Experion automated electrophoresis system. This shows an example of the electropherogram profiles illustrating the high quality of samples used. All preparations had RQI values of >9.5.
(DOCX)
Regulation of the Antigen presentation pathway in MBP-EAE. This figure shows the upregulation of MHC class I and MHC class II linked pathways of antigen presentation in MBP-EAE.
(TIF)
Regulation of the T Helper Cell Differentiation in MBP-EAE. This figure shows that there is upregulation of the pathways of Th0 cell differentiation, especially into Th1 and Th17 cells in MBP-EAE.
(TIF)
Regulation of Cytotoxic T-lymphocyte mediated apoptosis of target cells in MBP-EAE. This figure shows that there is upregulation of the pathways involved in T cell cytotoxicity of target cells in MBP-EAE.
(TIF)
Regulation of Fc gamma –mediated phagocytosis in macrophages and monocytes in MBP-EAE. This shows that in rats with MBP-EAE there is up-regulation of many of the genes in the pathways leading to phagosome formation after Fc gamma binding.
(TIF)
Regulation of Interferon signalling in MBP-EAE. This shows that there is upregulation of many of the genes in the JAK/STAT pathway of interferon signalling in rats with MBP-EAE.
(TIF)
Regulation of Dendritic cell maturation in MBP-EAE. This figure shows that many of the genes in the pathway of dendritic cell maturation are upregulated in MBP-EAE, except IL-12 which is downregulated.
(TIF)
B cell development in MBP-EAE. This shows that many cell surface receptors involved in the stages of B cell maturation are up- regulated in MBP-EAE.
(TIF)
Regulation of Trem1 Signalling in MBP-EAE. This shows that many of the intracellular signalling molecules involved in signalling by the triggering receptor expressed on myeloid cells 1 (TREM1) are upregulated in MBP-EAE. This pathway is involved in adaptive and innate immunity.
(TIF)
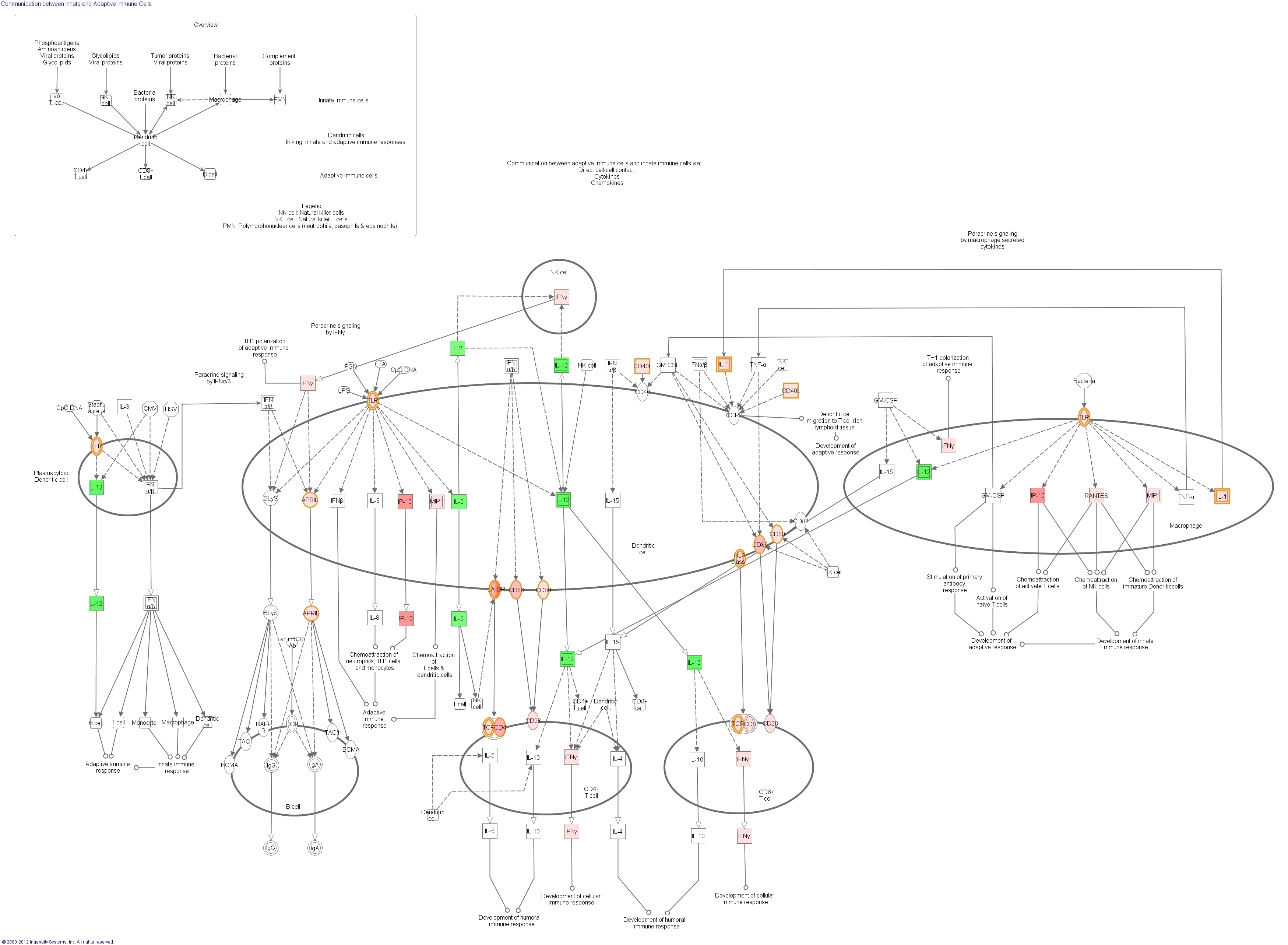
Regulation of Communication between innate and adaptive immune cells in MBP-EAE. This shows that many signalling molecules involved in this pathway are upregulated except for IL12 which is down regulated.
(JPG)
{kind=link}
Regulation of the Complement pathway in MBP-EAE. This shows that the classical pathway and the alternate pathway but not the common terminal pathway are regulated in MBP-EAE.
(TIF)
Regulation of genes in the pathway entitled Pathogenesis of Multiple Sclerosis. This is a pathway of chemokine receptors. The receptors CCR1, CCR5, CXCR3, CCL5, CXCL9 and CXCL10 are up-regulated in MBP-EAE.
(TIF)
Regulation of Synaptic long term potentiation in MBP-EAE. This shows that this pathway of signalling in response to glutamate is down-regulated in MBP-EAE.
(TIF)
Regulation of Dopamine-DARPP32 feedback in cAMP signalling in MBP-EAE. This shows that this pathway of signalling in response to dopamine is down-regulated in MBP-EAE.
(TIF)
Regulation of the Amyotrophic Lateral Sclerosis pathway in MBP-EAE. This pathway of intra-cellular signalling that leads to cell death and degeneration in response to glutamate is down-regulated in MBP-EAE.
(TIF)
Regulation of Creb signalling in neurons in MBP-EAE. This shows that this pathway of intracellular signalling leading to expression of cAMP response element-binding (Creb) is down-regulated in MBP-EAE.
(TIF)
Regulation Endothelin-1 signalling in MBP-EAE. This shows that this pathway of intracellular signalling in response to endothelin 1 (ET1) is downregulated in MBP-EAE.
(TIF)
Regulation the Biosynthesis of steroids in MBP-EAE. This shows that the squalene pathway involved in the bio-synthesis of steroids is down-regulated in MBP-EAE.
(TIF)
Regulation Taurine/hypotaurine metabolism in MBP-EAE. This shows downregulation of elements of the pathway that leads to synthesis of taurine and hypotaurine from cysteine in MBP-EAE.
(TIF)
(XLSX)
(XLSX)
Acknowledgments
We gratefully acknowledge Gareth Price, Renee Gallagher and Thu Nguyen from the Mater Medical Research Institute for help with the micro-array studies.
The data discussed in this publication have been deposited in NCBI’s Gene Expression
Omnibus and are accessible through GEO Series accession number GSE39119 (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE39119)
Funding Statement
This study was funded by a grant from Multiple Sclerosis Australia. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Sawcer S, Hellenthal G, Pirinen M, Spencer CC, Patsopoulos NA, et al. (2011) Genetic risk and a primary role for cell-mediated immune mechanisms in multiple sclerosis. Nature 476: 214–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Wilson HL (2012) B cells contribute to MS pathogenesis through antibody-dependent and antibody-independent mechanisms. Biologics 6: 117–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Smith KJ, McDonald WI (1999) The pathophysiology of multiple sclerosis: the mechanisms underlying the production of symptoms and the natural history of the disease. Philos Trans R Soc Lond B Biol Sci 354: 1649–1673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Rivers TM, Sprunt DM, Berry GP (1933) Observations on attempts to produce acute disseminated encephalomyelitis in monkeys. J Exp Med 58: 39–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Freund J, Stern ER, Pisani TM (1947) Isoallergic encephalomyelitis and radiculitis in guinea pigs after one injection of brain and mycobacteria in water-in-oil emulsion. J Immunol 57: 179–194. [PubMed] [Google Scholar]
- 6. Greer JM, Sobel RA, Sette A, Southwood S, Lees MB, et al. (1996) Immunogenic and encephalitogenic epitope clusters of myelin proteolipid protein. J Immunol 156: 371–379. [PubMed] [Google Scholar]
- 7. Chalk JB, McCombe PA, Smith R, Pender MP (1994) Clinical and histological findings in proteolipid-protein- induced experimental autoimmune encephalomyelitis (EAE) in the Lewis rat. Distribution of demyelination differs from that in EAE induced by other antigens. J Neurol Sci 123: 154–161. [DOI] [PubMed] [Google Scholar]
- 8. Slavin A, Ewing C, Liu J, Ichikawa M, Slavin J, et al. (1998) Induction of a multiple sclerosis-like disease in mice with an immunodominant epitope of myelin oligodendrocyte glycoprotein. Autoimmunity 28: 109–120. [DOI] [PubMed] [Google Scholar]
- 9. Encinas JA, Weiner HL, Kuchroo VK (1996) Inheritance of susceptibility to experimental autoimmune encephalomyelitis. J Neurosci Res 45: 655–669. [DOI] [PubMed] [Google Scholar]
- 10. Encinas JA, Lees MB, Sobel RA, Symonowicz C, Weiner HL, et al. (2001) Identification of genetic loci associated with paralysis, inflammation and weight loss in mouse experimental autoimmune encephalomyelitis. Int Immunol 13: 257–264. [DOI] [PubMed] [Google Scholar]
- 11. Stridh P, Thessen HM, Beyeen AD, Adzemovic MZ, Laaksonen H, et al. (2010) Fine-mapping resolves Eae23 into two QTLs and implicates ZEB1 as a candidate gene regulating experimental neuroinflammation in rat. PLoS ONE 5: e12716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Pender MP (1988) The pathophysiology of myelin basic protein-induced acute experimental allergic encephalomyelitis in the Lewis rat. J Neurol Sci 86: 277–289. [DOI] [PubMed] [Google Scholar]
- 13. Pender MP (1989) Recovery from acute experimental allergic encephalomyelitis in the Lewis rat. Early restoration of nerve conduction and repair by Schwann cells and oligodendrocytes. Brain 112: 393–416. [DOI] [PubMed] [Google Scholar]
- 14. McCombe PA, Fordyce BW, de Jersey J, Yoong G, Pender MP (1992) Expression of CD45RC and Ia antigen in the spinal cord in acute experimental allergic encephalomyelitis: an immunocytochemical and flow cytometric study. J Neurol Sci 113: 177–186. [DOI] [PubMed] [Google Scholar]
- 15. Pender MP, McCombe PA, Yoong G, Nguyen KB (1992) Apoptosis of alpha beta T lymphocytes in the nervous system in experimental autoimmune encephalomyelitis: its possible implications for recovery and acquired tolerance. J Autoimmun 5: 401–410. [DOI] [PubMed] [Google Scholar]
- 16. McCombe PA, Nickson I, Tabi Z, Pender MP (1996) Apoptosis of V beta 8.2+ T lymphocytes in the spinal cord during recovery from experimental autoimmune encephalomyelitis induced in Lewis rats by inoculation with myelin basic protein. J Neurol Sci 139: 1–6. [PubMed] [Google Scholar]
- 17. Herz J, Zipp F, Siffrin V (2010) Neurodegeneration in autoimmune CNS inflammation. Exp Neurol 225: 9–17. [DOI] [PubMed] [Google Scholar]
- 18. Ohgoh M, Hanada T, Smith T, Hashimoto T, Ueno M, et al. (2002) Altered expression of glutamate transporters in experimental autoimmune encephalomyelitis. J Neuroimmunol 125: 170–178. [DOI] [PubMed] [Google Scholar]
- 19. Tokuhara N, Namiki K, Uesugi M, Miyamoto C, Ohgoh M, et al. (2010) N-type calcium channel in the pathogenesis of experimental autoimmune encephalomyelitis. J Biol Chem 285: 33294–33306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Schneider C, Schuetz G, Zollner TM (2009) Acute neuroinflammation in Lewis rats - a model for acute multiple sclerosis relapses. J Neuroimmunol 213: 84–90. [DOI] [PubMed] [Google Scholar]
- 21. Brand-Schieber E, Werner P, Iacobas DA, Iacobas S, Beelitz M, et al. (2005) Connexin43, the major gap junction protein of astrocytes, is down-regulated in inflamed white matter in an animal model of multiple sclerosis. J Neurosci Res 80: 798–808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Matejuk A, Dwyer J, Hopke C, Vandenbark AA, Offner H (2003) 17Beta-estradiol treatment profoundly down-regulates gene expression in spinal cord tissue in mice protected from experimental autoimmune encephalomyelitis. Arch Immunol Ther Exp (Warsz ) 51: 185–193. [PubMed] [Google Scholar]
- 23. Ibrahim SM, Mix E, Bottcher T, Koczan D, Gold R, et al. (2001) Gene expression profiling of the nervous system in murine experimental autoimmune encephalomyelitis. Brain 124: 1927–1938. [DOI] [PubMed] [Google Scholar]
- 24. Mueller AM, Pedre X, Stempfl T, Kleiter I, Couillard-Despres S, et al. (2008) Novel role for SLPI in MOG-induced EAE revealed by spinal cord expression analysis. J Neuroinflammation 5: 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Denisov V, Strong W, Walder M, Gingrich J, Wintz H (2008) Development and validation of RQI: An RNA quality indicator for Experion ™ automated electrophoresis system. BioRad Bulletin 5761.
- 26. Benjamini Y, Hochberg Y (1995) Controllong the false discovery rate: a practical and powerful approach to multiple testing. Journal of the Royal Statistical Society Series B (Methodological) 57: 289–300. [Google Scholar]
- 27. McCombe PA, de Jersey J, Pender MP (1994) Inflammatory cells, microglia and MHC class II antigen-positive cells in the spinal cord of Lewis rats with acute and chronic relapsing experimental autoimmune encephalomyelitis. J Neuroimmunol 51: 153–167. [DOI] [PubMed] [Google Scholar]
- 28. Mueller AM, Pedre X, Stempfl T, Kleiter I, Couillard-Despres S, et al. (2008) Novel role for SLPI in MOG-induced EAE revealed by spinal cord expression analysis. J Neuroinflammation 5: 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Berard JL, Zarruk JG, Arbour N, Prat A, Yong VW, et al.. (2012) Lipocalin 2 is a novel immune mediator of experimental autoimmune encephalomyelitis pathogenesis and is modulated in multiple sclerosis. Glia. [DOI] [PubMed]
- 30. Murphy AC, Lalor SJ, Lynch MA, Mills KH (2010) Infiltration of Th1 and Th17 cells and activation of microglia in the CNS during the course of experimental autoimmune encephalomyelitis. Brain Behav Immun 24: 641–651. [DOI] [PubMed] [Google Scholar]
- 31. Barnum SR, Szalai AJ (2006) Complement and demyelinating disease: no MAC needed? Brain Res Rev 52: 58–68. [DOI] [PubMed] [Google Scholar]
- 32. Ahmed Z, Doward AI, Pryce G, Taylor DL, Pocock JM, et al. (2002) A role for caspase-1 and -3 in the pathology of experimental allergic encephalomyelitis : inflammation versus degeneration. Am J Pathol 161: 1577–1586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Furlan R, Martino G, Galbiati F, Poliani PL, Smiroldo S, et al. (1999) Caspase-1 regulates the inflammatory process leading to autoimmune demyelination. J Immunol 163: 2403–2409. [PubMed] [Google Scholar]
- 34. Kerfoot SM, Long EM, Hickey MJ, Andonegui G, Lapointe BM, et al. (2004) TLR4 contributes to disease-inducing mechanisms resulting in central nervous system autoimmune disease. J Immunol 173: 7070–7077. [DOI] [PubMed] [Google Scholar]
- 35. Andersson A, Covacu R, Sunnemark D, Danilov AI, Dal BA, et al. (2008) Pivotal advance: HMGB1 expression in active lesions of human and experimental multiple sclerosis. J Leukoc Biol 84: 1248–1255. [DOI] [PubMed] [Google Scholar]
- 36. Li C, Chan YR (2011) Lipocalin 2 regulation and its complex role in inflammation and cancer. Cytokine 56: 435–441. [DOI] [PubMed] [Google Scholar]
- 37. Windrem MS, Roy NS, Wang J, Nunes M, Benraiss A, et al. (2002) Progenitor cells derived from the adult human subcortical white matter disperse and differentiate as oligodendrocytes within demyelinated lesions of the rat brain. J Neurosci Res 69: 966–975. [DOI] [PubMed] [Google Scholar]
- 38. Dali-Youcef N, Lagouge M, Froelich S, Koehl C, Schoonjans K, et al. (2007) Sirtuins: the ‘magnificent seven’, function, metabolism and longevity. Ann Med 39: 335–345. [DOI] [PubMed] [Google Scholar]
- 39. Jastorff AM, Haegler K, Maccarrone G, Holsboer F, Weber F, et al. (2009) Regulation of proteins mediating neurodegeneration in experimental autoimmune encephalomyelitis and multiple sclerosis. Proteomics Clin Appl 3: 1273–1287. [DOI] [PubMed] [Google Scholar]
- 40. Singh NP, Hegde VL, Hofseth LJ, Nagarkatti M, Nagarkatti P (2007) Resveratrol (trans-3,5,4'-trihydroxystilbene) ameliorates experimental allergic encephalomyelitis, primarily via induction of apoptosis in T cells involving activation of aryl hydrocarbon receptor and estrogen receptor. Mol Pharmacol 72: 1508–1521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Stark GR, Darnell JE Jr (2012) The JAK-STAT pathway at twenty. Immunity 36: 503–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Selkoe DJ, Wolfe MS (2007) Presenilin: running with scissors in the membrane. Cell 131: 215–221. [DOI] [PubMed] [Google Scholar]
- 43. Pender MP (1986) Ascending impairment of nociception in rats with experimental allergic encephalomyelitis. J Neurol Sci 75: 317–328. [DOI] [PubMed] [Google Scholar]
- 44. Gheorghe CP, Goyal R, Mittal A, Longo LD (2010) Gene expression in the placenta: maternal stress and epigenetic responses. Int J Dev Biol 54: 507–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Bergsteinsdottir K, Yang HT, Pettersson U, Holmdahl R (2000) Evidence for common autoimmune disease genes controlling onset, severity, and chronicity based on experimental models for multiple sclerosis and rheumatoid arthritis. J Immunol 164: 1564–1568. [DOI] [PubMed] [Google Scholar]
- 46. de Graaf KL, Weissert R, Kjellen P, Holmdahl R, Olsson T (1999) Allelic variations in rat MHC class II binding of myelin basic protein peptides correlate with encephalitogenicity. Int Immunol 11: 1981–1988. [DOI] [PubMed] [Google Scholar]
- 47. Burrows GG, Bebo BF Jr, Adlard KL, Vandenbark AA, Offner H (1998) Two-domain MHC class II molecules form stable complexes with myelin basic protein 69–89 peptide that detect and inhibit rat encephalitogenic T cells and treat experimental autoimmune encephalomyelitis. J Immunol 161: 5987–5996. [PubMed] [Google Scholar]
- 48. Jelinsky SA, Miyashiro JS, Saraf KA, Tunkey C, Reddy P, et al. (2005) Exploiting genotypic differences to identify genes important for EAE development. J Neurol Sci 239: 81–93. [DOI] [PubMed] [Google Scholar]
- 49. Hisahara S, Yuan J, Momoi T, Okano H, Miura M (2001) Caspase-11 mediates oligodendrocyte cell death and pathogenesis of autoimmune-mediated demyelination. J Exp Med 193: 111–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Das A, Guyton MK, Matzelle DD, Ray SK, Banik NL (2008) Time-dependent increases in protease activities for neuronal apoptosis in spinal cords of Lewis rats during development of acute experimental autoimmune encephalomyelitis. J Neurosci Res 86: 2992–3001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Jagodic M, Kornek B, Weissert R, Lassmann H, Olsson T, et al. (2001) Congenic mapping confirms a locus on rat chromosome 10 conferring strong protection against myelin oligodendrocyte glycoprotein-induced experimental autoimmune encephalomyelitis. Immunogenetics 53: 410–415. [DOI] [PubMed] [Google Scholar]
- 52. Huitinga I, Bauer J, Strijbos PJ, Rothwell NJ, Dijkstra CD, et al. (1998) Effect of annexin-1 on experimental autoimmune encephalomyelitis (EAE) in the rat. Clin Exp Immunol 111: 198–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Jagodic M, Becanovic K, Sheng JR, Wu X, Backdahl L, et al. (2004) An advanced intercross line resolves Eae18 into two narrow quantitative trait loci syntenic to multiple sclerosis candidate loci. J Immunol 173: 1366–1373. [DOI] [PubMed] [Google Scholar]
- 54. Eltayeb S, Berg AL, Lassmann H, Wallstrom E, Nilsson M, et al. (2007) Temporal expression and cellular origin of CC chemokine receptors CCR1, CCR2 and CCR5 in the central nervous system: insight into mechanisms of MOG-induced EAE. J Neuroinflammation 4: 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Ransohoff RM, Liu L, Cardona AE (2007) Chemokines and chemokine receptors: multipurpose players in neuroinflammation. Int Rev Neurobiol 82: 187–204. [DOI] [PubMed] [Google Scholar]
- 56. Sunnemark D, Eltayeb S, Nilsson M, Wallstrom E, Lassmann H, et al. (2005) CX3CL1 (fractalkine) and CX3CR1 expression in myelin oligodendrocyte glycoprotein-induced experimental autoimmune encephalomyelitis: kinetics and cellular origin. J Neuroinflammation 2: 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Jagodic M, Marta M, Becanovic K, Sheng JR, Nohra R, et al. (2005) Resolution of a 16.8-Mb autoimmunity-regulating rat chromosome 4 region into multiple encephalomyelitis quantitative trait loci and evidence for epistasis. J Immunol 174: 918–924. [DOI] [PubMed] [Google Scholar]
- 58. Nedachi T, Kawai T, Matsuwaki T, Yamanouchi K, Nishihara M (2011) Progranulin enhances neural progenitor cell proliferation through glycogen synthase kinase 3beta phosphorylation. Neuroscience 185: 106–115. [DOI] [PubMed] [Google Scholar]
- 59. Thessen HM, Gillett A, Olsson T, Jagodic M, Harris RA (2009) Characterization of Multiple Sclerosis candidate gene expression kinetics in rat experimental autoimmune encephalomyelitis. J Neuroimmunol 210: 30–39. [DOI] [PubMed] [Google Scholar]
- 60. Rochman Y, Spolski R, Leonard WJ (2009) New insights into the regulation of T cells by gamma(c) family cytokines. Nat Rev Immunol 9: 480–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Jee Y, Kim G, Tanuma N, Matsumoto Y (2001) STAT expression and localization in the central nervous system during autoimmune encephalomyelitis in Lewis rats. J Neuroimmunol 114: 40–47. [DOI] [PubMed] [Google Scholar]
- 62. Szalai AJ, Hu X, Adams JE, Barnum SR (2007) Complement in experimental autoimmune encephalomyelitis revisited: C3 is required for development of maximal disease. Mol Immunol 44: 3132–3136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Becanovic K, Wallstrom E, Kornek B, Glaser A, Broman KW, et al. (2003) New loci regulating rat myelin oligodendrocyte glycoprotein-induced experimental autoimmune encephalomyelitis. J Immunol 170: 1062–1069. [DOI] [PubMed] [Google Scholar]
- 64. Almolda B, Costa M, Montoya M, Gonzalez B, Castellano B (2009) CD4 microglial expression correlates with spontaneous clinical improvement in the acute Lewis rat EAE model. J Neuroimmunol 209: 65–80. [DOI] [PubMed] [Google Scholar]
- 65. Chang TT, Jabs C, Sobel RA, Kuchroo VK, Sharpe AH (1999) Studies in B7-deficient mice reveal a critical role for B7 costimulation in both induction and effector phases of experimental autoimmune encephalomyelitis. J Exp Med 190: 733–740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Dahlman I, Wallstrom E, Weissert R, Storch M, Kornek B, et al. (1999) Linkage analysis of myelin oligodendrocyte glycoprotein-induced experimental autoimmune encephalomyelitis in the rat identifies a locus controlling demyelination on chromosome 18. Hum Mol Genet 8: 2183–2190. [DOI] [PubMed] [Google Scholar]
- 67. Dinarello CA (1996) Biologic basis for interleukin-1 in disease. Blood 87: 2095–2147. [PubMed] [Google Scholar]
- 68. Du C, Sriram S (2002) Increased severity of experimental allergic encephalomyelitis in lyn−/− mice in the absence of elevated proinflammatory cytokine response in the central nervous system. J Immunol 168: 3105–3112. [DOI] [PubMed] [Google Scholar]
- 69.Yang S, Hua Y, Nakamura T, Keep RF, Xi G (2006) Up-regulation of brain ceruloplasmin in thrombin preconditioning. Acta Neurochir Suppl 96: 203–206. [DOI] [PubMed]
- 70. Monshouwer M, Agnello D, Ghezzi P, Villa P (2000) Decrease in brain cytochrome P450 enzyme activities during infection and inflammation of the central nervous system. Neuroimmunomodulation 8: 142–147. [DOI] [PubMed] [Google Scholar]
- 71. Bedoui S, Miyake S, Lin Y, Miyamoto K, Oki S, et al. (2003) Neuropeptide Y (NPY) suppresses experimental autoimmune encephalomyelitis: NPY1 receptor-specific inhibition of autoreactive Th1 responses in vivo. J Immunol 171: 3451–3458. [DOI] [PubMed] [Google Scholar]
- 72. Marta M, Stridh P, Becanovic K, Gillett A, Ockinger J, et al. (2010) Multiple loci comprising immune-related genes regulate experimental neuroinflammation. Genes Immun 11: 21–36. [DOI] [PubMed] [Google Scholar]
- 73. Liu X, Mashour GA, Webster HF, Kurtz A (1998) Basic FGF and FGF receptor 1 are expressed in microglia during experimental autoimmune encephalomyelitis: temporally distinct expression of midkine and pleiotrophin. Glia 24: 390–397. [DOI] [PubMed] [Google Scholar]
- 74. Scherer SS, Xu YT, Nelles E, Fischbeck K, Willecke K, et al. (1998) Connexin32-null mice develop demyelinating peripheral neuropathy. Glia 24: 8–20. [DOI] [PubMed] [Google Scholar]
- 75. Diez M, Abdelmagid N, Harnesk K, Strom M, Lidman O, et al. (2009) Identification of gene regions regulating inflammatory microglial response in the rat CNS after nerve injury. J Neuroimmunol 212: 82–92. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
RNA Virtual Gel Image. Tissue samples were snap frozen in liquid nitrogen and stored at −80°C prior to total RNA preparation using the QIAGEN RNeasy Lipid tissue kit. RNA quality analysis was carried out on the BioRad Experion automated electrophoresis system. This shows an example of the virtual gel image, illustrate the high quality of samples used. All preparations had RQI values of >9.5.
(DOCX)
RNA Electropherogram Profile. Tissue samples were snap frozen in liquid nitrogen and stored at −80°C prior to total RNA preparation using the QIAGEN RNeasy Lipid tissue kit. RNA quality analysis was carried out on the BioRad Experion automated electrophoresis system. This shows an example of the electropherogram profiles illustrating the high quality of samples used. All preparations had RQI values of >9.5.
(DOCX)
Regulation of the Antigen presentation pathway in MBP-EAE. This figure shows the upregulation of MHC class I and MHC class II linked pathways of antigen presentation in MBP-EAE.
(TIF)
Regulation of the T Helper Cell Differentiation in MBP-EAE. This figure shows that there is upregulation of the pathways of Th0 cell differentiation, especially into Th1 and Th17 cells in MBP-EAE.
(TIF)
Regulation of Cytotoxic T-lymphocyte mediated apoptosis of target cells in MBP-EAE. This figure shows that there is upregulation of the pathways involved in T cell cytotoxicity of target cells in MBP-EAE.
(TIF)
Regulation of Fc gamma –mediated phagocytosis in macrophages and monocytes in MBP-EAE. This shows that in rats with MBP-EAE there is up-regulation of many of the genes in the pathways leading to phagosome formation after Fc gamma binding.
(TIF)
Regulation of Interferon signalling in MBP-EAE. This shows that there is upregulation of many of the genes in the JAK/STAT pathway of interferon signalling in rats with MBP-EAE.
(TIF)
Regulation of Dendritic cell maturation in MBP-EAE. This figure shows that many of the genes in the pathway of dendritic cell maturation are upregulated in MBP-EAE, except IL-12 which is downregulated.
(TIF)
B cell development in MBP-EAE. This shows that many cell surface receptors involved in the stages of B cell maturation are up- regulated in MBP-EAE.
(TIF)
Regulation of Trem1 Signalling in MBP-EAE. This shows that many of the intracellular signalling molecules involved in signalling by the triggering receptor expressed on myeloid cells 1 (TREM1) are upregulated in MBP-EAE. This pathway is involved in adaptive and innate immunity.
(TIF)
Regulation of Communication between innate and adaptive immune cells in MBP-EAE. This shows that many signalling molecules involved in this pathway are upregulated except for IL12 which is down regulated.
(JPG)
Regulation of the Complement pathway in MBP-EAE. This shows that the classical pathway and the alternate pathway but not the common terminal pathway are regulated in MBP-EAE.
(TIF)
Regulation of genes in the pathway entitled Pathogenesis of Multiple Sclerosis. This is a pathway of chemokine receptors. The receptors CCR1, CCR5, CXCR3, CCL5, CXCL9 and CXCL10 are up-regulated in MBP-EAE.
(TIF)
Regulation of Synaptic long term potentiation in MBP-EAE. This shows that this pathway of signalling in response to glutamate is down-regulated in MBP-EAE.
(TIF)
Regulation of Dopamine-DARPP32 feedback in cAMP signalling in MBP-EAE. This shows that this pathway of signalling in response to dopamine is down-regulated in MBP-EAE.
(TIF)
Regulation of the Amyotrophic Lateral Sclerosis pathway in MBP-EAE. This pathway of intra-cellular signalling that leads to cell death and degeneration in response to glutamate is down-regulated in MBP-EAE.
(TIF)
Regulation of Creb signalling in neurons in MBP-EAE. This shows that this pathway of intracellular signalling leading to expression of cAMP response element-binding (Creb) is down-regulated in MBP-EAE.
(TIF)
Regulation Endothelin-1 signalling in MBP-EAE. This shows that this pathway of intracellular signalling in response to endothelin 1 (ET1) is downregulated in MBP-EAE.
(TIF)
Regulation the Biosynthesis of steroids in MBP-EAE. This shows that the squalene pathway involved in the bio-synthesis of steroids is down-regulated in MBP-EAE.
(TIF)
Regulation Taurine/hypotaurine metabolism in MBP-EAE. This shows downregulation of elements of the pathway that leads to synthesis of taurine and hypotaurine from cysteine in MBP-EAE.
(TIF)
(XLSX)
(XLSX)