

Chronic low grade tissue inflammation is an important cause of systemic insulin resistance, and is a key component of the decreased insulin sensitivity which exists in obesity and type 2 diabetes 1,2. Adipose tissue inflammation has been established as an important factor in the etiology of insulin resistance. Immune cells such as macrophages, T–cells, B–cells, mast cells and eosinophils have all been implicated as playing a role in this process 3–8. Neutrophils are the first immune cells to respond to inflammation, and can promote a more chronic inflammatory state by helping to recruit macrophages and interacting with antigen presenting cells 9–11. Neutrophils secrete several proteases, one of which is neutrophil elastase (NE), which can promote inflammatory responses in several disease models 12. Here we demonstrate that treatment of hepatocytes with NE causes cellular insulin resistance and that deletion of NE in obese mice leads to decreased tissue inflammation associated with reduced adipose tissue neutrophil and macrophage content. This is accompanied by improved glucose tolerance and increased insulin sensitivity. Taken together, we demonstrate that neutrophils can be added to the extensive repertoire of immune cells that participate in metaflammation.
In obese adipose tissue, there is a marked increase in the number of proinflammatory, M1–like, macrophages that secrete cytokines such as Tnf-α, Il1-β, Il6, etc., which can directly lead to decreased insulin sensitivity 1. While the role of tissue macrophages in this process has been well documented, recent studies have indicated contributions from several other immune cell types, including T cells 3–5, B cells 6, mast cells 7, and eosinophils 8.
Granulocytes comprise 60–70% of blood leukocytes, and more than 90% of granulocytes are neutrophils, making up the largest fraction of white blood cells. Feeding a high fat diet (HFD) to mice causes an increase in neutrophil recruitment into adipose tissue 13. Consequently, it is possible that neutrophils could play a role in initiating the inflammatory cascade in response to obesity. Adipose tissue neutrophils (ATNs) produce chemokines and cytokines, facilitating macrophage infiltration, which could contribute to the chronic low–grade inflammation that characterizes obesity–induced insulin resistance.
In addition to host defense, neutrophil serine proteases such as elastase (NE) have also been implicated in various non–infectious, inflammatory processes 12. Therefore, we asked whether neutrophils contribute to the etiology of inflammation–induced insulin resistance, and if NE plays a mechanistic role in this process.
We determined the time course of neutrophil infiltration in adipose tissue upon HFD feeding using FACS analyses to identify cells positive for the neutrophil markers Ly6g and Cd11b, and negative for the macrophage markers F4/80 and Cd11c. We refer to these cells as adipose tissue neutrophils (ATNs). Consistent with a previous report 13, we detected a rapid increase in ATN content at 3 days, and also found that this increase remained constant for up to 90 days of HFD (Fig 1a). Immunohistochemistry (IHC) studies also showed higher content of Ly6g/Cd11b double positive cells in WAT of HFD mice, compared to chow–fed mice (Fig 1b). Consistent with the IHC results, FACS analysis demonstrated a 20–fold increase in Cd11b/Ly6g double positive, F4/80 and Cd11c negative, cells in adipose tissue stromal vascular cells (SVCs) in HFD compared to chow fed mice as seen in Fig 1c and S1a.
Figure 1. Neutophils infiltrate eWAT in HFD mice, and ablation of neutrophil elastase improves insulin sensitivity in HFD–fed obese mice.
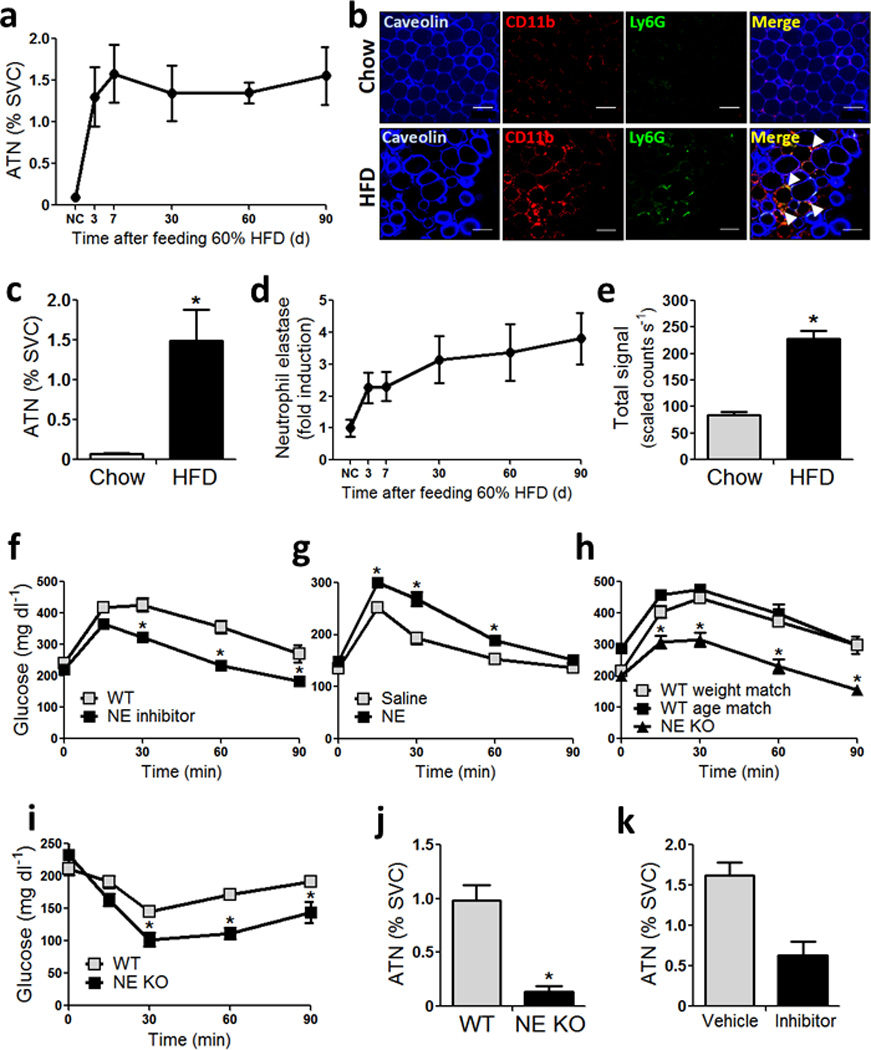
(a) Stromal vascular cells (SVCs) from epididymal white adipose tissue (eWAT) stained with Cd11b and Ly6g double positive, F4/80 and Cd11c negative cells analyzed by FACS. n = 3 – 4 mice per time point.
(b) Adipose tissue from C57BL/6J mice on chow and 60% high fat diet (HFD) stained with caveolin, Ly6g and Cd11b. The merged Ly6g and Cd11b double positive (yellow) cells indicated by white arrows are adipose tissue neutrophils (ATNs). Scale bar indicates 100 µm.
(c) FACS analyses showing ATN content (Ly6g/Cd11b double positive, F4/80 and Cd11c negative cells as % of SVCs) in 4 month old C57Bl/6J mice on chow and 20 week old mice fed HFD for 12 weeks. * indicates significance at p<0.05 using two–way ANOVA and Bonferroni post–test.
(d) mRNA abundance of NE in eWAT of C57Bl6/J mice, normalized to RNA PolII.
(e) NE activity in eWAT of male C57Bl/6J mice fed chow, or 60% HFD for 12 weeks. n = 5 for chow, and n = 8 for HFD. p value as indicated using two–way ANOVA and Bonferroni post–test.
(f) GTT on 7 h fasted C57BL/6J mice fed 60% HFD for 12 weeks, that were orally administered with vehicle and NE inhibitor (GW311616A) every day, for 2 weeks. n = 12 for vehicle, and n = 8 for drug treated group. * indicates significance at p<0.05 using two–way ANOVA and Bonferroni post–test.
(g) GTT in 5 month old C57Bl/6J mice on normal chow administered 1 mg kg−1 recombinant mouse NE for 7 days. n = 10 mice per group. * indicates significance at p<0.05 using two–way ANOVA and Bonferroni post–test.
(h) IP–GTT on 7 h fasted animals fed HFD for 10 weeks, using 1 mg kg−1 glucose, on WT weight, and age–matched WT mice with NE KO mice. The age matched mice are C57BL/6J mice obtained from JAX whose date of birth is matched with the NE KO mice. Weight matched mice are C57BL/6J mice obtained from JAX, that are approximately two and a half weeks younger than WT age matched, and NE KO mice. All WT mice were purchased at 6–7 weeks of age, acclimatized in our vivarium for 1–2 weeks and started on HFD at 8 weeks of age. n = 8 – 10 mice per group. * indicates significance at p<0.05 using two–way ANOVA and Bonferroni post–test.
(i) IP–ITT using 0.6 U kg−1 insulin in weight matched WT and NE KO mice fed HFD for 6 weeks. n = 8 – 10 mice per group. * indicates significance at p<0.05 using two–way ANOVA and Bonferroni post–test.
(j) FACS showing ATNs in WT and NE KO mice on HFD for 12 weeks. * indicates significance at p<0.05 using two–way ANOVA and Bonferroni post–test.
(k) FACS showing ATNs in 12 week HFD mice treated with vehicle of the NE inhibitor (GW311616A) for 2 weeks. * indicates significance at p<0.05 using two–way ANOVA and Bonferroni post–test.
Neutrophils secrete a proinflammatory proteinase termed neutrophil elastase (NE) 12, and this is higher in adipose tissue from HFD mice (Fig S1c). Expression of NE increased as early as 3 days and remained elevated at 12 weeks of HFD (Fig 1d), comparable to the pattern of increased ATN content. Consistent with the ATN content and NE expression, NE activity was also significantly higher in 12 week HFD–fed mice compared to lean chow–fed mice (Fig 1e, and Fig S2). HFD mice treated with the NE inhibitor, GW311616A 14, at a dose of 2 mg kg−1 per day for 14 days, showed significantly improved glucose tolerance (Fig 1f) with no change in body weight (Fig S3a). GW311616A also inhibited the onset of HFD–induced glucose intolerance when administered for 14 days (2 mg kg−1 day−1) at the start of HFD (Fig S3c). In contrast, recombinant mouse NE treatment (1 mg kg−1 day−1, 7 days) of chow–fed mice led to significant glucose intolerance (Fig 1g). Based on these treatment results and the fact that the number of ATNs is higher in HFD, we studied mice carrying a genetic deletion of NE (B6.129X1–Elanetm1Sds/J; NE KO). On HFD, we found that the NE KO mice gain somewhat less weight compared to WT (Fig S4a), consistent with their modestly higher core temperature (Fig S4b) and O2 consumption (Fig S4c). Liver weight was significantly lower, and WAT weight was significantly higher in NE KO mice compared to WT, both in absolute amounts, and when expressed as percent of body weight (Fig S4d). Ten weeks after HFD–feeding, we performed GTTs in NE KO mice and used age matched, and weight matched WT controls on the same diet. NE KO mice displayed a significant improvement in glucose tolerance (Fig 1h), and lower fasting insulin compared to WT (Fig S4e). Consistent with the GTT results, ITTs showed that the NE KO mice were more insulin sensitive compared to WT (Fig 1i).
ATN content was ~90% lower in NE KO mice compared to WT on HFD. In addition, mice treated with the NE inhibitor also displayed a marked lowering in ATN content (Fig 1k). Thus, both genetic and pharmacologic NE loss of function produced improved glucose tolerance with less ATNs, while pharmacologic gain of function led to glucose intolerance.
To quantify the overall magnitude of the insulin sensitivity and to determine tissue–specific contributions, we performed hyperinsulinemic–euglycemic clamp studies. The glucose infusion rate (GIR) required to maintain euglycemia was significantly higher in NE KO animals compared to WT (Fig 2a). There were no differences in total glucose disposal rate (Fig 2b), insulin stimulated glucose disposal rate (Is–GDR) (Fig 2c), or basal hepatic glucose production (HGP) (Fig 2d) between groups. However, the ability of insulin to inhibit HGP was significantly greater in NE KO mice compared to WT, as shown by the clamp HGP (Fig 2e) and % suppression of HGP (Fig 2f). Basal and clamp free fatty acid concentrations were significantly lower in NE KO mice (Fig 2g), and the effect of insulin to suppress FFA levels was greater in NE KO mice compared to WT (Fig 2h). Taken together, these results show that NE deletion leads to greater hepatic and adipose tissue insulin sensitivity.
Figure 2. Increased insulin sensitivity in NE KO mice.
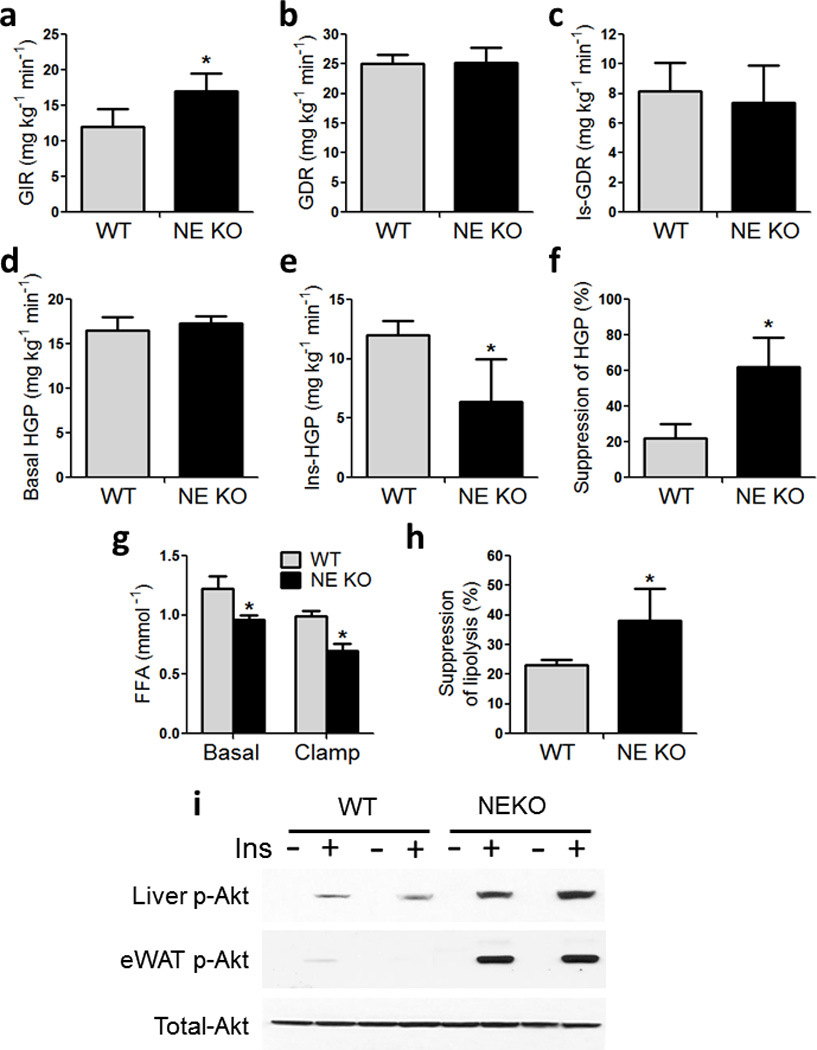
Hyperinsulinemic–euglycemic clamp studies in WT and NE KO mice on HFD for 10–11 weeks. (a) Glucose infusion rate, (b) total glucose disposal rate, (c) insulin–stimulated glucose disposal rate, (d) basal hepatic glucose production, (e) clamp hepatic glucose production, (f) percent suppression of hepatic glucose production,
(g) Free fatty acids (FFAs) 6 h fasted WT and NE KO mice (Basal), and at the end of the clamp procedure (clamp). * indicates significance at p<0.05 using two–way ANOVA and Bonferroni post–test.
(h) Percent suppression of lipolysis was calculated from (g). * indicates significance at p<0.05 using Student’s t test. * indicates significance at p<0.05 using two–way ANOVA and Bonferroni post–test.
(i) Acute insulin response in WT and NE KO mice using 0.35 U kg–1 insulin injected via inferior vena cava. Liver isolated at basal (−Ins) and 3 min (+Ins), adipose tissue (eWAT) harvested at basal (−Ins) and 7 min (+Ins). Western blots showing liver and adipose tissue phospho– and total– Akt.
Next, we performed acute insulin response studies by injecting insulin and harvesting liver and eWAT. Consistent with the ex vivo glucose clamp studies, biochemical measures of hepatic and adipose insulin signaling were higher in NE KO mice compared to WT, as evidenced by elevated insulin–stimulated Akt phosphorylation (Fig 2i).
Since studies have reported neutrophil infiltration of liver in obese humans with NASH 15, we assessed hepatic neutrophils by IHC in our experimental groups. Hepatic neutrophil content is higher in HFD compared to chow–fed WT mice with no elevation noted in HFD NE KO mice (Fig 3a). Hepatic NE activity was also significantly higher in HFD–fed mice compared to lean chow–fed mice (Fig 3b and S2). Previous reports have demonstrated that extracellular NE can gain access to the intracellular space and mediate degradation of Irs1 16,17. Therefore, we asked whether NE can directly inhibit hepatic Irs1–mediated insulin signaling. Four month old fasted C57Bl/6J mice were administered recombinant mouse NE (1mg kg−1, 2 h apart). Two h after the second dose, we performed an acute insulin injection and harvested liver at the indicated times. Administration of NE caused a significant decrease in liver Irs1 (Fig 3d) and p–Akt in the basal and insulin treated states (Fig 3e). Irs1 in eWAT from NE–treated mice was also significantly lower than in vehicle controls (Fig S5a). In contrast, Irs1 expression was higher in liver (Fig 3c) and adipose tissue (Fig S5d) of NE KO mice compared to WT.
Figure 3. Neutrophils infiltrate the liver in HFD mice and cause impaired insulin signaling via degradation of Irs1.
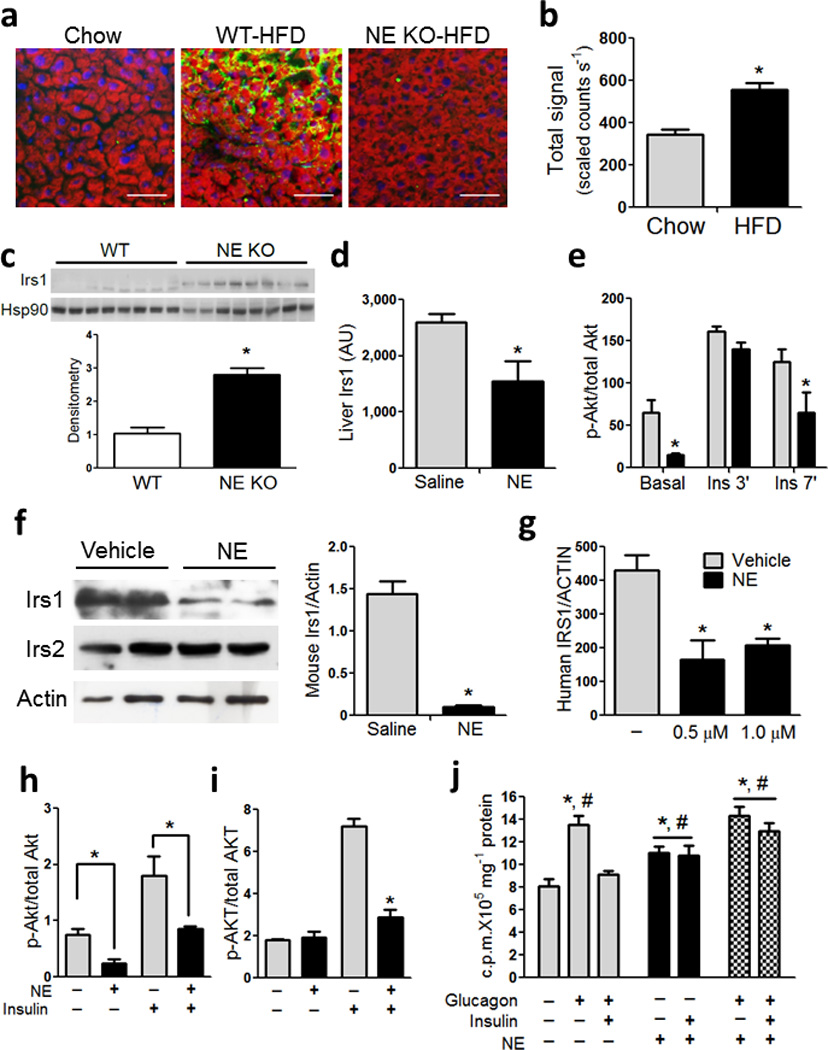
(a) Immunohistochemistry (IHC) on liver sections obtained from 4 month old chow, 20 week old WT and NE KO mice fed HFD for 12 weeks. Red color indicates lipid content in hepatocytes, stained with BODIPY, blue indicates nuclei stained with DAPI and green indicates neutrophils, stained with Ly6g (1A8). Scale bar indicates 50 µm.
(b) NE activity in liver of male C57Bl/6J mice fed chow, or 60% HFD for 12 weeks. n = 5 for chow, and n = 8 for HFD. * indicates significance at p<0.05 using two–way ANOVA and Bonferroni post–test.
(c) Western blots for total Irs1 and Hsp90 from WT and NE KO mice on HFD for 12 weeks. Densitometry analysis was performed and represented below the blot. * indicates significance at p<0.05.
(d) Liver Irs1 quantitated by MSD analyses from fasted 4–month old C57Bl6/J mice on chow diet. Mice were injected with saline or recombinant mouse NE. Tissues were harvested 2 h after NE injection.
(e) Acute insulin response in 4 month old C57Bl/6J mice on chow treated with recombinant mouse NE. A submaximal insulin dose (0.1 U–1 kg) was used and liver samples were obtained at the indicated times points. MSD was used to analyze phospho– and total–Akt. * indicates significance at p<0.05 using two–way ANOVA and Bonferroni post–test.
(f) Primary mouse hepatocytes were treated with recombinant mouse NE for 4 h and protein was harvested and western blots were performed according to standard protocol. * indicates significance at p<0.05 using two–way ANOVA and Bonferroni post–test.
(g) Cryopreserved human hepatocytes were treated with purified human NE for 6 h. Representative figure from at least 3 independent experiments. * indicates significance at p<0.05 using two–way ANOVA and Bonferroni post–test.
(h) Primary mouse hepatocytes were treated with NE for 4 h and spiked with insulin for 5 min to obtain insulin induction. Figure shows quantitation of western blots from at least 3 independent experiments performed in duplicate. * indicates significance at p<0.05 using two–way ANOVA and Bonferroni post–test.
(i) Primary human hepatocytes were treated with human NE for 6 h and spiked with insulin for 5 min to obtain insulin induction. Protein was harvested and MSD was used to determine human phospho– and total–AKT according to manufacturer’s protocol. Figure show quantitation data from least 2 independent experiments performed in triplicate. * indicates significance at p<0.05 using two–way ANOVA and Bonferroni post–test.
(j) Representative glucose output assay in primary mouse hepatocytes from at least 3 independent experiments performed in duplicate or triplicate. *, significantly higher than basal, and #, significantly higher than insulin + glucagon. Significance is at p<0.05 using Student’s t–test.
Next, we added NE directly to either primary mouse (Fig 3f) or human hepatocytes (Fig 3g), and observed a marked decrease in Irs1 protein content, consistent with enhanced degradation and a more modest decrease in mouse Irs2 expression. This lowering in Irs1 level resulted in reduced insulin stimulated Akt phosphorylation (Fig 3h, i).
To determine whether these changes in hepatocyte insulin signaling resulted in biologic effects, we measured glucose output in primary mouse hepatocytes with and without NE treatment. In this system, glucagon treatment leads to a near 2–fold increase in primary hepatocyte glucose output and insulin suppresses this to basal values. As seen (Fig 3j), NE treatment stimulated glucose output by 50% and insulin did not inhibit this effect. More importantly, NE treatment largely prevented the effect of insulin to inhibit glucagon–stimulated hepatocyte glucose output. Thus, NE directly leads to Irs degradation, lower insulin signaling, higher glucose production and cellular insulin resistance in primary mouse and human hepatocytes.
We also measured liver lipogenic and cholesterol synthesis genes, and found that Acc, Fas, Scd, Hmgcr, Hmgcs were significantly lower, while PpargC1a (PGC1α) and Nrf were higher in livers from NE KO mice compared to WT. There was no change in mRNA abundance of Srebp1c, Pepck, G6Pase and Cpt1a (Fig S6a). To confirm that decreased lipogenic gene expression resulted in functional changes in lipogenesis, we measured incorporation of 14C–actetate into liver lipids. Total incorporation (Fig S6b) and the rate of 14C–acetate incorporation (Fig S6c) into liver lipids were significantly lower in NE KO animals.
Since we observed improved adipose tissue insulin signaling in NE KO mice (Fig 2i), we treated 3T3–L1 adipocytes with recombinant mouse NE. The results show that NE led to decreased Irs1 expression (Fig S5b) and impaired insulin stimulated p–Akt in a dose dependent manner (Fig S5c).
We used intraperitoneal macrophages (IP–Macs) from WT and Tlr4 KO mice, and treated them with LPS and recombinant mouse NE. LPS caused higher mRNA abundance of Tnf-α, Il1-β, Cxcl1 (KC) and Il6 in WT, but not in Tlr4 KO IP–Macs. Similar to LPS, recombinant NE caused higher gene expression of Tnf-α, Il1-β, Cxcl1 (KC) and Il6 in WT, but not in Tlr4 KO IP–Macs, showing that the proinflammatory effects of NE are Tlr4 dependent (Fig 4a), consistent with previous reports 12. Gene expression analyses revealed that proinflammatory markers such as Tnf-α, Emr1 (F4/80), Cxcl1 (KC), Il1r1, Il1-β, and Ccl2 (Mcp1) were significantly lower, and the anti–inflammatory marker Arginase was significantly higher in NE KO mouse liver compared to WT (Fig 4b). Consistent with this decrease in proinflammatory gene expression, degradation of IkB was lower in NE KO mice compared to WT (Fig 4b inset).
Figure 4. NE KO mice have decreased inflammatory tone.
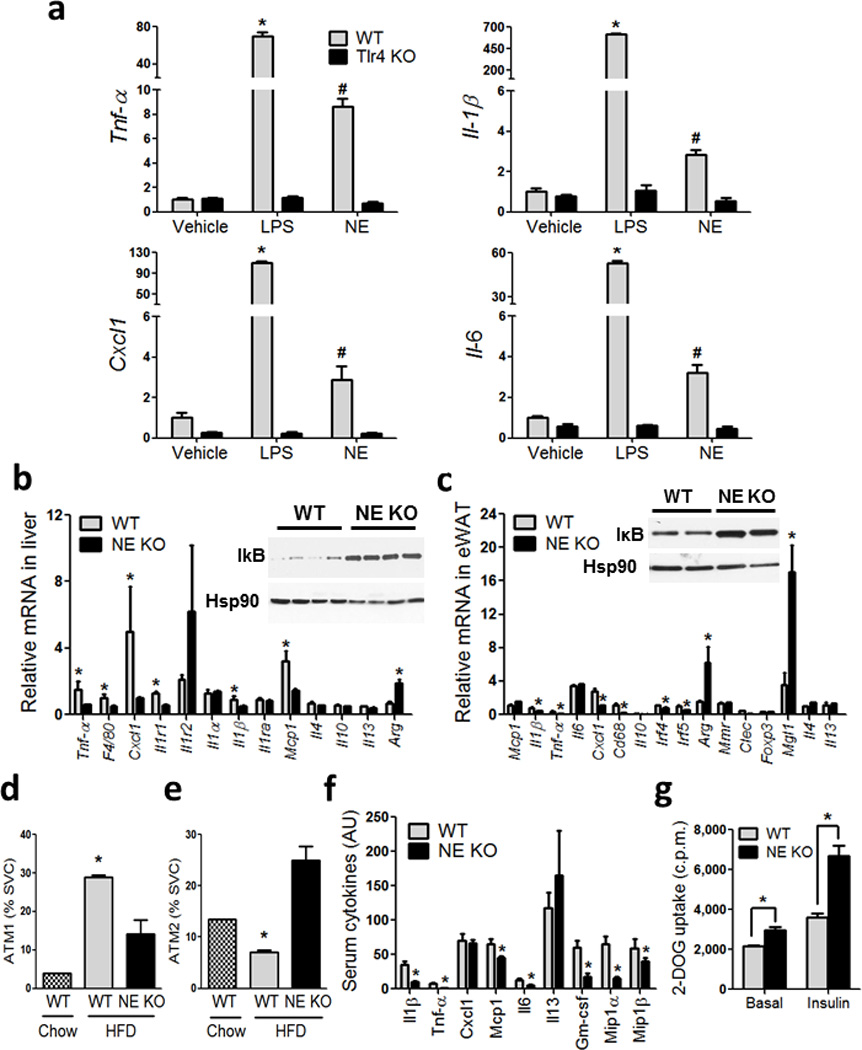
(a) qPCR of the indicated genes from IP–Macs harvested from WT and Tlr4 KO mice, treated with vehicle, LPS and recombinant mouse NE. *, represents significantly higher than all other treatment groups, and #, represents significantly lower than WT LPS, and significantly higher than all other treatment groups. * significance at p<0.05 using Student’s t test.
(b) qPCR analysis on inflammatory gene expression in liver, and (c) adipose tissue, from WT and NE KO mice on HFD. Western blot for liver IκB and Hsp90 (b) inset, and adipose IκB and Hsp90 (c) inset. * indicates significance at p<0.05 using Student’s t test.
(d) SVCs from eWAT stained with F4/80, Cd11b, and Cd11c and analyzed by FACS. Cells that are triple positive for all three markers are referred to as ATM1, and (e) cells that are positive for both F4/80 and Cd11b and negative for Cd11c are referred to as ATM2. * indicates significance at p<0.05 using two–way ANOVA and Bonferroni post–test.
(f) Serum cytokines measured using the Millipore Luminex assay from WT and NE KO mice on HFD. * indicates significance at p<0.05 using Student’s t test.
(g) Glucose uptake in eWAT explants harvested from WT and NE KO mice and incubated ex vivo in the absence and presence of insulin, followed by measurement of 2–deoxyglucose (2–DOG) uptake. * indicates significance at p<0.05 using two–way ANOVA and Bonferroni post–test.
Gene expression analyses in adipose tissue showed that Il1-β, Tnf-α, Cxcl1 (KC), Cd68, Irf4, Irf5 were lower, and Arg, Clec10a (Mgl1) and Il4 were higher in NE KO mice compared to WT (Fig 4c). IκB levels were also lower in NE KO mice compared to WT (Fig 4c inset). To determine the contribution of adipocytes vs immune cells to these changes in gene expression, SVCs and adipocytes from WT and NE KO animals were isolated, and each fraction displayed decreased inflammatory markers in NE KO compared to WT (Fig S7a). FACS analyses for adipose tissue macrophage (ATM) content showed that the Cd11c+, M1–like, cells were decreased (Fig 4d), while F4/80+, Cd11b+, Cd11c−, or M2–like, cells were increased in NE KO mice compared to WT (Fig 4e). This suggests that elastase secreted from neutrophils plays a role in recruiting these cells to adipose tissue and possibly in their polarization state. For example, NE functions as a Tlr4 activator 12,18–20, and stimulation of the Tlr4 proinflammatory pathway leads to increased chemokine release from a variety of adipose tissue cell types, including adipocytes and macrophages 1,2. Consistent with changes in inflammatory tone in liver and adipose tissue, serum Il1-β, Tnf-α, Mcp–1, Il6, Gm–csf, Mip1-α, Mip1-β (Fig 4f) and Resistin (Fig S7b) were significantly lower in NE KO compared to WT.
Gene expression analyses of adipose tissue revealed that Acc and Fas, Hsl, Atgl and Glut4 mRNA abundance was significantly increased in NE KO animals compared to WT (Fig S7c). Fat pads were excised for glucose uptake analyses, and both basal and insulin–stimulated glucose uptake was higher in NE KO mice compared to WT (Fig 4g), consistent with the in vivo and in vitro data showing improved adipose insulin sensitivity.
In these studies, we have shown a sustained increased in adipose tissue and liver neutrophil content in HFD/obesity. Since neutrophils are known to play a role in the early stages of inflammatory responses, it is likely that neutrophils participate in the inflammation which characterizes obesity. Furthermore, secreted elastase from neutrophils is the key effector in this process. Consistent with this, we find that direct addition of NE to hepatocytes or adipocytes causes cellular insulin resistance, and in the context of HFD/obesity, NE KO mice were protected from adipose tissue and liver inflammation, with a corresponding increase in adipose tissue and hepatic insulin sensitivity. We also treated mice with a small molecule NE inhibitor and found that this compound improved glucose tolerance in HFD/obese mice.
In addition to macrophages, a variety of immune cells, such as lymphocytes, eosinophils, and mast cells have been shown to participate in the complex intracellular communication network which organizes the chronic inflammatory response of obesity. Based on our studies in chronic HFD mice and that of others13, demonstrating the presence of adipose neutrophils upon acute HFD feeding, we suggest that neutrophils should be added as active participants in the immune cell–type conversation, which ultimately leads to obesity–induced inflammation and insulin resistance.
Methods
Mice
NE KO, JAX labs B6.129X1–Elanetm1Sds/J, Stock number 006112, and WT C57BL/6J, Stock Number 000664 mice were obtained from JAX. Only male mice were used in this study. We placed NE KO and age matched WT on 60% high fat diet (D12492, Research Diets) for 12 weeks, and monitored body weight and food intake. For weight matched controls, we used WT mice on 60% HFD that were about 2–3 weeks younger than NE KO mice. We housed all animals according to UCSD and IACUC approved protocols.
All mouse procedures conformed to the Guide for Care and Use of Laboratory Animals of the US National Institutes of Health, and were approved by the Animal Subjects Committee of UCSD.
Metabolic studies
For ITT and GTTs, we fasted mice for 7 h. For GTT, we administered an IP dose of 1 gm kg−1 dextrose, and measured blood glucose at the indicated time points. We measured fasting insulin after 6 h of fasting on the day of GTT. For ITT, we used an IP dose of 0.6 U kg−1 insulin, and measured blood glucoseat the indicated time points.
We performed hyperinsulinemic euglycemic clamp studies as described 21. We only used mice that lost <4% of their precannulation weight after 4–5 d of recovery. We administered a constant infusion (5 µCi h−1) of D–[3–3H] glucose (Du Pont–NEN) 6h after fasting. After 90 min of tracer equilibration and basal sampling, we infused 50% dextrose; Abbott and tracer 5 µCi h−1 plus insulin (6 mU kg−1min−1) via the jugular vein cannula. We sampled blood from tail clips at 10 min intervals and steady–state conditions (120 mg dl−1 ± 5 mg dl−1) were achieved at the end of the clamp by maintaining glucose infusion and plasma glucose concentration for a minimum of 20 min.
Acute insulin response
We fasted mice either overnight, or 7 h. We performed this procedure on WT and NE KO mice on HFD at an insulin dose of 0.35 U kg−1. We also performed his procedure on black 6 mice on chow injected with vehicle and recombinant mouse NE (4517–SE) at a submaximal insulin dose of 0.1 U kg−1. Briefly, we anesthetized mice, and suture sealed a small lobe of the liver to prevent bleeding, and excised. We also obtained an eWAT biopsy and flash froze in liquid N2. These samples are referred to as basal. We injected insulin at the mentioned dose into the inferior vena cava and obtained tissue biopsies as described, at the indicated time points and flash froze in liquid N2. We prepared lysates and ran western blots or MSD according to standard protocol.
Indirect calorimetry and core temperature
We performed indirect calorimetry between week 7–8 of HFD as described 22. We individually house mice in metabolic cages for measurements. We allowed all mice to adapt to the new environment for 48 h before study. We normalized oxygen consumption (VO2) and carbon dioxide production (VCO2) with respect to body weight. We calculated energy expenditure on the basis of the formula energy expenditure = 3.815 × VO2 + 1.232 × VCO2.
We measured core temperature by insertion of a rectal probe, and took all temperature measurements under fed conditions at about 3 PM. We took the temperature measurements between 8–9 weeks of HFD feeding.
Western blotting and gene expression analyses
We performed western blotting as described 23. All antibodies are from Cell Signaling Technology. We performed quantitative PCR (qPCR) as described 24.
Fluorescence-activated cell sorting (FACS) analyses
We weighed epididymal fat pads rinsed in phosphate–buffered saline, and then minced in FACS buffer (phosphate–buffered saline containing 1% BSA). We prepared adipocytes and stromal vascular cells from collagenase digested adipose tissue. We performed FACS analysis of stromal vascular cells (SVCs) for macrophage content and subtypes as described, and estimation of macrophage subsets numbers g−1 of fat was performed as described 21. We incubated stromal vascular cells with Fc Block (BD Biosciences, San Jose, CA) for 20 min at 4 °C prior to staining with fluorescently labeled primary antibodies and unstained, single stains, and fluorescence minus one controls were used for setting compensation and gates. We purchased the neutrophil marker Ly6G antibody from BD, clone 1A8.
Confocal microscopy of mouse adipose tissue
We excised finger nail sized fat pad samples and blocked for 1 h in 5% BSA in PBS with gentle rocking at RT. For detection of intracellular antigens, blocking and subsequent incubations were done in 5% BSA in PBS with 0.3% Triton X–100. We diluted primary antibodies in blocking buffer to 0.5–1 mg ml−1 and added to fat samples for overnight at 4 °C. After three washes, we added fluorochrome–conjugated secondary antibodies for 1 h at RT. We imaged fat pads on an inverted confocal microscope (Olympus Fluoview 1000). Anti–mouse antibodies used were against Cd11b (Abcam); Caveolin, and Ly6g (1A8) (BD Biosciences).
Lipogenesis in liver explants
We quickly sliced liver samples into 1–2 mm sizes and transferred into 0.5 ml/well phosphate–salt–bicarbonate buffer (10 mM Hepes, 4 mM KCl, 125 mM NaCl, 0.85 mM KH2PO4, 1.25 m Na2HPO4, 1 mM MgCl2, 1 mM CaCl2 and 15 mM NaHCO3) containing 0.5% fatty acid free BSA and 0.2 mM unlabeled sodium acetate in 12–well culture plates. We added 1 µCi 2–14C acetate to each well and incubated for 30 and 60 min at 37°C in a 5% CO2 incubator. We terminated incubation by adding 0.5 ml of 2 N HCl to each well. We transferred explants with the incubation buffer to 5 ml polyethylene tubes and centrifuged and supernatants removed. Next, we added 0.5 ml 1 N HCl to each tube and homogenized the explant suspensions, followed by the addition of 1.5 ml of a mixture of chloroform and methanol (2:1) to each homogenate. We vortexed the tubes to obtain a monophasic mixture. Next, 0.5 ml chloroform and 0.5 ml 1 M NaCl were added to the homogenates, and the tubes were vortexed and centrifuged to obtain separate two layers. We collected the lower chloroform layer, evaporated and counted for incorporation of labeled acetate into lipids. We saved an aliquot of the homogenates for protein assay before adding organic solvent cocktail. Specific activity in the incubation was 22,000 c.p.m. nmol–1 acetate.
Glucose uptake in adipose explants
We quickly sliced adipose tissue samples were quickly sliced into 1–2 mm sizes. Adipose explants prepared from half of each fat pad were taken into 0.5 ml/well phosphate–salt –bicarbonate buffer (10 mM Hepes, 4 mM KCl, 125 mM NaCl, 0.85 mM KH2PO4, 1.25 mM Na2HPO4, 1mM MgCl2, 1 mM CaCl2 and 15 mM NaHCO3) containing 0.5% fatty acid free BSA in 12–well culture plates. We incubated plates for 15 min in 5% CO2 incubator at 37 °C. We added insulin to some wells at a final concentration of 17 nM, and incubated for an additional 30 min. Next, we added 0.2 µCi 3H–2 deoxy–glucose (final concentration 0.1 mM) to each well, and incubated for 10 min at 37°C in a 5% CO2 incubator. We terminated the incubation by placing the plates on an ice tray followed by addition of Cytochalasin B (final concentration 0.01 mM) to stop further uptake. We carefully removed the radiolabeled buffer avoiding pieces of the explants, and washed the explants 3 times with chilled PBS. Next, we added 0.5 ml of 1 N NaOH to each well and plates were shaken for 30 min in room temp. We transferred the alkaline suspensions to 5 ml culture tubes and homogenized. We saved an aliquot for protein assay and transferred the rest of the homogenate to 10 ml scintillation vials. We neutralized the alkaline homogenates with 0.5 ml 1 N HCl added into the vials, and counted radioactivity using 10 ml scintillation cocktail.
Intraperitoneal macrophage (IP-Mac) isolation and culture
We harvested IP–macs from WT and Tlr 4KO mice as described 21. Three days after harvest and plating, we treated cells with 100 nM recombinant mouse NE (R&D Systems: 4517-S E) and LPS (100 ng ml−1) for 6 h prior to RNA isolation, and qPCR analyses. We used heat inactivated NE as control.
Human hepatocytes culture
We used primary cryopreserved plateable human hepatocytes in this experiment from BD Bioscience (Lot310), and used BD Cryohepatocytes recovery protocol to recover the cells and plated overnight. On the second day, we treated the cells in triplicate with human neutrophil elastase (Innovative Research, Lot M77514) at 1000, 500nM in low glucose medium (Gibco 10567) and 0.5% charcoal stripped FBS (Gibco Lot 934492) for 6 h, and washed once with ice cold PBS. We used Meso Scale Discovery (MSD) used to assay for Irs1, phopho–Akt and total Akt. 20 µl of sample was assayed for Irs1 or pAkt/tAkt) using the instructions provided by the kit manufacturer (Catalog number for Total IRS1 is N450HLA–1; Catalog number for pAkt/tAkt is K15100D–2).
Glucose output assay in mouse hepatocytes
We plated primary mouse hepatocytes on collagen–coated plates, and maintained in Medium E with 10 % FBS and 100 nM dexamethasone. We washed cultures with Krebs–Henseleit bicarbonate buffer with 2.5 mM calcium and 1% BSA, and incubated in the same buffer containing hormones and substrates in 5% CO2 incubator. We used 2 mM pyruvate (0.5 µCi Pyruvate/incubation) as substrate, and performed incubations out in 0.5 ml buffer in 12–well plate containing 0.25–0.5 million cells/well (12–well plate). We incubated insulin (0.1 µM) or Glucagon (100 ng ml−1) in the absence of substrate, followed by 3 h with substrates. At the end of incubation, we transferred buffers to 1.7 ml microfuge tubes and added 0.25 ml 5% ZnSO4 and 0.25 ml 0.3 N Ba (OH)2 suspensions to each tube followed by 0.5 ml water. We collected supernatants in fresh set of tubes and assayed for radiolabeled glucose released into the media. We treated cells in the culture plates with 0.5 ml 10% HClO4, shaken for 15 min and transferred the acidic extracts to microfuge tubes. We dissolved cellular debris with proteins remaining on the plates in 1 NaOH and subjected to protein assay. We neutralized the acidic extracts in the tubes were with 0.5 ml of 2 M K2CO3. We removed precipitates by centrifugation and collected the supernatants for determination of intracellular radiolabeled glucose concentrations.
We performed the glucose assay by using the supernatants from the culture buffer and the assayed cell extracts for radioactive glucose by mixed (cation and anion) ion–exchange chromatography using AG–501x8 resins (BIO–RAD), using a batch treatment method. We added 150–200 mg resins to each tube and vortexed intermittently for 15 min. We centrifuged the tubes and the supernatants were transferred to scintillation vials for counting radioactivity.
Neutrophil elastase activity
We used six month–old C57Bl6/J on chow diet and 20 week old C57Bl6/J mice on 60% HFD for these studies. On the day of the experiment we administered all animals with 100 µl Neutrophil Elastase 680 FAST (Boston MA, Perkin Elmer), via tail vein injection. The 680 FAST comprises of two NIR fluorochromes (VivoTag–S680, PerkinElmer, Boston, MA) linked to N– and C–terminus of the peptide PMAVVQSVP, a highly NE–selective sequence25. NE680 FAST is optically silent in its native state and becomes highly fluorescent upon cleavage by neutrophil elastase, and can therefore be used as a direct sensing agent for neutrophil elastase activity.
Five h post–injection, we sacrificed the animals, and harvested the liver and eWAT. We immediately performed ex vivo liver and eWAT imaging on a CRi Maestro optical imaging platform using the Red filter sets (Excitation Range: 616–660 nm, Emission: 675 nm longpass). We spectrally unmixed fluorescent data to obtain signal specific to NE680 FAST. We manually drew regions of interest (ROI) encompassing the whole organs and the resulting signal computed in the units of scaled counts second−1. Particular care was taken to ensure that the size of the ROIs drawn across animals was constant.
3T3–L1 adipocyte culture
3T3–L1 cells were differentiated and cultured as described21. On the day of the experiments, we treated cells with the indicated amount of recombinant mouse NE for 6 h. Prior to the addition of NE, we serum–starved cells for 2 h. We added insulin at 10 nM for 5 min after which total protein was harvested according to manufacturer’s protocols and ran on MSD plates.
Statistical analyses
We performed two–way ANOVA with Bonferroni’s post test on GraphPad Prsim5 (San Diego, CA), to determine significance between experimental and control groups.
Supplementary Material
Acknowledgements
This work was supported by grants to J.M.O. from the National Institution of Health: DK033651, DK074868, T32 DK 007494, DK 090962 and DK063491. This work was also supported by the Eunice Kennedy Shriver NICHD/NIH through cooperative agreement of U54 HD 012303–25 as part of the specialized Cooperative Centers Program in Reproduction and Infertility Research.
We wish to sincerely thank S. Shapiro (University of Pittsburgh) for helpful comments over the course of the project, C. Pham (Washington University in St. Louis) for providing Cathepsin G and NE double KO mice, S. Nalbandian at UCSD for breeding and caring for the NE KO mice, P. Bansal (Pfizer), B. Ghosh (Pfizer) and J. Wellen (Pfizer) for the NE activity imaging studies.
Footnotes
Contributions
S.T. and D.Y.O designed and performed the experiments. S.T., D.Y.O., and J.M.O. analyzed, interpreted data, and co–wrote the manuscript. All other authors performed experiments and contributed to discussions. This work was supported by grants to J.M.O. as detailed above.
Competing financial interests
The authors declare no competing financial interests.
References
- 1.Olefsky JM, Glass CK. Macrophages, inflammation, and insulin resistance. Annu Rev Physiol. 2010;72:219–246. doi: 10.1146/annurev-physiol-021909-135846. [DOI] [PubMed] [Google Scholar]
- 2.Gregor MF, Hotamisligil GS. Inflammatory mechanisms in obesity. Annu Rev Immunol. 2011;29:415–445. doi: 10.1146/annurev-immunol-031210-101322. [DOI] [PubMed] [Google Scholar]
- 3.Nishimura S, et al. CD8+ effector T cells contribute to macrophage recruitment and adipose tissue inflammation in obesity. Nat Med. 2009;15:914–920. doi: 10.1038/nm.1964. [DOI] [PubMed] [Google Scholar]
- 4.Winer S, et al. Normalization of obesity–associated insulin resistance through immunotherapy. Nat Med. 2009;15:921–929. doi: 10.1038/nm.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Feuerer M, et al. Lean, but not obese, fat is enriched for a unique population of regulatory T cells that affect metabolic parameters. Nat Med. 2009;15:930–939. doi: 10.1038/nm.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Winer DA, et al. B cells promote insulin resistance through modulation of T cells and production of pathogenic IgG antibodies. Nat Med. 2011;17:610–617. doi: 10.1038/nm.2353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Liu J, et al. Genetic deficiency and pharmacological stabilization of mast cells reduce diet–induced obesity and diabetes in mice. Nat Med. 2009;15:940–945. doi: 10.1038/nm.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wu D, et al. Eosinophils sustain adipose alternatively activated macrophages associated with glucose homeostasis. Science. 2011;332:243–247. doi: 10.1126/science.1201475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mantovani A, Cassatella MA, Costantini C, Jaillon S. Neutrophils in the activation and regulation of innate and adaptive immunity. Nat Rev Immunol. 2011;11:519–531. doi: 10.1038/nri3024. [DOI] [PubMed] [Google Scholar]
- 10.Nathan C. Neutrophils and immunity: challenges and opportunities. Nat Rev Immunol. 2006;6:173–182. doi: 10.1038/nri1785. [DOI] [PubMed] [Google Scholar]
- 11.Borregaard N. Neutrophils, from marrow to microbes. Immunity. 2010;33:657–670. doi: 10.1016/j.immuni.2010.11.011. [DOI] [PubMed] [Google Scholar]
- 12.Pham CT. Neutrophil serine proteases: specific regulators of inflammation. Nat Rev Immunol. 2006;6:541–550. doi: 10.1038/nri1841. [DOI] [PubMed] [Google Scholar]
- 13.Elgazar–Carmon V, Rudich A, Hadad N, Levy R. Neutrophils transiently infiltrate intra–abdominal fat early in the course of high–fat feeding. J Lipid Res. 2008;49:1894–1903. doi: 10.1194/jlr.M800132-JLR200. [DOI] [PubMed] [Google Scholar]
- 14.Macdonald SJ, et al. The discovery of a potent, intracellular, orally bioavailable, long duration inhibitor of human neutrophil elastase—GW311616A a development candidate. Bioorg Med Chem Lett. 2001;11:895–898. doi: 10.1016/s0960-894x(01)00078-6. [DOI] [PubMed] [Google Scholar]
- 15.Rensen SS, et al. Increased hepatic myeloperoxidase activity in obese subjects with nonalcoholic steatohepatitis. Am J Pathol. 2009;175:1473–1482. doi: 10.2353/ajpath.2009.080999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Houghton AM, et al. Neutrophil elastase–mediated degradation of IRS-1 accelerates lung tumor growth. Nat Med. 2010;16:219–223. doi: 10.1038/nm.2084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Houghton AM. The paradox of tumor–associated neutrophils: fueling tumor growth with cytotoxic substances. Cell Cycle. 2010;9:1732–1737. doi: 10.4161/cc.9.9.11297. [DOI] [PubMed] [Google Scholar]
- 18.Walsh DE, et al. Interleukin–8 up–regulation by neutrophil elastase is mediated by MyD88/IRAK/TRAF–6 in human bronchial epithelium. J Biol Chem. 2001;276:35494–35499. doi: 10.1074/jbc.M103543200. [DOI] [PubMed] [Google Scholar]
- 19.Snelgrove RJ, et al. A critical role for LTA4H in limiting chronic pulmonary neutrophilic inflammation. Science. 2010;330:90–94. doi: 10.1126/science.1190594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chua F, Laurent GJ. Neutrophil elastase: mediator of extracellular matrix destruction and accumulation. Proc Am Thorac Soc. 2006;3:424–427. doi: 10.1513/pats.200603-078AW. [DOI] [PubMed] [Google Scholar]
- 21.Oh DY, et al. GPR120 is an omega–3 fatty acid receptor mediating potent anti–inflammatory and insulin–sensitizing effects. Cell. 2010;142:687–698. doi: 10.1016/j.cell.2010.07.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lu M, et al. Brain PPAR–gamma promotes obesity and is required for the insulin–sensitizing effect of thiazolidinediones. Nat Med. 2011;17:618–622. doi: 10.1038/nm.2332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li P, et al. Functional heterogeneity of CD11c–positive adipose tissue macrophages in diet–induced obese mice. J Biol Chem. 2010;285:15333–15345. doi: 10.1074/jbc.M110.100263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Talukdar S, Hillgartner FB. The mechanism mediating the activation of acetyl–coenzyme A carboxylase–alpha gene transcription by the liver X receptor agonist T0–901317. J Lipid Res. 2006;47:2451–2461. doi: 10.1194/jlr.M600276-JLR200. [DOI] [PubMed] [Google Scholar]
- 25.Kalupov T, et al. Structural characterization of mouse neutrophil serine proteases and identification of their substrate specificities: relevance to mouse models of human inflammatory diseases. J Biol Chem. 2009;284:34084–34091. doi: 10.1074/jbc.M109.042903. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.