

Abstract
The dialysis disequilibrium syndrome is a rare but serious complication of hemodialysis. Despite the fact that maintenance hemodialysis has been a routine procedure for over 50 years, this syndrome remains poorly understood. The signs and symptoms vary widely from restlessness and headache to coma and death. While cerebral edema and increased intracranial pressure are the primary contributing factors to this syndrome and are the target of therapy, the precise mechanisms for their development remain elusive. Treatment of this syndrome once it has developed is rarely successful. Thus, measures to avoid its development are crucial. In this review, we will examine the pathophysiology of this syndrome and discuss the factors to consider in avoiding its development.
Keywords: Reverse urea effect, Idiogenic osmoles, Uremia, Hemodialysis, Urea kinetics
Introduction
The dialysis disequilibrium syndrome is defined as a clinical syndrome of neurologic deterioration that is seen in patients who undergo hemodialysis [1–3]. It is more likely to occur in patients during or immediately after their first treatment, but can occur in any patient who receives hemodialysis. The symptoms involve the neurologic system and are similar to symptoms that occur with increased intracranial pressure or acute hyponatremia, such as restlessness, headache, mental confusion and coma. Because these symptoms are not specific for the disequilibrium syndrome, other diagnoses that must be considered and ruled out are listed in Table 1 [4].
Table 1.
Differential diagnosis for signs and symptoms of dialysis disequilibrium
| 1. Subdural hematoma |
| 2. Uremia |
| 3. Nonketotic hyperosmolar coma |
| 4. Acute cerebrovascular event |
| 5. Dialysis dementia |
| 6. Excessive ultrafiltration and seizure |
| 7. Hypoglycemia |
| 8. Malignant hypertension |
| 9. Hyponatremia |
The exact incidence of dialysis disequilibrium is not known, but seems to be decreasing since the early days of hemodialysis, most likely due to the fact that current patients are initiated on dialysis at much lower urea concentrations than previously [3]. While the exact etiology of the syndrome remains unknown, there are many factors that have been examined and will be reviewed here.
The first description of dialysis disequilibrium was reported in 1962 and presents many of the concepts of the syndrome that will be discussed [5]. The authors noted that while most patients showed improvement in their mental status after hemodialysis, there were several patients that worsened despite the fact that their blood chemistries had improved. Symptoms that were reported included mental confusion, headache and occasional muscle twitching. Measurement of urea in the blood and cerebrospinal fluid (CSF) showed that after hemodialysis treatment, there was a substantial gradient, with the urea concentration in the CSF being higher than that in the blood [5]. The authors concluded that this urea gradient caused water to move into the central nervous system (CNS) and that the increased intracranial pressure that resulted accounted for the symptoms that were described.
The issues raised by the authors are prescient and will serve as the basis for this discussion. First, the movement of urea between the blood and the CSF and its influence on water movement will be discussed. Second, because water movement is primarily responsible for the increased intracranial pressure, we will examine the factors contributing to the osmotic gradient between the blood and the CNS as well as the water permeability of the blood–brain barrier (BBB) and the osmotic pressure exerted by urea. Third, the impact of the rate of removal of urea will be discussed. Last, we will review measures that can be taken to avoid the dialysis disequilibrium syndrome or to minimize its consequences if it has developed.
The CNS and urea
The composition and volume of the neural microenviroment is controlled by the exchange of solutes and water across the BBB, which comprises plasma membranes of neurons and glia, and across the pia and ependyma [6, 7]. The microvasculature of the brain and the choroid plexus of the lateral and fourth ventricles form the blood–brain and blood–CSF barriers, respectively. They control the chemical composition of the brain’s extracellular fluid (ECF) and the CSF.
The true BBB is the microvasculature’s unfenestrated endothelial cells with their complex tight junctions, which regulate the movement of small organic solutes and ions between the blood and the extracellular space (Fig. 1a). Astrocytic foot processes encircle the microvessels but are not sealed, allowing the access of the interstitial fluid to the endothelial cell basement membrane. However, the endothelial cells of the BBB have a low ion permeability, high solute reflection coefficient and high electrical resistance compared to other vascular beds [8, 9].
Fig. 1.

A schematic diagram of the blood brain barrier. a The endothelial cell of the microvasculature has tight junctions that prevent paracellular movement of solute and water. Thus, it is the true blood brain barrier [14]. (Reproduced from [37], with permission). b In the presence of hypertonicity, the astrocyte cell–cell junction is no longer intact. This will allow a more rapid entry of water into the brain tissue. (Reproduced from [37], with permission)
The reflection coefficient of a solute is the measure of the solute’s ability to exert an osmotic force. It is defined as the ratio of its measured osmotic pressure to the ideal osmotic pressure predicted by the van’t Hoff equation. Values range from 0 to 1, with a value of 1 meaning that it exerts its maximal osmotic force and a value of zero meaning that it exerts no osmotic force. The reflection coefficient for urea is 0.44–0.59 (vs. 0.48 for glycerol and 0.90 for mannitol), demonstrating its ability to exert some osmotic pressure across the BBB [10, 11].
With respect to non-electrolyte substances, the tight intercellular junctions and unfenestrated endothelial cells prevent water-soluble and polar substances from rapidly entering the brain interstitial space and the synaptic space. As mentioned previously, the permeability of urea in the brain is relatively low compared to other organic solutes and enters the brain more slowly than in other tissues [12].
The tight junctions of the cells limit paracellular transport of solutes and water so that transcellular transport is required and consequently can be regulated. Recent evidence shows that specific transporters for both urea (UT-B1) and water (AQP4 and AQP9) are present in these cells and that their expression is altered in uremia [13]. The expression of UT-B1 was found to be decreased by half in the brains of rats that were nephrectomized while the expression of the aquaporins was increased [13]. In addition, hyperosmolality can disrupt the BBB and thus decrease the regulation of transport through this space (Fig. 1b) [14].
The transport of urea through the BBB has been examined in normal rabbits [11, 12]. It was demonstrated that while urea can enter the CNS, it does so much more slowly in comparison to it entering muscle tissue. The reflection coefficient for raffinose across the BBB was found to be 1.0 compared to the urea reflection coefficient that was shown to be 0.44, indicating that raffinose exerts more of an effect on osmotic water movement than urea at the same concentration [11].
In those humans who have been studied, the predialysis urea concentration in the CSF was slightly lower than that in the blood [5, 15]. This indicates that there was a slight blood–CSF gradient for entry of urea into the CNS. After hemodialysis, the urea concentration in the CSF was higher than that in the blood, thus setting up an osmotic gradient for the movement of water into the CNS. In their study, Rosen et al. noted that the difference in the CSF–blood urea concentration was greater in those patients with higher predialysis blood urea concentrations (Fig. 2) [15]. In other words, those patients that were more uremic developed a larger osmotic gradient for water to move into the CNS [15].
Fig. 2.

Urea concentration gradients between the cerebrospinal fluid (CSF) and blood in patients undergoing hemodialysis. Vertical arrows represent the difference in the urea concentrations between the blood and CSF, horizontal axis represents the initial blood urea concentration. As can be seen, the patients with higher initial blood urea concentrations had larger post-dialysis blood–CSF urea gradients. (Reproduced from [15] with permission)
Development of cerebral edema
In recent years it has become clear that aquaporins (AQPs) regulate the movement of water in many tissues. The aquaporin present in the blood brain barrier, AQP4, resides in the cell membrane of the astrocytes [16]. Mice that are null for AQP4 do not develop cerebral edema to the same degree that wild-type mice do [16]. Thus, this AQP plays a critical role in the development of cerebral edema.
Although AQP4 provides a route for the movement of water from the blood stream into the CNS, there must be an osmotic gradient driving the movement of water. In guinea pigs, the osmotic gradient that was needed to drive significant amounts of water into the CNS was found to be 45 mOsm/kg water [17]. These studies were performed by acutely loading the guinea pigs with water and inhibiting water excretion by giving the animals vasopressin. This osmotic gradient is central to the understanding of the development of cerebral edema during the dialysis disequilibrium syndrome. The key question has been whether or not the gradient produced by the difference in urea concentration alone will account for the water movement into the brain [10]. Current hypotheses to explain the osmotic gradient are the “Reverse Urea Effect” and the “Idiogenic Osmoles” hypothesis.
The “Reverse Urea Effect” is based on the fact that the urea concentration in the CNS remains elevated because of its slower diffusion from the CNS to the blood than the diffusion of urea from the blood into the dialysate compartment. This is essentially the finding that the initial investigators found when they measured urea in the CSF of patients after the hemodialysis procedure and found it to be significantly higher than that of the blood stream. Careful measurements of blood and brain electrolyte, urea and water content in rats that were made uremic by ureteral ligation showed similar results [18, 19]. Rats that were dialyzed rapidly had a much higher urea concentration in the brain than in the blood. The increase in the urea concentration could account for the increased osmolality as there was no evidence of any new osmoles being produced.
The “Idiogenic Osmole” hypothesis is supported by experiments performed in dogs that were also made uremic by ureteral ligation [20]. In these studies, animals were dialyzed rapidly (over 100 min) or slowly (over 200 min) to achieve the same decline in urea concentration in the blood. In the animals that were rapidly dialyzed, the brain tissue had a significantly higher osmolality (27 mOsm/kg water) than the blood at the end of the dialysis procedure. This difference in osmolality could not be explained totally by the changes in electrolytes and urea. Thus, the authors proposed that the increase of brain osmolality was secondary to the new formation of organic molecules called “idiogenic osmoles” [20].
While there are some disparities between the two hypotheses, there are a number of similarities. First, both investigators demonstrated that the urea concentration in the CNS was much higher than that in the blood at the end of dialysis and that there was an osmotic gradient for water movement into the brain. Secondly, there was ample evidence that there was an increased brain water content after the dialysis procedure, indicating that water had moved down the osmotic gradient into the brain. The differences revolve around the argument of the magnitude of the osmotic gradient and whether or not the high urea concentration could account for this.
The focus of attention on the magnitude of the gradient was the osmotic gradient found in a study of guinea pigs that showed a gradient of 45 mOsm/kg water would cause the development of cerebral edema [17, 20]. However, with recent data showing that the expression of both AQP4 and AQP9 is increased in the brains of uremic rats, it might be possible that a lower gradient would cause water movement, leading to the development of cerebral edema in uremic patients [13]. It is also possible that the urea gradient might have a larger influence in uremic patients. The reflection coefficient of urea in the CNS has been measured in normal rabbits [11]. With recent data showing that UT-B1 transporter expression in the brain decreases in uremic rats, the reflection coefficient could be higher so that a smaller gradient of urea would have a larger osmotic force [13]. The increased expression of AQP4 and AQP9 and decreased expression of UT-B1 in uremic animals compared to non-uremic ones could result in a higher urea reflection coefficient for urea. This would allow for an increased water movement, with a smaller gradient of urea contributing to the disparity of these hypotheses.
The models used by the investigators were also slightly different. The model used by Arieff et al. was ureteral ligation in the dog [20], and the animals were then studied 3 days later. The model used by Silver et al. was the rat, and these animals were studied 42 h after ureteral ligation [18]. The difference in the timing of uremia leaves open the possibility that uremic toxins that would lead to the production of “idiogenic osmoles” could occur after 48 h of uremia. It is also possible that the uremic toxins themselves could be the “idiogenic osmoles.” These are questions that remain unanswered at the current time.
Evidence for cerebral edema in patients who had developed the dialysis disequilibrium syndrome comes from autopsy data and from radiologic imaging of the brain [1, 21, 22]. In careful studies of rats that had been nephrectomized, magnetic resonance imaging (MRI) confirmed the presence of cerebral edema after the rats received hemodialysis [23]. More interestingly, the authors concluded from the results of their diffusion-weighted MRI study that the edema was interstitial and not intracellular [23]. Similar conclusions were drawn by Chen et al. in their study of hemodialysis patients who underwent diffusion-weighted MRI after their dialysis treatment; these investigators found evidence of interstitial edema and not intracellular edema [24].
The effect of cerebral acidosis
The role of acidosis in the development of the disequilibrium syndrome is not entirely clear. It has been shown that despite the rapid elevation of blood pH with the bicarbonate infusion of the dialysate, the brain intracellular pH (pHi) and CSF pH are significantly lower in the rapidly dialyzed group [25]. The arterial partial pressure of CO2 remained unchanged, suggesting that the production of the paradoxical CSF acidosis after rapid hemodialysis was not secondary to systemic hypoventilation.
The increased acidosis of the CNS can alter its ability to regulate solute and water transport through the BBB. In addition, the resulting changes in intracellular organic acids could impact the intracellular osmolality by displacing cations from their binding sites on intracellular proteins [25]. Thus, the role of changes in CNS acid–base levels is very complex and will need further investigation.
The impact of the rate of removal of urea
Arieff et al. examined directly the effect of the rate of urea removal [20] by making the animals (dogs) uremic by ureteral ligation and studying them 3 days later. The initial urea concentration was about 70 mmol/l [approximately 200 mg/dl of blood urea nitrogen (BUN)]. One group of animals was dialyzed over 100 min with a blood flow rate of 12 ml/kg/min and another group was dialyzed over 200 min with a blood flow rate of 5 ml/kg/min. Both groups had identical urea concentrations at the end of dialysis of 25 mmol/l (approximately 70 mg/dl of BUN). These researchers showed that the fast dialysis group developed seizures and increased intracranial pressure, whereas the slow dialysis group did not develop seizures or cerebral edema. These results clearly demonstrate that the rate of urea removal is crucial to the development of the syndrome. Both groups of animals had the same decrease in urea (65 % reduction ratio of urea), but only the fast dialysis group had symptoms.
The difference in osmolality between the brain and blood was larger in the group that received the fast dialysis and led to the idea of “idiogenic osmoles” being created [3, 20]. What is not clear is whether or not the slow dialysis group had idiogenic osmoles present in their brain tissue before the dialysis procedure which were subsequently removed more efficiently because of the slower rate of dialysis.
Unfortunately, there are no human studies comparing various rates of removal of urea to determine the rate at which the disequilibrium syndrome will develop. One case report described a very high urea reduction ratio (70 %) for the initial dialysis that led to the syndrome and ultimately to death of the patient [21]. However, another case report described symptoms occurring with a urea reduction ratio as low as 17 % over 2 h [26]. This patient had a very high urea concentration prior to dialysis (299 mg/dl or about 100 mmoles/l). Thus, not only is the rate of removal of urea critical to the development of the syndrome but also the initial urea concentration. This is most likely due to the fact that the blood–brain urea gradient at the end of dialysis will be higher in those patients with higher urea concentrations prior to dialysis (see Fig. 2) [15].
In addition, other factors that predispose patients to the development of the syndrome will have an impact on which rate of removal will lead to disequilibrium. For example, patients with known seizure disorders or other neurologic conditions are more prone to develop symptoms during dialysis [2, 3]. Another concern for the pediatric nephrologist is the fact that younger patients are more prone to develop disequilibrium [27]. It is unknown why this is true, but it could be related to the smaller volume of distribution of urea in these patients. Thus, while it is not clear what the maximal rate of removal of urea is safe, it is critical to initiate hemodialysis with a low rate of urea removal.
Prevention of the dialysis disequilibrium syndrome
Since the principle factor leading to the disequilibrium syndrome is the development of an osmotic gradient causing water to move into the brain, preventing the development of this gradient should prevent the syndrome. The simplest way to do this is to perform hemofiltration on the patient instead of dialysis [28]. This method of treatment relies on the convective removal of solute from the patient in place of diffusive removal. Thus, the osmolalities of the body fluid compartments will not change as rapidly as they do during standard hemodialysis. This method was shown to reduce some of the symptoms that are related to the disequilibrium syndrome [28].
Using standard hemodialysis, the approach that is widely recommended is to slowly lower the blood urea concentration. A goal of reducing the urea concentration by 40 % over 2 h for the first treatment is reasonable [29]. This would be a urea reduction ratio [URR = (pre-dialysis BUN − post-dialysis BUN)/predialysis BUN] of 0.4. It must be emphasized that there are no controlled trials demonstrating that this will be safe for all patients. The prescription for achieving this goal will depend on the size of the patient, which determines the volume of distribution of urea. Once this has been estimated, the blood flow rate and time of dialysis can be determined using urea kinetic modeling [30].
Briefly, the decline in the blood urea concentration can be modeled as a first order kinetic problem and would thus be described by an exponential decay. The rate constant for this process can be approximated by an expression known as KT/V. “K” represents the dialyzer clearance of urea that is a function of the size of the dialyzer and the blood flow rate in milliliters per minute. “T” is the time of the treatment (in min), and “V” is the volume of distribution of urea (in ml), which can be approximated by the total body water. Because of the exponential relationship of this expression to the blood urea concentration, the equation that relates the urea reduction ratio (URR) to the KT/V is the following:
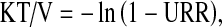 |
where ln is the natural logarithm. Thus, a urea reduction ratio of 0.4 would be the same as a KT/V of 0.5. It should be pointed out that this is a very simplified version of urea kinetic modeling, but it will serve as a good approximation for initial dialysis treatments. More complex forms of the modeling equations have been developed [30, 31].
It is important to emphasize that while the degree of reduction in urea is critical, the time of the treatment can also be critical. In the experiments with the dogs, the reduction of urea was the same in both the slow dialysis group and the fast dialysis group. However, only the fast dialysis group developed symptoms. Thus, the “T” in the above expression is usually set at 120 min and then the “K” can be determined after the “V” has been calculated.
In addition to the slow removal of urea, another way to prevent the syndrome is to add an osmotic agent to the blood stream, as this will help prevent the development of a blood–brain osmotic gradient and consequently help prevent cerebral edema. The easiest agent to add to the blood stream is sodium. Most of the modern hemodialysis machines allow for sodium modeling so that the sodium concentration of the dialysate can be set at a much higher concentration throughout the treatment. Using this approach, it has been shown that symptoms related to the syndrome could be prevented in patients undergoing dialysis compared to those who were treated using the standard sodium concentration [32]. The investigators also monitored the electroencephalogram (EEG) of these patients and found abnormalities in only two of the nine patients treated with the higher sodium dialysate compared to ten of the 13 patients treated with the standard sodium dialysate.
Other osmotic agents that have been used include mannitol and glucose [33]. Patients that were dialyzed using either a high glucose concentration in the bath (717 mg/dl) or were given intravenous mannitol (1 gm/kg body weight) had fewer symptoms related to dialysis disequilibrium, and the measured blood osmolality change was significantly reduced. When the two maneuvers were combined, the change in the blood osmolality was reduced even further and the incidence of symptoms was reduced to only 10 % of the control group. When the two maneuvers were compared individually, mannitol infusion had a better outcome than the high glucose dialysate. This has become the traditional agent used in most centers during the initiation of hemodialysis to prevent the disequilibrium syndrome [29].
Another agent that has been studied is glycerol [34]. When added to the dialysate in the uremic dog model, it was found to be superior to mannitol in preventing the syndrome. In particular, the EEG remained normal when glycerol was used as compared to the addition of mannitol to the dialysate. This is a theoretical treatment since these experiments were only performed in animals.
Lastly, urea itself can be used in the dialysate to prevent the development of the blood–brain urea gradient. This approach has been used in many of the animal models discussed above and has been shown to prevent the development of the osmotic gradient and signs or symptoms of the syndrome [3, 18, 20]. Kennedy, who first described the syndrome in 1962, used urea in the dialysate of patients who were undergoing hemodialysis and reported his findings in 1964 [35].
Treatment of the dialysis disequilibrium syndrome
The treatment of disequilibrium after it has developed is aimed at reducing the intracranial pressure of the patient. Standard maneuvers are to give mannitol or hypertonic saline to raise the blood osmolality and to hyperventilate the patient. These attempts have been described in many of the case reports of dialysis disequilibrium; however, they may be futile [21]. Thus, it is crucial for the nephrologist to consider the above options for the prevention of the syndrome.
Conclusion
The dialysis disequilibrium syndrome was initially described over 50 years ago. Although our understanding of the syndrome is more complete now, many details remain unknown. Controlled trials examining various rates of urea reduction will never be performed, and thus most nephrologists err on the side of very slowly reducing the urea concentration in a new patient. In addition, few details are available regarding the treatment and prevention in children. Once the syndrome has developed, it is very difficult to reverse and has a very high mortality rate. This is truly a condition where “an ounce of prevention is worth a pound of cure.”
Key points
1. Dialysis disequilibrium can occur in any patient undergoing hemodialysis, but it is more often seen when patients are undergoing their first treatment.
Slow removal of urea during the first several treatments is critical for avoiding this syndrome.
If a patient shows signs or symptoms of dialysis disequilibrium, steps to lower intracranial pressure can help reduce morbidity and mortality.
Research points
The role of acidosis and intracellular pH changes remain ill defined. Studies to determine how acidosis should be corrected could help eliminate this syndrome.
While it is clear that there is a urea gradient from the blood into the CSF, the existence and role of idiogenic osmoles remain elusive. Could these represent uremic toxins?
The optimal rate of removal of urea remains unknown. Other methods of preventing rapid changes in osmolality should be investigated.
Questions (answers are provided following the reference list)
1. What is/are the crucial element(s) involved in the development of dialysis disequilibrium syndrome
Pre-dialysis concentration of urea
Rate of urea removal
Pre-dialysis blood pressure
Patient’s volume status
a and b
2. What component of the neural microenviroment is responsible for regulating movement of small organic solutes and ions between the blood and the CNS extracellular space?
Choroid plexus
Microvacular unfenestrated endothelial cells with tight junctions
Astrocytes
Neurons
Glia
3. In uremic rats AQP4, AQP9 and UTB-1 are:
AQP4↑, AQP9↓, UTB-1↑
AQP4↑, AQP9↑, UTB-1↓
AQP4↓, AQP9↑, UTB-1↓
AQP4↑, AQP9↑, UTB-1↑
AQP4↓, AQP9↓, UTB-1↓
4. A patient presents with signs and symptoms of uremia and is found to have a BUN of 200 mg/dl. A reasonable plan to avoid disequilibrium for the first dialysis treatment would be to lower his BUN to:
40 mg/dl over 2 hours
80 mg/dl over 4 hours
120 mg/dl over 2 hours
120 mg/dl over 1 hour
5. In the above patient, an additional maneuver to avoid disequilibrium would be to:
infuse mannitol (1 gm/kg) over the first hour of treatment
use a dialysate sodium concentration of 130 mEq/liter
raise the dialysate bicarbonate concentration to 40 mEq/liter
avoid any ultrafiltration of fluid
Footnotes
Answers
1. E. Disequilibrium has been reported in a patient with a urea reduction ratio of 17 %. This patient had an initial urea concentration of 299 mg/dl. Thus, the initial BUN is critical. In addition, animal models have shown that the rate of reduction of urea is a crucial element in development of dialysis disequilibrium syndrome [15, 26, 36].
2. B. The microvascular endothelial cells of the BBB have low ion permeability, high reflection coefficient and high electrical resistance compared to other vascular beds. They regulate the movement of small organic solutes and ions [8, 9].
3. B. There is increased expression of AQP9 and AQP4 and decreased expression of UTB-1 [13].
4. C. This is a reduction of 40 % from the initial BUN over a time period of 2 h. While there are no studies to prove this is safe, there is consensus that this is a reasonable plan.
5. A. Infusing mannitol has been shown to reduce the symptoms of disequilibrium. Using a high dialysate sodium concentration (not low) would also help in preventing it.
References
- 1.Harris CP, Townsend JJ. Dialysis disequilibrium syndrome. West J Med. 1989;151:52–55. [PMC free article] [PubMed] [Google Scholar]
- 2.Arieff AI. More on the dialysis disequilibrium syndrome. West J Med. 1989;151:74–76. [PMC free article] [PubMed] [Google Scholar]
- 3.Arieff AI. Dialysis disequilibrium syndrome: current concepts on pathogenesis and prevention. Kidney Int. 1994;45:629–635. doi: 10.1038/ki.1994.84. [DOI] [PubMed] [Google Scholar]
- 4.Mahoney CA, Arieff AI. Uremic encephalopathies: clinical, biochemical, and experimental features. Am J Kidney Dis. 1982;2:324–336. doi: 10.1016/s0272-6386(82)80090-5. [DOI] [PubMed] [Google Scholar]
- 5.Kennedy AC, Linton AL, Eaton JC. Urea levels in cerebrospinal fluid after haemodialysis. Lancet. 1962;1:410–411. doi: 10.1016/S0140-6736(62)91365-X. [DOI] [PubMed] [Google Scholar]
- 6.Liebner S, Czupalla CJ, Wolburg H. Current concepts of blood–brain barrier development. Int J Dev Biol. 2011;55:467–476. doi: 10.1387/ijdb.103224sl. [DOI] [PubMed] [Google Scholar]
- 7.Paolinelli R, Corada M, Orsenigo F, Dejana E. The molecular basis of the blood brain barrier differentiation and maintenance. Is it still a mystery? Pharmacol Res. 2011;63:165–171. doi: 10.1016/j.phrs.2010.11.012. [DOI] [PubMed] [Google Scholar]
- 8.Abbott NJ. Astrocyte-endothelial interactions and blood–brain barrier permeability. J Anat. 2002;200:629–638. doi: 10.1046/j.1469-7580.2002.00064.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li G, Simon MJ, Cancel LM, Shi ZD, Ji X, Tarbell JM, Morrison B, 3rd, Fu BM. Permeability of endothelial and astrocyte cocultures: in vitro blood–brain barrier models for drug delivery studies. Ann Biomed Eng. 2010;38:2499–2511. doi: 10.1007/s10439-010-0023-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Silver SM, Sterns RH, Halperin ML. Brain swelling after dialysis: old urea or new osmoles? Am J Kidney Dis. 1996;28:1–13. doi: 10.1016/S0272-6386(96)90124-9. [DOI] [PubMed] [Google Scholar]
- 11.Fenstermacher JD, Johnson JA. Filtration and reflection coefficients of the rabbit blood–brain barrier. Am J Physiol. 1966;211:341–346. doi: 10.1152/ajplegacy.1966.211.2.341. [DOI] [PubMed] [Google Scholar]
- 12.Kleeman CR, Davson H, Levin E. Urea transport in the central nervous system. Am J Physiol. 1962;203:739–747. doi: 10.1152/ajplegacy.1962.203.4.739. [DOI] [PubMed] [Google Scholar]
- 13.Trinh-Trang-Tan MM, Cartron JP, Bankir L. Molecular basis for the dialysis disequilibrium syndrome: altered aquaporin and urea transporter expression in the brain. Nephrol Dial Transplant. 2005;20:1984–1988. doi: 10.1093/ndt/gfh877. [DOI] [PubMed] [Google Scholar]
- 14.Rapoport SI, Robinson PJ. Tight-junctional modification as the basis of osmotic opening of the blood–brain barrier. Ann N Y Acad Sci. 1986;481:250–267. doi: 10.1111/j.1749-6632.1986.tb27155.x. [DOI] [PubMed] [Google Scholar]
- 15.Rosen SM, O’Connor K, Shaldon S. Haemodialysis disequilibrium. Br Med J. 1964;2:672–675. doi: 10.1136/bmj.2.5410.672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Verkman AS. Aquaporins: translating bench research to human disease. J Exp Biol. 2009;212:1707–1715. doi: 10.1242/jeb.024125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Stern WE, Coxon RV. Osmolality of brain tissue and its relation to brain bulk. Am J Physiol. 1964;206:1–7. doi: 10.1152/ajplegacy.1964.206.1.1. [DOI] [PubMed] [Google Scholar]
- 18.Silver SM, DeSimone JA, Jr, Smith DA, Sterns RH. Dialysis disequilibrium syndrome (DDS) in the rat: role of the "reverse urea effect". Kidney Int. 1992;42:161–166. doi: 10.1038/ki.1992.273. [DOI] [PubMed] [Google Scholar]
- 19.Silver SM. Cerebral edema after rapid dialysis is not caused by an increase in brain organic osmolytes. J Am Soc Nephrol. 1995;6:1600–1606. doi: 10.1681/ASN.V661600. [DOI] [PubMed] [Google Scholar]
- 20.Arieff AI, Massry SG, Barrientos A, Kleeman CR. Brain water and electrolyte metabolism in uremia: effects of slow and rapid hemodialysis. Kidney Int. 1973;4:177–187. doi: 10.1038/ki.1973.100. [DOI] [PubMed] [Google Scholar]
- 21.Bagshaw SM, Peets AD, Hameed M, Boiteau PJ, Laupland KB, Doig CJ. Dialysis Disequilibrium Syndrome: brain death following hemodialysis for metabolic acidosis and acute renal failure—a case report. BMC Nephrol. 2004;5:9. doi: 10.1186/1471-2369-5-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sheth KN, Wu GF, Messe SR, Wolf RL, Kasner SE. Dialysis disequilibrium: another reversible posterior leukoencephalopathy syndrome? Clin Neurol Neurosurg. 2003;105:249–252. doi: 10.1016/S0303-8467(03)00039-8. [DOI] [PubMed] [Google Scholar]
- 23.Galons JP, Trouard T, Gmitro AF, Lien YH. Hemodialysis increases apparent diffusion coefficient of brain water in nephrectomized rats measured by isotropic diffusion-weighted magnetic resonance imaging. J Clin Invest. 1996;98:750–755. doi: 10.1172/JCI118847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen CL, Lai PH, Chou KJ, Lee PT, Chung HM, Fang HC. A preliminary report of brain edema in patients with uremia at first hemodialysis: evaluation by diffusion-weighted MR imaging. AJNR Am J Neuroradiol. 2007;28:68–71. [PMC free article] [PubMed] [Google Scholar]
- 25.Arieff AI, Guisado R, Massry SG, Lazarowitz VC. Central nervous system pH in uremia and the effects of hemodialysis. J Clin Invest. 1976;58:306–311. doi: 10.1172/JCI108473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lopez-Almaraz E, Correa-Rotter R. Dialysis disequilibrium syndrome and other treatment complications of extreme uremia: a rare occurrence yet not vanished. Hemodial Int. 2008;12:301–306. doi: 10.1111/j.1542-4758.2008.00270.x. [DOI] [PubMed] [Google Scholar]
- 27.Grushkin CM, Korsch B, Fine RN. Hemodialysis in small children. JAMA. 1972;221:869–873. doi: 10.1001/jama.1972.03200210017005. [DOI] [PubMed] [Google Scholar]
- 28.Kishimoto T, Yamagami S, Tanaka H, Ohyama T, Yamamoto T, Yamakawa M, Nishino M, Yoshimoto S, Maekawa M. Superiority of hemofiltration to hemodialysis for treatment of chronic renal failure: comparative studies between hemofiltration and hemodialysis on dialysis disequilibrium syndrome. Artif Organs. 1980;4:86–93. doi: 10.1111/j.1525-1594.1980.tb03911.x. [DOI] [PubMed] [Google Scholar]
- 29.Patel N, Dalal P, Panesar M. Dialysis disequilibrium syndrome: a narrative review. Semin Dial. 2008;21:493–498. doi: 10.1111/j.1525-139X.2008.00474.x. [DOI] [PubMed] [Google Scholar]
- 30.Daugirdas JT. Second generation logarithmic estimates of single-pool variable volume Kt/V: an analysis of error. J Am Soc Nephrol. 1993;4:1205–1213. doi: 10.1681/ASN.V451205. [DOI] [PubMed] [Google Scholar]
- 31.Goldstein SL, Sorof JM, Brewer ED. Natural logarithmic estimates of Kt/V in the pediatric hemodialysis population. Am J Kidney Dis. 1999;33:518–522. doi: 10.1016/S0272-6386(99)70189-7. [DOI] [PubMed] [Google Scholar]
- 32.Port FK, Johnson WJ, Klass DW. Prevention of dialysis disequilibrium syndrome by use of high sodium concentration in the dialysate. Kidney Int. 1973;3:327–333. doi: 10.1038/ki.1973.51. [DOI] [PubMed] [Google Scholar]
- 33.Rodrigo F, Shideman J, McHugh R, Buselmeier T, Kjellstrand C. Osmolality changes during hemodialysis. Natural history, clinical correlations, and influence of dialysate glucose and intravenous mannitol. Ann Intern Med. 1977;86:554–561. doi: 10.7326/0003-4819-86-5-554. [DOI] [PubMed] [Google Scholar]
- 34.Arieff AI, Lazarowitz VC, Guisado R. Experimental dialysis disequilibrium syndrome: prevention with glycerol. Kidney Int. 1978;14:270–278. doi: 10.1038/ki.1978.119. [DOI] [PubMed] [Google Scholar]
- 35.Kennedy AC, Linton AL, Luke RG, Renfrew S, Dinwoodie A. The pathogenesis and prevention of cerebral dysfunction during dialysis. Lancet. 1964;1:790–793. doi: 10.1016/S0140-6736(64)92981-2. [DOI] [PubMed] [Google Scholar]
- 36.Arieff AI, Massry SG, Barrientos A, Kleeman CR. Brain water and electrolyte metabolism in uremia: effects of slow and rapid hemodialysis. Kidney Int. 1973;4:177–187. doi: 10.1038/ki.1973.100. [DOI] [PubMed] [Google Scholar]
- 37.Kleeman CR. Metabolic coma. Kidney Int. 1989;36:1142–1158. doi: 10.1038/ki.1989.313. [DOI] [PubMed] [Google Scholar]