

Abstract
We have studied the conformational dynamics associated with the nanoscale DNA bending induced by human immunodeficiency virus type 1 (HIV-1) nucleocapsid (NC) protein using single-molecule Förster Resonance Energy Transfer (SM-FRET). To gain molecular-level insights into how HIV-1 NC locally distorts the structures of duplexed DNA segments, the dynamics, reversibility, and sequence specificity of NC’s DNA bending behavior have been systematically studied. We have performed SM-FRET measurements on a series of duplexed DNA segments with varying sequences, lengths, and local structures in the presence of the wide-type HIV-1 NC and NC mutants lacking either the basic N-terminal domain or the zinc fingers. Based on the SM-FRET results, we have proposed a possible mechanism for the NC-induced DNA bending in which both NC’s zinc fingers and N-terminal domain are found to play crucial roles. The SM-FRET results reported here add new mechanistic insights to the biological behaviors and functions of HIV-1 NC as a retroviral DNA-architectural protein which may play critical roles in the compaction, nuclear import, and integration of the proviral DNA during the retroviral life-cycle.
Keywords: single-molecule spectroscopy, FRET, HIV-1, nucleocapsid, DNA bending, nucleic acid chaperone
Introduction
Double-stranded DNA (dsDNA) has long been considered as a relatively stiff polymer with a bending-persistence length of ~ 50 nm (140–150 bp) in salted buffer solutions.1, 2 A short dsDNA segment within the persistence length is thought to behave like a rigid rod that requires large force and energy-cost to bend.1, 2 Although energetically unfavorable, sharp DNA bending over length scales as small as a few nanometers occurs spontaneously with the help of DNA-bending proteins.3–14 DNA in vivo is often sharply distorted away from its classic B-form conformations, giving rise to highly compacted DNA structures in nucleoprotein complexes, such as eukaryotic nucleosomes,7, 10 viral capsids,11 and transcription initiation complexes.12–14 The molecular conformations and conformational dynamics of these nucleoprotein complexes are essentially key factors that determine the thermodynamics and kinetics of various biomolecular processes involving such nanoscale DNA bending.4, 6, 7, 10
Probing the conformational dynamics associated with local DNA bending over nanometer length scale, however, has been a significant challenge. Such protein-induced DNA bending usually involves various intermediates along multiple reaction pathways and exhibits heterogeneous molecular dynamics over multiple time-scales 8, 9, 15–20 that cannot be easily synchronized and thereby resolved by conventional ensemble measurements. Using single-molecule approaches turns out to be an extremely powerful way to characterize these complex biomolecular processes without the ensemble averaging effects.
Here we use single-molecule Förster Resonance Energy Transfer (SM-FRET) 21 to probe the conformational dynamics and heterogeneity associated with the nanoscale DNA bending induced by the HIV-1 nucleocapsid (NC) protein. HIV-1 NC is a small viral protein (55 amino-acid in length) composed of a basic N-terminal domain and two nonequivalent CCHC-type zinc fingers.22, 23 In spite of its structural simplicity, NC behaves as a multifunctional viral protein that has long been recognized as both a structural protein that stabilizes the virion and a nucleic acid chaperone that facilitates critical structural rearrangements of nucleic acids, such as the strand transfers during the reverse transcription.22, 23 While still lacking molecular-level understanding, NC’s roles in proviral DNA compaction and integration have also been proposed.24–26 Whether the NC-mediated proviral DNA compaction involves sharp, nanoscale DNA bending had been an open question until recently we discovered that HIV-1 NC could indeed induce dynamic sharp bending of a short proviral DNA segment over nanometer length scale.27 The biological roles of NC as a nanoscale DNA-bending protein in retrovirus are believed to be related to several key events during the retroviral life-cycle, such as the protection of the proviral DNA from nuclease digestion, nuclear import of the proviral DNA, and the integration of proviral DNA into the host cell genome.24–27
SM-FRET is a powerful spectroscopic tool that one can use to unravel the complex structural dynamics of biomolecules.18, 21, 28–39 In this paper, we use SM-FRET as a nanoscale molecular ruler to study in great detail the conformational dynamics of the DNA-NC nucleoprotein complexes over multiple time scales from milliseconds to minutes, based on which molecular-level understanding of how NC’s binding locally distorts the structures of DNA has been developed. We first compare the conformational dynamics of a fully duplexed 59-base pair (bp) DNA segment corresponding to the HIV-1 Trans Activating Response (TAR) sequences as a model system in the absence and presence of NC. Several important issues regarding the DNA-NC interactions, such as the bending dynamics, reversibility and sequence specificity, have been addressed using SM-FRET. We then present SM-FRET results on DNA duplex segments with mismatching bases to show how the presence of internal loops modifies the energy landscape and kinetics of the DNA bending process. To gain further insights into the mechanisms, we have also compared the DNA bending behaviors of the wide-type HIV-1 NC and NC mutants lacking either the basic N-terminal domain or the zinc fingers. To correlate the DNA bending with the NC binding stoichiometry, we have further investigated the effects of NC concentrations and the bending of DNA segments with varying lengths. Finally in the summary section, we propose a possible mechanism based on the SM-FRET results for the NC-induced DNA bending in which NC’s zinc fingers and N-terminal domain are both found to play crucial roles
Experimental Details
Sample preparation
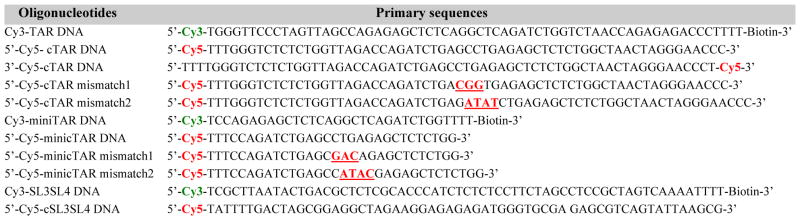
DNA oligonucleotides containing appropriate dye-labeling and biotin-functionalization were purchased from Integrated DNA Technologies (Coralville, IA) and were purified by the supplier using high-performance liquid chromatography (HPLC). All the DNA oligonucleotides used in the present studies are listed in Table 1. The dyes were attached to the thymine at the 5′ or 3′ end of the DNA molecules through a spacer composed of 6 saturated carbon–carbon bonds to ensure the rotational freedom of the dye molecules such that all relative dipole orientations were sampled much faster than the timescale of the measurements, giving rising to an average orientation factor (κ2) of 2/3.28 The DNA duplexes were formed through the well-studied NC-chaperoned strand annealing between the Cy3-labeled DNA oligonucleotides and their complimentary Cy5-labeled DNA oligonucleotides as described previously.40, 41 Some mismatched nucleotides were included in several Cy5-labeled DNA oligonucleotides (highlighted with underlines and marked in red in Table 1) to form DNA duplexes with internal loops at the mid-point of the duplexed DNA segments upon strand annealing. Biotin functionalization was added to the 3′ end of the Cy3-labeled oligonucleotides for the immobilization of the molecules on coverslips through biotin-streptavidin interactions. The HIV-1 NC protein for these experiments was prepared following the protocol reported previously. 42, 43
Table 1.
Molecular constructs for SM-FRET measurements. Primary sequences of the DNA oligonucleotides used in the present studies are listed, along with appropriate Cy3/Cy5 and biotin functionalization for each oligonucleotide.
|
Flow cell system for SM-FRET measurements
The short segments of DNA duplexes labeled with Cy3 and Cy5 at the two ends were formed through the NC-chaperoned annealing reactions carried out in a home-built flow cell system. Typically, 10 nM Cy5-labeled oligonucleotides and 1μM NC protein solutions were co-flowed into the flow cell to react with the Cy3-labeled oligonucleotides that were immobilized on the coverslip surface to form the DNA duplexes. The kinetics of the annealing reactions could be studied in great detail using SM-FRET as described in previous papers.40, 41 The commercial coverslips (Fisher Scientific) were cleaned with piranha (sulfuric acid : hydrogen peroxide, 7:3) and then treated with Vectabond/acetone 1% w/v solutions (Vector Laboratories, Burlingame, CA) for 5 min. Each coverslip was subsequently PEGylated and biotinylated, after which a reaction chamber with inlet and outlet ports (Nanoport, Upchurch Scientific, Oak Harbor, WA) was assembled. The details of the chamber assembly process and the immobilization of DNA oligonucleotides on coverslip surface have been described previously.27, 43 All the reactions were carried out at room temperature in HEPES buffer (25 mM HEPES, 40 mM NaCl, and 0.2 mM MgCl2, pH = 7.3) in an oxygen scavenger system 33 containing β-D(+)glucose 3% w/v (Sigma-Aldrich, St. Louis, MO), glucose oxidase 0.1 mg/mL, catalase 0.02 mg/mL (Roche Applied Science, Hague Road, IN) and 1% v/v 2-mercaptoethanol (Sigma-Aldrich, St. Louis, MO).
Confocal fluorescence microscope for SM-FRET measurements
A home-built scanning confocal optical microscope system based on a Zeiss inverted microscope 27, 40, 41, 44 was used in these SM-FRET experiments. A Queensgate X,Y scanning stage (NPW-XY-100A, Queensgate, Torquay, U.K.) was used to scan the samples. An oil immersion, high numerical aperture objective (Zeiss Fluar, 100×, NA 1.3) was used for excitation (514nm or 633nm) and signal collection. An argon gas laser (514 nm) (Melles Griot) and a HeNe laser (633 nm) (Melles Griot) were used for excitations and the laser power focused on the samples was typically 8 μW for the argon laser or 1 μW for the HeNe laser. The donor and acceptor fluorescence were separated by a dichroic beam splitter (Chroma 630 DCXR, Chroma Tech., VT) into two beams, and each was detected by an avalanche photodiode (APD) (Perkin-Elmer Optoelectronics SPCM-AQR-15, Vaudreuil, QC, Canada).
Data collection and analysis
The time-resolved FRET trajectories of individual molecules were recorded using the scanning confocal microscope in two data collection modes: image scanning mode and individual trajectory mode. In the image scanning mode, SM-FRET data were collected synchronously through separate detection channels for Cy3 and Cy5 fluorescence intensities while rapidly switching the laser excitation between 514 nm and 633 nm (29 KHz), which selectively excited Cy3 and Cy5, respectively. For each scanned sample region, three confocal images were obtained simultaneously including a donor image (514 nm excitation), an acceptor image (514 nm excitation), and a red channel image (633 nm excitation). The SM-FRET images were acquired at several times, t, with typical time intervals of ~ 230 s between adjacent images. A custom-written Matlab routine was used to find individual molecules, calibrate stage-drift, subtract image background, correct donor/acceptor signal cross-talks, and calculate FRET efficiencies for each molecule. The corrected donor and acceptor intensities, ID(t) and IA(t), respectively, were used to calculate the trajectories of apparent FRET efficiency, EA(t), using the following equation:
| (1) |
The actual FRET efficiency, EFRET(t), is related to EA(t) by the inclusion of the dye quantum yields, ϕi, and detector quantum efficiencies, ηi, as in
| (2) |
To accurately calculate EFRET, the changes in the quantum yields of both the donor (ϕD) and the acceptor (ϕA) due to protein binding should also be taken into consideration.27 It was determined that EA(t) EFRET(t) 43, 45 in the case of the current experimental setup. Since we used SM-FRET to probe the conformational dynamics rather than the absolute end-to-end distances of the DNA segments, the EA values obtained from the SM-FRET images were used to plot all the FRET trajectories and histograms.
In the individual trajectory mode, the laser (514 nm excitation) was focused on one individual immobilized molecule each time to continuously record the donor and acceptor trajectories typically for several seconds until the dyes were photobleached. The raw data of the SM-FRET trajectories were recorded with 1 ms time resolution and the individual EA(t) trajectories were subsequently boxcar time averaged, or “smoothed” using different bin times, τB, of for example 2, 10, 50, and 250 ms. During boxcar averaging, a group of N adjacent EA(t) points were averaged together where N = τB/τD. Here τD was the time spacing in the original non-time-averaged data, which was 1 ms in the present case. The boxcar time-averaged SM-FRET trajectories were then combined to yield ensemble EA histograms. The EA autocorrelations were calculated with 1 ms time resolution using the following equation:
| (3) |
The ensemble SM-FRET histograms were obtained by combining all the spectroscopic occurrences within SM-FRET trajectories from many individual molecules under the same condition. The data from molecules that were photobleached within 2 s were discarded. The ensemble FRET autocorrelation curves were obtained by averaging SM-FRET autocorrelations of many individual molecules under the same condition.
Results and Discussions
Scheme 1 illustrates the typical molecular constructs that were used in SM-FRET measurements, which included some modifications to those reported in our previous paper.27 The donor and acceptor dyes were labeled at the two ends of the duplexed DNA segment and attached to the different DNA strands in the duplex. Biotin was attached to the end of the DNA segment instead of being in the middle of the duplex such that the interference from surface immobilization can be in principle minimized. The surface of the coverslip was functionalized with a self-assembled monolayer of PEG to suppress the nonspecific interactions between the biomolecules of interest and the coverslip surface. The bending of the DNA segments upon NC binding, which was instantaneously accompanied by changes in the end-to-end distance of the DNA, was monitored in real time using FRET. The time-resolved FRET trajectories of individual molecules were recorded using a combination of the image scanning mode and the individual trajectory mode. The image scanning mode was used to globally image the samples to study relatively slow dynamics on time scale of minutes while the individual trajectory mode was used to study relatively fast dynamics on the time scales from milliseconds to seconds.
Scheme 1.

Schematics illustrating the NC-induced nanoscale bending of a dual dye-labeled, fully duplexed DNA segment.
Conformational dynamics of fully duplexed TAR-cTAR segments
Using SM-FRET, we first set out to study how the binding of NC modifies the conformational dynamics of a 59-bp fully duplexed DNA segment corresponding to the TAR sequences of the HIV-1 proviral DNA. The duplexed DNA segment under current investigations was formed though NC-chaperoned annealing of a Cy5-labeled cTAR DNA hairpin to a Cy3-labeled TAR DNA hairpin immobilized on a coverslip, which has been previously studied in great detail.27, 40, 41 Figure 1A shows the SM-FRET trajectories (obtained in image scanning mode) of 120 DNA duplex molecules found in a 30 μm × 30 μm region and the corresponding molecularly-averaged EA trajectory during two different time periods. During time period I, the immobilized DNA duplexes were exposed to 2 μM NC, while during time period II the DNA molecules were exposed to buffer. The dissociation constant (Kd) values for NC’s binding to various duplexed DNA segments and DNA hairpins have been measured to be in the range of 50 nM to 400 nM under various buffer conditions.22 A NC concentration of 2 μM, which was well above the Kd, was used in the present case to ensure saturated binding of NC to the DNA substrates. The significantly reduced end-to-end distances of the NC-bound DNA duplexes in comparison to those of the naked DNA duplexes in buffer were a direct result of NC-induced DNA bending. Since this dsDNA segment is shorter than the DNA’s persistence length, the naked duplexes can be regarded as rigid rods with an end-to-end distance of ~20 nm, which should correspond to FRET close to 0. However, when bound with NC during time period I, the DNA duplexes were sharply bent to give much smaller end-to-end distances that fell into the regime where FRET efficiency became sensitive to the distance between Cy3 and Cy5. The rapid fluctuation in the EA values of individual molecules and the large standard deviations of the molecularly-averaged EA values indicated that multiple conformations of the DNA-NC nucleoprotein complexes interconverted dynamically at room temperature on the time scale of minutes.
Figure 1.

Conformational dynamics of TAR-cTAR DNA duplexes in the presence and absence of NC. (A) SM-FRET trajectories (obtained in the image scanning mode) of 120 molecules found in a 30 μm × 30 μm region (each colored line corresponds to a single molecule) and the corresponding molecularly-averaged FRET trajectory. The dual dye-labeled TAR-cTAR DNA duplexes were immobilized on a coverslip. During time periods I and II, 2μM NC and buffer were flowed into the reaction chamber, respectively. (B) FRET histograms of all spectroscopic occurrences (upper panel) and time-averaged FRET (lower panel) of the TAR-cTAR duplex molecules in 2μM NC and buffer. Donor, acceptor and the corresponding FRET trajectory (obtained in the individual trajectory mode) collected on one representative molecule in buffer (C) and in 2 μM NC (D) with a bin time of 10 ms. (E) The ensemble FRET histograms obtained from FRET trajectories of 64 molecules in buffer with a bin time τB of 2 ms, 10 ms, 50 ms, and 250 ms. (F) The ensemble FRET autocorrelation obtained from FRET trajectories of the 64 molecules with a time resolution of 1 ms. (G) The ensemble FRET histograms obtained from FRET trajectories of 64 molecules in 2 μM NC with a bin time τB of 2 ms, 10 ms, 50 ms, and 250 ms. (H) The ensemble FRET autocorrelation obtained from FRET trajectories of the 102 molecules with a time resolution of 1 ms.
During time period II, the bound NC molecules were washed off the DNA segments by flowing buffer into the reaction chamber. As shown in previous papers,27, 40 the bound NC can be completely removed from DNA duplexes or DNA hairpin structures by performing this buffer wash step for ~ 100 s. It was observed that the FRET values of individual molecules became very close to zero after buffer wash. This clearly indicated an increase in the end-to-end distances of the DNA duplexes because the possibility of acceptor photobleaching or strand dissociation was ruled out by the measurements of Cy5 emission excited by the HeNe laser (633 nm excitation). In contrast to the NC-bound DNA segments, the naked DNA segments in buffer showed relatively static extended conformations. There were relatively small fluctuations in FRET with very small probability of having FRET values larger than 0.3 (< 3 %). The standard deviations of the molecularly-averaged EA values also significantly decreased in comparison to those of the duplexes in 2 μM NC. These indicated that the equilibrium distribution of naked DNA duplexes strongly favored the extended conformations with end-to-end distances much larger than the Förster Radius of Cy3–Cy5 pair (~6 nm)38 and the probability of observing conformational dynamics associated with sharp bending of the TAR-cTAR duplexes in the absence of NC was extremely rare. These results are in line with our previous observations on DNA segments with biotin-functionalization added in the middle of the duplexes.27
One straightforward way to illustrate the conformational dynamics of the DNA-NC complexes is to compare the EA histograms of all spectroscopic occurrences (Figure 1B, upper panel) with those of the time-averaged EA (Figure 1B, lower panel). Here the time-averaged EA refers to the average EA of each molecule in all scanned images. For the DNA duplexes in buffer, there was only one peak centered at ~ 0.05 in the EA histogram for all spectroscopic occurrences. This peak remained at the same position but became narrower in the time-averaged EA histograms simply due to the signal averaging. For the DNA duplexes in 2 μM NC, multiple peaks showed up in the EA histogram of all spectroscopic occurrences, indicating the coexistence of several highly bent (EA > 0.4) and less bent (0.1< EA <0.4) conformations. These conformations dynamically interconverted in a highly reversible manner on time scale of minutes, giving rise to a single-peaked distribution centered at ~0.4 in the time-averaged EA histogram. The fully reversible conformational changes over time scale of minutes can be further verified by “sub-ensemble” analysis on the EA trajectories as shown in Figure S1 in supporting information.
Figures 1C and 1D show representative SM-FRET trajectories, obtained in the individual trajectory mode (bin time of 10 ms), of TAR-cTAR DNA duplex in buffer and in 2 μM HIV-1 NC, respectively. One of the simplest ways to represent SM-FRET data is to show the ensemble EA histograms. To construct ensemble EA histograms, the individual EA(t) trajectories were boxcar time-averaged with varying bin times, τB, of 2, 10, 50, and 250 ms. The boxcar time-averaged single molecule trajectories were combined to yield ensemble EA histograms as shown in Figures 1E and 1G. As shown in Figure 1C, the naked DNA displayed very small FERT fluctuations on time scales ranging from milliseconds to seconds, indicating a relatively static conformation of the DNA duplex in buffer. The single-peaked EA distribution centered at ~0.05 agreed very well with the EA histogram obtained using the image scanning mode shown in Figure 2B. The τB = 2 ms EA histogram showed a relatively broad peak corresponding to the predominant extended conformation of the DNA duplexes. The width of the peak was primarily due to photon shot noise rather than EA fluctuations, which was verified by the fact that as τB increased, the single-peak feature in the ensemble EA histograms became significantly narrower without any peak position shift.
Figure 2.

Conformational dynamics of TAR-cTAR mismatch1 DNA duplexes in 2μM NC and in buffer. (A) SM-FRET trajectories (obtained in the image scanning mode) of 197 molecules found in a 30 μm × 30 μm region and the corresponding molecularly-averaged FRET trajectory. The dual dye-labeled TAR-cTAR mismatch1 DNA duplexes were immobilized on a coverslip. During time periods I and II, 2μM NC and buffer were flowed into the reaction chamber, respectively. (B) FRET histograms of all spectroscopic occurrences (upper panel) and time-averaged FRET (lower panel) of the DNA duplex molecules in 2μM NC and buffer. Donor, acceptor and the corresponding FRET trajectory (obtained in the individual trajectory mode) collected on one representative molecule in 2μM NC (C) and in buffer (D) with a bin time of 10 ms. (E) The ensemble FRET histograms obtained from FRET trajectories of 70 molecules in 2μM NC with a bin time τB of 2 ms, 10 ms, 50 ms, and 250 ms. (F) The ensemble FRET autocorrelation obtained from FRET trajectories of the 70 molecules with a time resolution of 1 ms. (G) The ensemble FRET histograms obtained from FRET trajectories of 60 molecules in buffer with a bin time τB of 2 ms, 10 ms, 50 ms, and 250 ms. (H) The ensemble FRET autocorrelation obtained from FRET trajectories of the 60 molecules with a time resolution of 1 ms.
Ensemble EA autocorrelation is another way to characterize EA dynamics. Figure 1F shows the ensemble EA autocorrelation for the naked TAR-cTAR DNA duplexes in buffer, which clearly shows no observable conformational dynamics over time scales form milliseconds to seconds, further verifying the static, extended conformation of the naked DNA duplexes. The SM-FRET results obtained in both the image scanning and individual trajectory modes are in line with the classic picture of duplexed DNA in which dsDNA shorter than the persistence length is considered to behave like a rigid rod.1 Although some recent experimental observations have suggested that duplexed DNA might be more flexible, especially on the nanometer length scale,19, 46–48 than described in the classic picture, SM-FRET results reported here apparently show that the probability of observing sharp bending of TAR-cTAR duplexes, which is evident by a significant increase in EA, is extremely rare for DNA molecules in absence of NC.
In striking contrast to the naked DNA in buffer, the DNA-NC nucleoprotein complexes showed highly complex conformational dynamics over multiple time scales. As shown in Figure 1D, the EA trajectory of one representative DNA-NC complex molecule in the presence of 2μM NC displays dynamic conformational fluctuations. The ensemble EA histograms (Figure 1G) built from individual molecular trajectories clearly showed the multi-peaked features, corresponding to the presence of multiple DNA bent conformations. The dynamics of the conformational fluctuations were found to very complicated. At τB = 2 ms, the EA histogram showed a very broad feature that possibly contained several photon shot noise-dominated broad peaks that overlapped with each other. As the τB values were progressively increased, each feature gradually got narrower and became distinguishable from each other. It is noteworthy that as τB increased, in addition to the increase in the signal-to-noise ratio of the data due to time averaging, the EA fluctuations were, however, also smoothed, or averaged, out of the data by this process if the conformational dynamics of the molecules occurred on a time scale faster than τB. Although some fast conformational dynamics on time-scale of milliseconds were averaged out as τB was increased all the way from 2 ms to 250 ms, several conformations with apparent life-times longer than sub-seconds, which were evident by the presence of several distinguishable features in the EA histograms with τB of 250 ms, could be clearly observed. The ensemble EA autocorrelation curve could be well fit using at least a three-step exponential decay with relaxation times over multiple time scales from milliseconds to subseconds. Such complicated multiple time-scale decay profile of the EA autocorrelation further suggested that in addition to the existence of multiple conformations, there might also be multiple reaction pathways for the interconversion between the various conformations. The complicated heterogeneity in the conformational dynamics of the DNA-NC complexes is likely a result of the heterogeneous NC-nucleic acid interactions.
The EA fluctuations over the multiple time-scales observed here are direct consequences of the dynamic conformational changes of the biomolecules. The possible complication due to photophysics of the labeling dyes was ruled out by SM-FRET measurements performed on TAR-cTAR duplexes with the donor and acceptor dyes at the same end of the DNA duplex. As shown in Figure S2 in the supporting information, the EA trajectory for each individual molecule showed very small fluctuations around EA of 1 both in the presence of 2 μM NC and in buffer, which indicated that the interference from photoblinking or other processes associated with dye photophysics causing EA fluctuations was not a significant issue under the current experimental conditions. The ensemble EA autocorrelation amplitudes shown in Figure S2 were extremely small in comparison to that illustrated in Figure 1. These small EA autocorrelation amplitudes are presumably due to the localized base-pair breathing in dsDNA, which typically occurs on millisecond time scale.48–51
Reversibility and sequence specificity of NC’s DNA bending behavior
The dynamic nanoscale DNA bending observed here is a direct consequence of NC’s binding to DNA, which has been reported to be highly dynamic and reversible.22, 40, 42 Here, we have performed multiple cycles of switching between 2 μM NC and buffer solutions and the DNA bending/unbending was observed to be fully reversible (Figure S3 in supporting information). As demonstrated previously,27 it is highly likely that the reversible bending/unbending of the DNA segments essentially arises from the dynamic association/dissociation of NC with the DNA substrates.
Although HIV-1 NC has higher binding affinity to GT-rich sequences, it binds to various nucleic acid sequences and structures in a nonsequence-specific manner.22 Therefore, NC’s DNA bending behavior is expected to be nonsequence-specific at high NC concentrations when the GT-rich regions and the lower affinity binding sites on the DNA are all saturated with NC. The most straightforward way to test this is to perform SM-FRET measurements on DNA segments with sequences different from the TAR-cTAR model sequences. Figure S4 in supporting information shows the SM-FRET trajectories (obtained in the image scanning mode) of 210 molecules revealing the conformational dynamics of a fully duplexed 59-bp proviral DNA segment corresponding to the transcripts of the SL3-SL4 ψ recognition element region of HIV-1 genome (see the sequences in Table 1). Very similar to the TAR-cTAR sequences, dynamic nanoscale DNA bending was observed in the presence of 2 μM NC whereas the DNA segments showed relatively static, extended conformations in buffer solution. The NC-induced DNA bending could be effectively inhibited in the presence of excessive non-specific binding competitors, such as Mg2+ (Figure S5 in supporting information), further verifying the nonsequence-specificity of NC’s DNA bending behavior. Such nonsequence-specific DNA bending behavior suggests that NC can induce the formation of many sharp DNA kinks all the way along the proviral DNA chain, leading to the effective proviral DNA condensation. It is also likely that the bound NC molecules are mobile rather than localized on the proviral DNA due to lack of sequence-specificity, giving rise to the complicated and heterogeneous conformational dynamics of the DNA-NC complexes.
Conformational dynamics of DNA duplexes with internal loops
One of the key features of NC’s nucleic acid chaperone activity is NC’s capability to partially melt the Watson-Crick base pairs in the duplexed regions of nucleic acids.22, 42, 44, 45, 52–54 Such duplex-melting behavior of NC is essentially a result of NC’s stronger binding preference to single-stranded than double-stranded regions.22, 42, 44, 45, 52–54 It is essentially these locally melted regions, also known as “bubbles”, that have been reported to serve as the nucleation sites for the initiation of the annealing reactions during the minus strand transfer in reverse transcription.40, 41, 43 It is hypothesized that the NC-induced sharp DNA bending is directly related to NC’s abilities to shift the secondary structure of duplexed DNA toward partially melted conformations with transient bubbles distributed along the DNA chain that locally disrupt the DNA duplexes, giving rise to the significantly increased structural flexibility of the DNA duplexes.
In comparison to fully duplexed DNA segments, the presence of internal loops near the mid-point of a short DNA duplex segment is hypothesized to help stabilize the highly bent conformation of DNA upon NC binding. The presence of such internal loops should also in principle profoundly change the energy landscape of the DNA bending process, thereby shifting the relative population of the various bent conformations at equilibrium and modifying the conformational dynamics. To test this hypothesis, we have performed SM-FRET measurements on duplexed DNA segments of mutated TAR-cTAR sequences with either a symmetric or an asymmetric looped region, also called as a “bubble”, at the mid-point of the segments.
SM-FRET data shown in Figure 2 illustrate the conformational dynamics of the TAR-cTAR mismatch1 DNA duplexes (see the sequences in Table 1) in 2μM NC and in buffer over multiple time scales. This duplexed DNA segment has a symmetric looped region composed of three nucleotide mismatches at the mid-point of the segment. Figure 2A shows the SM-FRET trajectories (obtained in the image scanning mode) of 197 molecules found in a 30 μm × 30 μm region and the corresponding molecularly-averaged FRET trajectory. During time period I when 2μM NC was flowed into the reaction chamber, the EA values of each molecule fluctuated around 0.6, giving rise to a single-peaked feature in the EA histogram of all spectroscopic occurrences as shown in the upper panel of Figure 2B. The profile of the EA distribution was far from a Gaussian line-shape and was asymmetrically broadened with a shoulder extending to the low EA region. The histogram of time-averaged EA (lower panel of Figure 2B) showed that the EA peak became significantly narrower without significant changes in its asymmetric profile after time-averaging, indicating that the peak broadening was not only dominated by photon shot noise but due to the presence of multiple conformations as well. In contrast to the fully duplexed TAR-cTAR segments in 2μM NC (see Figure 1) which showed several highly bent and less bent conformations with comparable populations at equilibrium, the highly bent conformation corresponding to EA of 0.6 dominated the distribution of conformations at equilibrium for this internally bubbled DNA segment. After a buffer wash step (time period II), the naked bubbled DNA duplexes in buffer showed much larger EA fluctuations than the fully duplexed DNA segments in buffer. As shown in the upper panel in Figure 2B, the EA histogram of all spectroscopic occurrences for the TAR-cTAR mismatch1 DNA duplexes in buffer displayed an asymmetric peak at around 0.2 with an asymmetric tail on the large EA side, indicating bent conformations for this DNA segment. An internally looped region in a DNA duplex is structurally flexible and kinking of DNA segments typically occurs at the loop regions.46, 55–57 It is likely that the bubble at the mid-point of the segments provide a flexible region where a kink is formed with two rigid duplexed arms jointed at the bubble region, forming a V-shaped structure. Since the bubble is structurally flexible, the two arms can move around flexibly, giving rise to the EA fluctuations observed here. The looped DNA duplex already adopts bent conformations in buffer and becomes more sharply bent upon NC binding possibly due to the increased structurally flexible of the two arms and/or increased kinking angles at the bulge region. In analogy to the fully duplexed DNA segments, the binding/unbinding of NC to this internally looped DNA segment is also reversible. Therefore, NC also exhibits reversible DNA bending behavior to TAR-cTAR mismatch1 DNA segments as shown in Figure S6 in supporting information.
SM-FRET measurements obtained in the individual trajectory mode provided more information about the conformational dynamics of this looped DNA segment on time scales from milliseconds to seconds. Figures 2C and 2D show the SM-FRET trajectories of one representative molecule for the bulged DNA segment in 2μM NC and buffer, respectively. When bound with NC, the bulged DNA segments were mostly highly bent with EA values fluctuating around 0.6 but sometimes adopted less bent conformations with EA values below 0.5. For naked DNA in buffer, EA fluctuated around 0.2 for most of the time and switched to some high EA over 0.4 occasionally. The EA transitions between the different bent conformations were not instantaneous but rather gradual, indicating that the conformational changes were rather continuous than abrupt. Figures 2E and 2G show the ensemble EA histograms of the TAR-cTAR mismatch1 DNA segments in 2μM NC and buffer, respectively. As we gradually increased the τB, the dominating EA peaks at 0.6 and 0.2 became sharper while some less populated EA states became better resolved. There were multiple conformations and interconversion pathways in these dynamic molecular systems as multiple exponential decays were used to fit the ensemble EA autocorrelations (Figures 2F and 2H) and the conformational dynamics spanned time scales from milliseconds to subseconds. The EA autocorrelation amplitude of the looped DNA in NC was smaller than that of the fully duplexed DNA in NC, indicating a reduced structural fluctuation of highly bent DNA conformations.
We have also performed SM-FRET measurements on a mutated TAR-cTAR duplex with an asymmetric bubble in the mid-point of the segment. SM-FRET data shown in Figure 3 illustrate the conformational dynamics of the TAR-cTAR mismatch2 DNA duplexes (see the sequences in Table 1) in 2μM NC and in buffer. Very similar conformations and conformational dynamics were observed for this asymmetrically looped DNA segments in comparison to the DNA segments with a symmetric internal loop shown in Figure 2, except that breaking the loop symmetry resulted in a further increased structural flexibility of naked DNA segments in buffer and a decreased structural flexibility of the highly bent conformations for DNA bound with NC according to the peak widths of EA histograms (Figures 3B, 3E, and 3G) and EA autocorrelation amplitudes (Figures 3F and 3H). NC also exhibits highly reversible DNA bending behavior to TAR-cTAR mismatch2 DNA segments, as shown in Figure S7 in supporting information.
Figure 3.

Conformational dynamics of TAR-cTAR mismatch2 DNA duplexes in 2μM NC and in buffer. (A) SM-FRET trajectories (obtained in the image scanning mode) of 158 molecules found in a 30 μm × 30 μm region and the corresponding molecularly-averaged FRET trajectory. The dual dye-labeled TAR-cTAR mismatch2 DNA duplexes were immobilized on a coverslip. During time periods I and II, 2μM NC and buffer were flowed into the reaction chamber, respectively. (B) FRET histograms of all spectroscopic occurrences (upper panel) and time-averaged FRET (lower panel) of the DNA duplex molecules in 2μM NC and buffer. Donor, acceptor and the corresponding FRET trajectory (obtained in the individual trajectory mode) collected on one representative molecule in 2μM NC (C) and in buffer (D) with a bin time of 10 ms. (E) The ensemble FRET histograms obtained from FRET trajectories of 72 molecules in 2μM NC with a bin time τB of 2 ms, 10 ms, 50 ms, and 250 ms. (F) The ensemble FRET autocorrelation obtained from FRET trajectories of the 72 molecules with a time resolution of 1 ms. (G) The ensemble FRET histograms obtained from FRET trajectories of 60 molecules in buffer with a bin time τB of 2 ms, 10 ms, 50 ms, and 250 ms. (H) The ensemble FRET autocorrelation obtained from FRET trajectories of the 60 molecules with a time resolution of 1 ms.
There are two major conclusions we can draw based on the SM-FRET data shown in Figures 2 and 3. First, internally looped DNA duplexes are structurally much more flexible than the fully duplexed DNA segments in buffer and the internal bubble provides a flexible region which is largely responsible for the dynamic kinking of the DNA segments observed here. Second, the presence of a bubble at the mid-point of a DNA segment can help stabilize the highly bent DNA conformations and shift the equilibrium toward the highly bent conformations upon NC binding.
DNA bending behavior of NC mutants
The two main features of NC’s nucleic acid chaperone activity are its abilities to aggregate nucleic acids and to destabilize nucleic acid duplexes,22 both of which are hypothesized to be crucial to NC’s DNA bending capability as well. To test our hypothesis, we have performed SM-FRET measurements to systematically compare the DNA bending behaviors of the wild-type HIV-1 NC protein and NC mutants lacking either the zinc fingers or the basic N-terminal domain.
It has been known that NC’s zinc fingers are responsible for the destabilization of the duplexed regions of nucleic acids.22, 42, 44, 45, 52–54, 58 The destabilization free energy per base pair for HIV-1 NC protein has been estimated to be ~ 0.26 kcal/mol/bp.59 This is a relatively moderate effect, considering that the average melting free energy per base pair at 37°C is 2 kcal/mol/bp for DNA duplexes.60 However, it may be sufficient to significantly shift the equilibrium toward the partially melted DNA conformations and increase the probability of having transient bubbles along the DNA segments, leading to a significant increase of the DNA’s structural flexibility. To examine the role of the zinc fingers in the nanoscale DNA bending, SM-FRET measurements were carried out with NC (SSHS),53, 54 a zinc-free mutant of the HIV-1 NC. This NC mutant is a variant in which all the three cysteines in each zinc finger were changed to serine (illustrated in Figure 4A). This is the smallest change one can make to NC that will essentially destroy the metal ion binding sites, while still allowing retention of neighboring residues such as certain basic and aromatic amino acids, which are also important for HIV-1 NC nucleic acid binding. It has been previously reported that this NC (SSHS) mutant is incapable of destabilizing DNA duplex structures due to lack of properly folded zinc finger structures.52–54 As shown in the top panel of Figure 4B, no apparent nanoscale bending of the TAR-cTAR DNA duplex was observed even in the presence of NC (SSHS) at concentration as high as 2 μM. In dramatic contrast to the wild-type NC which can increase the flexibility of duplexed DNA segments, the binding of NC (SSHS) to the internally looped DNA duplexes resulted in a significant increase in the rigidity of the DNA segments as indicated by the decrease in the EA values and the narrowing of the EA distribution (see the middle and bottom panels of Figure 4B). Such increased rigidity of DNA can be interpreted as a result of saturated binding of NC (SSHS) to the DNA segments, forming a rigid coaxial filament composed of a positively charged protein shell surrounding a negatively charged DNA core. This clearly verifies that NC’s zinc fingers, which play crucial roles in the duplex-destabilization, are indispensible to NC’s DNA bending capability.
Figure 4.

(A) Primary structures of the wild-type HIV-1 NC and the mutant NC (SSHS). (B) FRET histograms of all spectroscopic occurrences (obtained in image scanning mode) for DNA duplexes in 2μM NC (SSHS) and in buffer: (top panel) TAR-cTAR duplexes; (middle panel) TAR-cTAR mismatch1 duplexes; and (bottom panel) TAR-cTAR mismatch2 duplexes. The insets in each panel show the cartoons of each DNA construct used in the SM-FRET measurements.
We have also studied the DNA bending behavior of NC (11–55),22, 26, 61–63 an N-terminus truncated mutant that lacks the first ten residues (see Figure 5A). It is well-known that the cationic N-terminal domain is a major factor in NC’s nucleic acid binding and aggregation activity.22, 26, 61–63 Although NC (11–55) can also bind to nucleic acids with sub-micromolar Kd and locally melt the duplexed regions of the nucleic acids,26, 61, 64 it is incapable of aggregating nucleic acids due to lack of the basic N-terminal domain. As shown in the top panel of Figure 5B, no apparent nanoscale bending of the TAR-cTAR DNA duplex was observed in 2 μM NC (11–55). For the two looped DNA duplexes, the EA distributions in 2 μM NC (11–55) and in buffer were extremely similar as shown in the middle and bottom panels of Figure 5B. These clearly indicate that the N-terminal domain is also indispensible to NC’s DNA bending capability. We hypothesize that the N-terminal domain may contribute to the DNA bending through the charge neutralization of the phosphate backbone and probably more importantly through the creation of intramolecular interactions. The cationic N-terminal domain can effectively neutralize the phosphate backbone of the DNA to reduce the rigidity of the DNA segments and asymmetric charge neutralization of the phosphate backbone leads to the generation of a lateral force perpendicular to the DNA axis 52 which may induce the DNA bending. More importantly, strong intramolecular attractive interactions, which may provide additional force for the DNA bending, can be created when multiple copies of NC are bound to the DNA segments due to NC’s unique nucleic acid aggregating capability.
Figure 5.

(A) Primary structures of the mutant NC (11–55). (B) FRET histograms of all spectroscopic occurrences (obtained in image scanning mode) for DNA duplexes in 2μM NC (11–55) and in buffer: (top panel) TAR-cTAR duplexes; (middle panel) TAR-cTAR mismatch1 duplexes; and (bottom panel) TAR-cTAR mismatch2 duplexes. The insets in each panel show the cartoons of each DNA construct used in the SM-FRET measurements.
The SM-FRET measurements on the two NC mutants provide strong evidence that both the basic N-terminal domain and the zinc fingers of NC are crucial to NC’s DNA bending behavior. The intramolecular attraction force generated due to the nucleic acid aggregation propensity of NC’s N-terminal domain may provide the primary driving force for the DNA bending, however, it does not fully account for the sharp bending of short dsDNA segments observed in the present case. In order to allow the sharp bending of short dsDNA segments to occur at reduced energy-cost, localized duplex destabilization induced by NC’s zinc fingers is crucial as well. Although the melted/kinked regions might occur upon NC binding with enhanced probability at some locations along the DNA segments, they might also be free to diffuse along the DNA chain when the conformational interconversion of the nucleoprotein complexes occurs, which may essentially contribute to the highly complex conformational dynamics and heterogeneity associated with this nanoscale DNA bending process.
Correlation between DNA bending and NC binding stoichiometry
We have characterized the bending of TAR-cTAR DNA duplexes in the presence of varying NC concentrations, which provided important evidence that effective bending of a DNA duplex required the binding of multiple copies of NC to the DNA substrates. As reported previously 27 (also shown in Figure S8 in supporting information), with increase in NC concentration bent DNA conformations became increasingly populated in comparison to the extended conformations and the fraction of highly bent versus less bent conformations also gradually increased. The effective bending of the DNA segments, which was evident by the characteristic multi-peaked features of the EA histograms, was achieved at NC concentrations above 1 μM. It indicated that effective DNA bending required saturated binding of multiple copies of NC to the DNA substrates, which could be achieved only at high NC concentrations. As shown in Figure 6, the sharp increase in the molecularly-averaged FRET (<EA>) over a narrow NC concentration window implies that the DNA bending process is highly cooperative, involving multiple NC molecules bound to one DNA segment. We fit the experimental data using a least square fitting process with the following modified Hill equation
Figure 6.

Molecularly-averaged FRET, <EA>, as a function of NC concentration. The error bars represent the standard deviations of the <EA> values obtained from three experimental runs. The curve shows the results of least square fitting using a modified Hill equation shown in Equation (4).
| (4) |
where <EA>0 is the <EA> value in the absence of NC, <EA>max is the <EA> value upon saturated NC binding, K is the NC concentration producing an <EA> value that equals (<EA>0 + <EA>max )/2, and n is the Hill coefficient. A best fit with an R-square value of 0.9917 was obtained when <EA>max - <EA>0 = 0.3326, n = 4.203, and K = 704.2 nM (see the red curve in Figure 6). This n value was much large than 1, indicating the high cooperativity of the DNA bending process.
The binding footprint size of NC on nucleic acids was estimated to be 7–8 nucleotides for single-stranded and 3–4 base pairs for double-stranded regions,23, 24, 59 respectively. Since the TAR-cTAR DNA duplex is 59-nt in length, it is hypothesized that the efficient bending of this DNA segment involves the binding of roughly 15–20 copies of NC to the DNA under saturating NC binding conditions. To gain further insights, we have performed SM-FRET measurements on DNA segments with shorter lengths. Figure 7A shows FRET histograms of (obtained in image scanning mode) for the miniTAR-minicTAR DNA duplexes (see the sequences in Table 1) in 2μM NC and in buffer. This DNA segment is a truncated TAR-cTAR segment with 27 base pairs, which only allows binding of roughly 6–9 copies of NC under saturating NC binding conditions. It is apparent that 6–9 copies of NC were not enough to bend this DNA segment efficiently as evident by the fact that the fraction of bent conformations with EA larger than 0.4 was very small (<5%) at NC concentration of 2 μM. We have also measured the bending of short duplexed DNA segments with either a symmetric bubble (miniTAR-minicTAR mismatch1 duplexes, see the sequences in Table 1) or an asymmetric bubble (miniTAR-minicTAR mismatch2 duplexes, see the sequences in Table 1) at the mid-point of the segments (see Figures 7B and 7C). In both cases, we hypothesized that 6–9 copies of NC were bound to one DNA segment under saturating NC binding conditions with one copy of NC bound to the loop region and 3–4 copies of NC bound to each duplexed arm adjacent to the bulge. The characteristic peaks in the EA histograms became broader upon NC binding, indicating that NC could induce an increase in the structural flexibility of the DNA segments. However, the DNA segments bound with NC showed small shifts of EA toward higher values with respect to the EA values of naked DNA segments in buffer. It is apparent that NC does not bend these shorter DNA segments as sharply as those longer segments shown in Figures 1, 2, and 3.
Figure 7.

FRET histograms of all spectroscopic occurrences (obtained in image scanning mode) for DNA duplexes in 2μM NC and in buffer for: (A) miniTAR-minicTAR duplexes; (B) miniTAR-minicTAR mismatch1 duplexes; and (C) miniTAR-minicTAR mismatch2 duplexes. The insets in each panel show the cartoons of each DNA construct used in the SM-FRET measurements.
This less effective bending of these short DNA segments can be interpreted as a result of having fewer NC proteins bound on each DNA segment. Since there are only 3–4 copies of NC allowed to bind to each arm, the intramolecular interactions, or the “bridging” effects, between the two arms arising from the NC’s nucleic acid-aggregating behavior are rather limited in comparison to those longer sequences on which 7–9 copies of NC are bound on each arm. Therefore, it is not surprising that these shorter DNA segments are less sharply bent than the longer segments under saturating NC binding conditions. This strongly indicates that the sharp DNA bending over nanometer length scale is correlated to NC’s binding stiochiometry. Binding of multiple copies of NC to the DNA substrates under saturating NC binding conditions favors the sharp DNA bending.
Summary
We have studied the highly complex conformational dynamics associated with the HIV-1 NC induced nanoscale DNA bending using SM-FRET. In striking contrast to fully duplexed DNA segments in buffer which show relatively static extended conformations, the DNA bound with multiple copies of NC shows multiple bent conformations which dynamically interconvert over a broad distribution of time-scales from milliseconds to minutes. NC’s DNA bending behavior has two interesting features. First, NC distorts the DNA local structures dynamically and reversibly over nanometer length scale. Second, NC bends DNA in a nonsequence-specific manner at μM level NC concentrations.
In comparison to fully duplexed DNA segments, the presence of an internal loop in a DNA segment significantly changes the energy landscape and conformational dynamics of the DNA bending process. While the bubbled DNA duplexes readily adopt bent conformations in buffer, they become more sharply bent upon binding of multiple copies of NC. The presence of an internal bubble at the midpoint of a DNA segment can shift the equilibrium towards the highly bent conformations upon NC binding and help stabilize the highly bent conformations of the DNA.
The binding of NC to DNA is accompanied by two major consequences both of which provide crucial contributions to the nanoscale DNA bending. First, the flexibility of the DNA duplexes dramatically increases due to duplex destabilization induced by NC’s zinc fingers and charge neutralization of the phosphate backbone. Second, upon binding of multiple copies of NC to a DNA segment, intramolecular interactions, which provide the driving force for DNA bending, are created due to the nucleic acid aggregation propensity of the N-terminal domain. Comparative studies on DNA bending behaviors of the wild-type HIV-1 NC and NC mutants show that both the basic N-terminal domain and the zinc fingers of NC are indispensible to NC’s DNA bending capability. By varying the NC concentrations or the length of DNA segments, we have found that binding of multiple copies of NC to the DNA segments under saturating NC binding conditions is required for the sharp DNA bending over nanometer length scale.
Based on all these mentioned above, we propose that the dynamic nanoscale DNA bending observed here involves the formation of NC-induced non-base-paired bubbles that locally disrupt the DNA duplexes. Each bubble may hypothetically act as a flexible hinge where a sharp DNA kink is created. NC-induced bubbles in a DNA duplex may be further stabilized or enhanced by additional “bridging” NC molecules between the two “arms” of the duplex jointed by the bubble. The intramolecular interactions arising from the nucleic acid-aggregating propensity of NC may be generated among the multiple copies of NC bound to the “arms”, which in this case favors a sharply bent DNA structure. These bubbles are transient and mobile in nature due to the dynamic association/dissociation of NC and NC’s nonsequence-specific interactions with the DNA substrates, which in turn gives rise to the complex conformational dynamics of the DNA-NC nucleoprotein complexes over broad distribution of time scales. The highly dynamic conformations of the DNA-NC complexes imply that NC can induce a large number of flexible sharp DNA kinks along the proviral DNA, giving rise to highly compacted but dynamic conformations of the proviral DNA. In this way, NC can reduce the proviral DNA’s rigidity globally without forming rigid local kink structures at specific sequences, which allows a series of viral or cellular proteins to access the proviral DNA for a multitude of biological tasks, such as the protection, nuclear import, and integration of the proviral DNA, during the retroviral life-cycle.
Supplementary Material
Acknowledgments
H. W. would like to acknowledge the Startup funds provided by the College of Arts and Sciences of University of South Carolina, the funds from Advanced Support Programs for Innovative Research Excellence (ASPIRE) provided by the USC Office of Research, and the financial support from Welch Foundation (awarded to P. F. B) on the last year of his postdoctoral research. K. M.-F. acknowledges funding from the National Institutes of Health (GM65056). The authors would also like to thank Robert Gorelick of NCI-Frederick for providing the SSHS NC mutant used in this work.
Footnotes
The single-molecule spectroscopic measurements reported in this paper were performed by H.W. during the last six months of his postdoctoral research with P.F.B. at the University of Texas at Austin. Data analysis and manuscript preparation were accomplished after H.W. joined the faculty of Chemistry and Biochemistry at University of South Carolina.
Supporting Information: Additional information including 8 figures as noted in the text. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Hagerman PJ. Annu Rev Biophys Biophys Chem. 1988;17:265–286. doi: 10.1146/annurev.bb.17.060188.001405. [DOI] [PubMed] [Google Scholar]
- 2.Crothers DM, Drak J, Kahn JD, Levene SD. Methods Enzymol. 1992;212:3–29. doi: 10.1016/0076-6879(92)12003-9. [DOI] [PubMed] [Google Scholar]
- 3.Schleif R. Annu Rev Biochem. 1992;61:199–223. doi: 10.1146/annurev.bi.61.070192.001215. [DOI] [PubMed] [Google Scholar]
- 4.Grosschedl R, Giese K, Pagel J. Trends Genet. 1994;10:94–100. doi: 10.1016/0168-9525(94)90232-1. [DOI] [PubMed] [Google Scholar]
- 5.Agresti A, Bianchi ME. Curr Opin Genet Dev. 2003;13:170–178. doi: 10.1016/s0959-437x(03)00023-6. [DOI] [PubMed] [Google Scholar]
- 6.Garcia HG, Grayson P, Han L, Inamdar M, Kondev J, Nelson PC, Phillips R, Widom J, Wiggins PA. Biopolymers. 2007;85:115–130. doi: 10.1002/bip.20627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kornberg RD, Lorch YL. Cell. 1999;98:285–294. doi: 10.1016/s0092-8674(00)81958-3. [DOI] [PubMed] [Google Scholar]
- 8.Kuznetsov SV, Sugimura S, Vivas P, Crothers DM, Ansari A. Proc Natl Acad Sci U S A. 2006;103:18515–18520. doi: 10.1073/pnas.0608394103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sugimura S, Crothers DM. Proc Natl Acad Sci U S A. 2006;103:18510–18514. doi: 10.1073/pnas.0608337103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Workman JL, Kingston RE. Annu Rev Biochem. 1998;67:545–579. doi: 10.1146/annurev.biochem.67.1.545. [DOI] [PubMed] [Google Scholar]
- 11.Knobler CM, Gelbart WM. Annu Rev Phys Chem. 2009;60:367–383. doi: 10.1146/annurev.physchem.59.032607.093728. [DOI] [PubMed] [Google Scholar]
- 12.Lee TI, Young RA. Annu Rev Genet. 2000;34:77–137. doi: 10.1146/annurev.genet.34.1.77. [DOI] [PubMed] [Google Scholar]
- 13.Vandervliet PC, Verrijzer CP. Bioessays. 1993;15:25–32. doi: 10.1002/bies.950150105. [DOI] [PubMed] [Google Scholar]
- 14.Coulombre B, Burton ZF. Microbiol Mol Biol Rev. 1999;63:457–478. doi: 10.1128/mmbr.63.2.457-478.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sass LE, Lanyi C, Weninger K, Erie DA. Biochemistry. 2010;49:3174–3190. doi: 10.1021/bi901871u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Parkhurst KM, Richards RM, Brenowitz M, Parkhurst LJJ. Mol Biol. 1999;289:1327–1341. doi: 10.1006/jmbi.1999.2835. [DOI] [PubMed] [Google Scholar]
- 17.Gansen A, Valeri A, Hauger F, Felekyan S, Kalinin S, Toth K, Langowski J, Seidel CAM. Proc Natl Acad Sci U S A. 2009;106:15308–15313. doi: 10.1073/pnas.0903005106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tomschik M, Zheng HC, van Holde K, Zlatanova J, Leuba SH. Proc Natl Acad Sci U S A. 2005;102:3278–3283. doi: 10.1073/pnas.0500189102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wiggins PA, Van der Heijden T, Moreno-Herrero F, Spakowitz A, Phillips R, Widom J, Dekker C, Nelson PC. Nat Nanotechnol. 2006;1:137–141. doi: 10.1038/nnano.2006.63. [DOI] [PubMed] [Google Scholar]
- 20.Zhang JY, McCauley MJ, Maher LJ, Williams MC, Israeloff NE. Nucleic Acids Res. 2009;37:1107–1114. doi: 10.1093/nar/gkn1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Roy R, Hohng S, Ha T. Nat Methods. 2008;5:507–516. doi: 10.1038/nmeth.1208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Levin JG, Guo JH, Rouzina I, Musier-Forsyth K. Progress in Nucleic Acid Research and Molecular Biology. Vol. 80. Vol. 80. Elsevier Academic Press Inc; San Diego: 2005. Nucleic acid chaperone activity of HIV-1 nucleocapsid protein: Critical role in reverse transcription and molecular mechanism; pp. 217–286. [DOI] [PubMed] [Google Scholar]
- 23.Rein A, Henderson LE, Levin JG. Trends Biochem Sci. 1998;23:297–301. doi: 10.1016/s0968-0004(98)01256-0. [DOI] [PubMed] [Google Scholar]
- 24.Lapadat-Tapolsky M, De Rocquigny H, Van Gent D, Roques B, Plasterk R, Darlix JL. Nucleic Acids Res. 1993;21:831–839. doi: 10.1093/nar/21.4.831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Buckman JS, Bosche WJ, Gorelick RJJ. Virol. 2003;77:1469–1480. doi: 10.1128/JVI.77.2.1469-1480.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Krishnamoorthy G, Roques B, Darlix JL, Mely Y. Nucleic Acids Res. 2003;31:5425–5432. doi: 10.1093/nar/gkg738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang H, Yeh YS, Barbara PFJ. Am Chem Soc. 2009;131:15534–15543. doi: 10.1021/ja9070046. [DOI] [PubMed] [Google Scholar]
- 28.Gell C, Brockwell D, Smith A. Handbook of single molecule fluorescence spectroscopy. Oxford University Press Inc; New York, NY: 2006. [Google Scholar]
- 29.Weiss S. Nat Struct Biol. 2000;7:724–729. doi: 10.1038/78941. [DOI] [PubMed] [Google Scholar]
- 30.Schuler B, Lipman EA, Eaton WA. Nature. 2002;419:743–747. doi: 10.1038/nature01060. [DOI] [PubMed] [Google Scholar]
- 31.Zhuang XW, Bartley LE, Babcock HP, Russell R, Ha TJ, Herschlag D, Chu S. Science. 2000;288:2048–2051. doi: 10.1126/science.288.5473.2048. [DOI] [PubMed] [Google Scholar]
- 32.Hohng S, Zhou RB, Nahas MK, Yu J, Schulten K, Lilley DMJ, Ha TJ. Science. 2007;318:279–283. doi: 10.1126/science.1146113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ha T. Curr Opin Struct Biol. 2001;11:287–292. doi: 10.1016/s0959-440x(00)00204-9. [DOI] [PubMed] [Google Scholar]
- 34.Liu RC, Hu DH, Tan X, Lu HPJ. Am Chem Soc. 2006;128:10034–10042. doi: 10.1021/ja057005m. [DOI] [PubMed] [Google Scholar]
- 35.Myong S, Rasnik I, Joo C, Lohman TM, Ha T. Nature. 2005;437:1321–1325. doi: 10.1038/nature04049. [DOI] [PubMed] [Google Scholar]
- 36.Xie Z, Srividya N, Sosnick TR, Pan T, Scherer NF. Proc Natl Acad Sci USA. 2004;101:534–539. doi: 10.1073/pnas.2636333100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liu SX, Abbondanzieri EA, Rausch JW, Le Grice SFJ, Zhuang XW. Science. 2008;322:1092–1097. doi: 10.1126/science.1163108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ha T, Rasnik I, Cheng W, Babcock HP, Gauss GH, Lohman TM, Chu S. Nature. 2002;419:638–641. doi: 10.1038/nature01083. [DOI] [PubMed] [Google Scholar]
- 39.Karunatilaka KS, Solem A, Pyle AM, Rueda D. Nature. 2009;467:935–939. doi: 10.1038/nature09422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zeng YN, Liu HW, Landes CF, Kim YJ, Ma XJ, Zhu YJ, Musier-Forsyth K, Barbara PF. Proc Natl Acad Sci USA. 2007;104:12651–12656. doi: 10.1073/pnas.0700350104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Liu HW, Zeng YN, Landes CF, Kim YJ, Zhu YJ, Ma XJ, Vo MN, Musier-Forsyth K, Barbara PF. Proc Natl Acad Sci USA. 2007;104:5261–5267. doi: 10.1073/pnas.0700166104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cosa G, Harbron EJ, Zeng YN, Liu HW, O’Connor DB, Eta-Hosokawa C, Musier-Forsyth K, Barbara PF. Biophys J. 2004;87:2759–2767. doi: 10.1529/biophysj.104.043083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Liu HW, Cosa G, Landes CF, Zeng YN, Kovaleski BJ, Mullen DG, Barany G, Musier-Forsyth K, Barbara PF. Biophys J. 2005;89:3470–3479. doi: 10.1529/biophysj.105.065326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wang H, Ma XJ, Yeh YS, Zhu YJ, Daugherty MD, Frankel AD, Musier-Forsyth K, Barbara PF. Biophys J. 2010;99:3454–3462. doi: 10.1016/j.bpj.2010.09.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cosa G, Zeng YN, Liu HW, Landes CF, Makarov DE, Musier-Forsyth K, Barbara PFJ. Phys Chem B. 2006;110:2419–2426. doi: 10.1021/jp054189i. [DOI] [PubMed] [Google Scholar]
- 46.Cloutier TE, Widom J. Proc Natl Acad Sci U S A. 2005;102:3645–3650. doi: 10.1073/pnas.0409059102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mathew-Fenn RS, Das R, Harbury PAB. Science. 2008;322:446–449. doi: 10.1126/science.1158881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yuan CL, Chen HM, Lou XW, Archer LA. Phys Rev Lett. 2008;100:018102. doi: 10.1103/PhysRevLett.100.018102. [DOI] [PubMed] [Google Scholar]
- 49.Cao CY, Jiang YL, Stivers JT, Song FH. Nat Struct Mol Biol. 2004;11:1230–1236. doi: 10.1038/nsmb864. [DOI] [PubMed] [Google Scholar]
- 50.Spies MA, Schowen RLJ. Am Chem Soc. 2002;124:14049–14053. doi: 10.1021/ja012272n. [DOI] [PubMed] [Google Scholar]
- 51.O’Neil LL, Wiest OJ. Am Chem Soc. 2005;127:16800–16801. doi: 10.1021/ja056274+. [DOI] [PubMed] [Google Scholar]
- 52.Cruceanu M, Gorelick RJ, Musier-Forsyth K, Rouzina I, Williams MC. J Mol Biol. 2006;363:867–877. doi: 10.1016/j.jmb.2006.08.070. [DOI] [PubMed] [Google Scholar]
- 53.Williams MC, Gorelick RJ, Musier-Forsyth K. Proc Natl Acad Sci U S A. 2002;99:8614–8619. doi: 10.1073/pnas.132128999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Williams MC, Rouzina I, Wenner JR, Gorelick RJ, Musier-Forsyth K, Bloomfield VA. Proc Natl Acad Sci U S A. 2001;98:6121–6126. doi: 10.1073/pnas.101033198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yuan CL, Rhoades E, Lou XW, Archer LA. Nucleic Acids Res. 2006;34:4554–4560. doi: 10.1093/nar/gkl394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yan J, Marko JF. Phys Rev Lett. 2004;93:108108. doi: 10.1103/PhysRevLett.93.108108. [DOI] [PubMed] [Google Scholar]
- 57.Gohlke C, Murchie AIH, Lilley DMJ, Clegg RM. Proc Natl Acad Sci USA. 1994;91:11660–11664. doi: 10.1073/pnas.91.24.11660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Urbaneja MA, Wu M, Casas-Finet JR, Karpel RLJ. Mol Biol. 2002;318:749–764. doi: 10.1016/S0022-2836(02)00043-8. [DOI] [PubMed] [Google Scholar]
- 59.Stewart-Maynard KM, Cruceanu M, Wang F, Vo MN, Gorelick RJ, Williams MC, Rouzina I, Musier-Forsyth KJ. Virol. 2008;82:10129–10142. doi: 10.1128/JVI.01169-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.SantaLucia J, Hicks D. Annu Rev Biophys Biomolec Struct. 2004;33:415–440. doi: 10.1146/annurev.biophys.32.110601.141800. [DOI] [PubMed] [Google Scholar]
- 61.Godet J, de Rocquigny H, Raja C, Glasser N, Ficheux D, Darlix JL, Mely YJ. Mol Biol. 2006;356:1180–1192. doi: 10.1016/j.jmb.2005.12.038. [DOI] [PubMed] [Google Scholar]
- 62.Mirambeau G, Lyonnais S, Coulaud D, Hameau L, Lafosse S, Jeusset J, Justome A, Delain E, Gorelick RJ, Le Cam EJ. Mol Biol. 2006;364:496–511. doi: 10.1016/j.jmb.2006.08.065. [DOI] [PubMed] [Google Scholar]
- 63.Vo MN, Barany G, Rouzina I, Musier-Forsyth KJ. Mol Biol. 2006;363:244–261. doi: 10.1016/j.jmb.2006.08.039. [DOI] [PubMed] [Google Scholar]
- 64.Urbaneja MA, Kane BP, Johnson DG, Gorelick RJ, Henderson LE, Casas-Finet JRJ. Mol Biol. 1999;287:59–75. doi: 10.1006/jmbi.1998.2521. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.