

Abstract
BACKGROUND:
Rubinstein-Taybi syndrome (RSTS) is a rare congenital neurodevelopmental disorder, characterized by postnatal growth deficiency, typical dysmorphic features, broad thumbs and toes, and mental retardation. Very few cases are reported in literature from developing countries. Diagnosis is often delayed due to non-familiarity with the characteristic features of this syndrome.
AIMS:
To report 11 cases of RSTS and to review the current literature.
SETTINGS AND DESIGN:
Retrospective study conducted in genetic and metabolic unit of a tertiary care teaching hospital in north India over a period of 3½ years.
MATERIALS AND METHODS:
11 patients with diagnosis of RSTS were identified, and their case sheets were reviewed.
RESULTS:
Developmental delay was presenting complaint in 10 patients, and seizure in 1 case. 7 patients had microcephaly (head circumference below −3 SD), and a prominent beaked nose was seen in 9 patients. The intelligence quotient (IQ) varied from 22-62 in 7 patients who had mental retardation. The most notable features in hands were broadness, shortening, and flattening of the distal phalanx of thumbs or great toes. Additionally, we also noted webbing of neck, microphthalmia, and pachygyria (on MRI brain) in 1 patient each.
CONCLUSIONS:
The diagnosis of RSTS is primarily clinical and based on characteristic phenotype that is often combined with a variety of somatic anomalies. An early diagnosis facilitates appropriate genetic counseling and in planning the management.
Keywords: Beaked nose, broad thumbs, broad toes, dysmorphism, mental retardation
Introduction
Rubinstein-Taybi syndrome (RSTS) is a multiple congenital anomalies-mental retardation syndrome, characterized by postnatal growth failure, microcephaly, distinctive facial features, broad and often angulated thumbs and great toes, short stature, and mental retardation.[1,2] The symptom complex was first described by Michail et al. in 1957[3] and was precisely delineated later as a syndrome in 1963 by Rubinstein and Taybi.[1] The characteristic craniofacial features are downslanting palpebral fissures, columella extending below the nares, highly arched palate, grimacing smile, and talon cusps of permanent incisors.[2] Other variable findings are coloboma, cataract, congenital heart defects, renal abnormalities, and cryptorchidism.[2] Birth prevalence is 1 in 100,000 to 125,000.[2,4] The diagnosis of RSTS is primarily based on clinical features. CREBBP and EP300 are the only genes currently known to be associated with RSTS.[2,5,6] RSTS typically occurs as the result of a de novo mutation. In most instances, the parents of an individual with RSTS are not affected. Prenatal testing for pregnancies at increased risk is possible if the disease-causing mutation or deletion in the family is known.[4] In this paper, we present 11 cases of RSTS from a tertiary care center in north India seen over a period of 3½ years.
Materials and Methods
Between May 2008 and October 2011, a total of 973 patients registered in genetic and metabolic unit of a tertiary care teaching hospital in north India were screened, and 11 patients with diagnosis of RSTS were identified and their case sheets were reviewed. Information about demographics profile, presenting complaints were obtained. Detailed clinical examination findings, anthropometric profile, abnormal facial features, anomalies, associated features, and complications were noted. The diagnosis of RSTS was made on basis of clinical features and database search by an expert clinical geneticist.
Results
During 3½ years of study period, 11 patients with diagnosis of RSTS were identified out of 973 patients registered in genetic clinic. The mean age at diagnosis was 4.3 years (Range: 4 months - 15 years). There were more males than females (9 males and 2 females). The mean maternal age at time of birth was 25.4 years (Range: 21-31 years); the mean paternal age was 27.4 years (Range: 24-33 years). There was no history of consanguinity. None of the family members had history of mental retardation or obvious somatic malformations.
Clinical features
The clinical features of our patients are listed in Table 1. In this series, 7 patients had short stature (length or height below 3rd centile) and 8 had postnatal growth failure. Developmental delay was presenting complaint in 10 patients, and seizure in 1. 7 patients were mentally retarded. The intelligence quotient (IQ) varied from 22-62 with mean value of 39 using standard approved scales. All of our patients had broad thumbs and/or broad great toes. The thumbs were disproportionately broad in relation to size of hand, but degree of broadness varied from patient to patient. The most consistent feature were broadness, shortening, and flattening of the distal phalanx of thumb or great toe [Figure 1a]. In addition, 2 patients had radial displacement of the terminal phalanx of the thumb [Figure 1b], 2 had persistent fetal finger pads, and 1 had broadness and shortening (brachydactyly) of distal phalanx of other fingers. The main facial features were anti-mongoloid slant of the eyes (in 7 patients), heavy and high arched eyebrows (4 patients), hypertelorism (5 patients), low set ears (7 patients), a prominent beaked nose (9 patients), and narrow high-arched palate, so-called ‘steeple palate’ (6 patients) [Figure 2]. In addition, 5 had retrognathia/micrognathia, 4 had epicanthal folds, 4 had thick upper lip, 3 had malformed ears, and 2 had long eye lashes. Hypoplastic maxilla, malpositioned crowded teeth, and strabismus were noted in 1 patient each. Talon cusps of permanent incisors were seen in 3 patients. 7 patients had microcephaly (head circumference below -3 SD), and broad anterior fontanel was noted in 5 patients. 3 out of 9 male patients had cryptorchidism and 1 had micropenis. Hydronephrosis (left-sided) and absent kidney (left-sided) was noted in 1 patient each. 2 patients had cardiac malformations (ventricular septal defect and patent ductus arteriosus in 1 patient each), 1 had bilateral nasolacrimal duct obstruction, 1 had corneal opacity, 1 had tracheo-esophageal fistula (operated at 1 month of age), 2 had scoliosis, 1 had cervical dysraphism, and 2 had constipation. Skin involvement was noted in 3 patients (2 had hirsutism and 1 had capillary malformation). We also documented some unusual features like webbing of neck, micro-ophthalmia, and pachygyria (on MRI brain) in 1 patient each.
Table 1.
Clinical characteristics of present cohort of patients with Rubinstein-Taybi syndrome
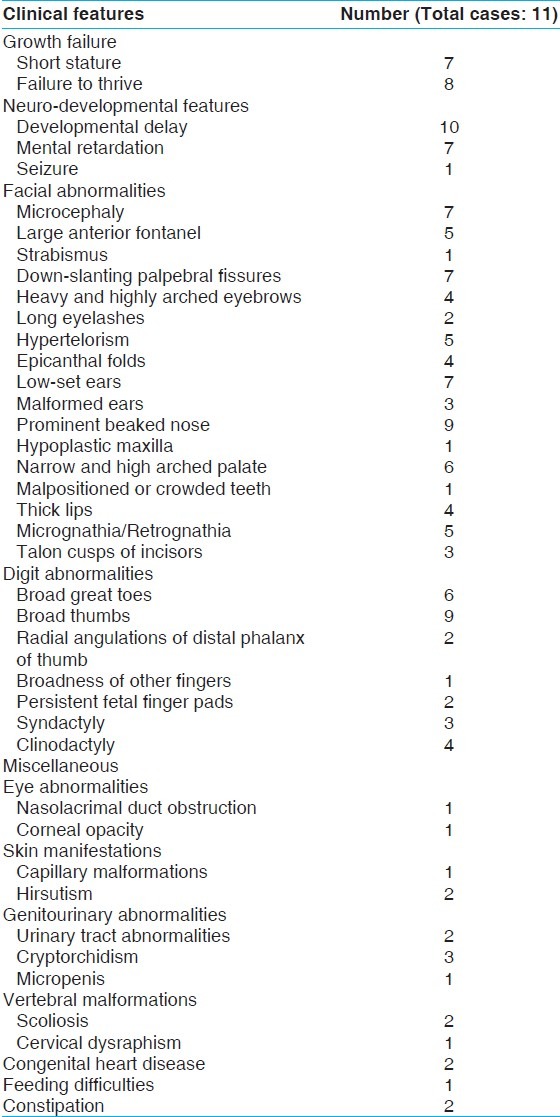
Figure 1.
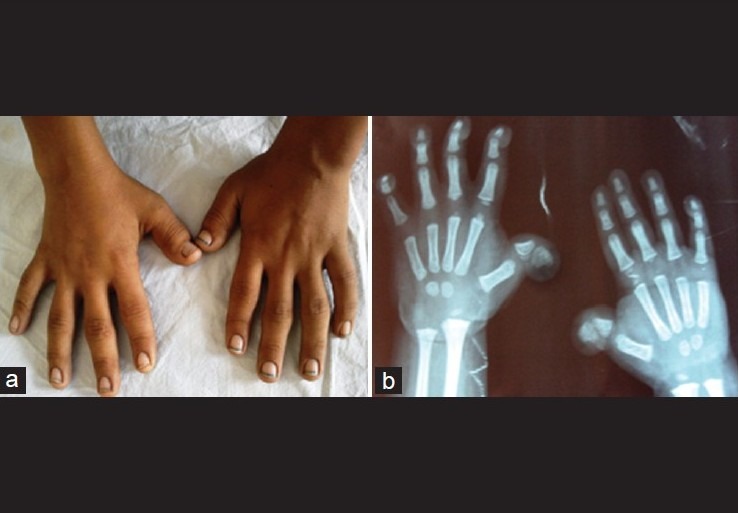
(a) Showing broad thumbs, (b) X-ray of hands of another patient showing broad and radially- deviated thumbs
Figure 2.

(a-c) Characteristic facial profile of two patients
Discussion
Rubinstein-Taybi syndrome (RSTS) (broad thumb-hallux syndrome) is a rare multiple congenital anomalies–mental retardation syndrome, characterized by postnatal growth deficiency, microcephaly, characteristic facies, broad thumbs and big toes, and mental retardation.[1,2,7] Till date, over 1000 cases have been described in literature.[8] RSTS is a prototype of diseases with genetic heterogeneity.
The main features that are characteristics of RSTS are noted on the face and limbs. The characteristic craniofacial features are down-slanting palpebral fissures, highly-arched and thick eyebrows, long eyelashes, beaked nose with columella extending well below the nares, a high arched palate, micrognathia, and a grimacing smile.[2,9] The finding of talon cusps at the permanent incisors is a characteristic feature.[10] Broad thumbs and broad big toes are present in almost all cases.[2] Thumbs are radially deviated in about 1/3rd of patients. In addition, terminal broadening of the distal phalanges of the fingers, persistent fetal pads, and clinodactyly of the 5th finger can be present.[2] Most of the patients in current series had characteristic facial features and broad thumbs and/or broad great toes. 2 patients had peculiar radial deviation of thumb and 4 had clinodactyly.
Postnatal growth failure was noted in 72.7% (8 of 11) patients in this study. Prenatal growth is usually normal; however, there is marked growth retardation with poor weight gain during infancy, often replaced by overweight in later childhood or in puberty.[11] Mental retardation is characteristic with an average IQ between 35 and 50, but a cognitive functioning outside these limits does occur.[12] The mean IQ of patients in our series was 39. Their behavior is characterized by short attention span, poor coordination, impulsivity, distractibility, instability of moods and stereotypies, which seem to increase with age.[2,13] Although the cognitive delay is usually present, they have a marked ability to establish excellent social contacts.[2]
Additional features reported are eye abnormalities including nasolacrimal duct obstruction, ptosis of eyelids, congenital or juvenile glaucoma, and refractive errors; a variety of congenital heart defects like VSD, ASD, PDA; genitourinary abnormalities; dental problems; joint hypermobility; obesity; life-long constipation; and skin anomalies such as hirsutism, nevus flammeus on the forehead, and keloid formation.[2,5,9] Few patients in our study had some of these features.
In the neonatal period, accompanying problems include respiratory and feeding difficulties, congenital heart defects, seizures, and growth retardation. Individuals with RSTS can be easily diagnosed early in life with the typical stigmata.[2,14,15] People with RSTS have an increased risk of developing malignancies, including brain tumors (meningioma, medulloblastoma) and hematologic malignancies (leukemia).[16] They tend to occur before 15 years of age, although meningioma also occurs in adulthood.[16] This necessitates regular follow- up and diagnostic evaluation in these cases. During the short follow-up, none of the patient in current series was found to have any tumor or malignancy. The severity of signs and symptoms vary among affected individuals.[2] Life expectancy seems to be normal.
There are no established diagnostic criteria for RSTS. Careful history taking, family history, and physical examination are the cornerstones to the diagnosis. The diagnosis is essentially clinical and relies on recognition of the characteristic features. The major features to be look for are the beaked nose with low hanging septum, broad thumbs and big toes, and mental retardation. Additional studies may include radiographies of hands and feet to check for (partial) duplications of the first rays. Database search is an essential tool to reach the diagnosis and exclude similar syndromes.
Several conditions including FG syndrome, Floating Harbor syndrome, and Tricho-rhino-phalangeal syndrome were considered in the differential diagnosis, but were subsequently excluded. FG syndrome (FGS) is an X-linked recessive disorder, characterized by mental retardation, hypotonia, particular dysmorphic facial features, broad thumbs and halluces, anal anomalies, constipation, and abnormalities of the corpus callosum. Clinical diagnosis of FGS is difficult clinically because of broad spectrum of signs and symptoms.[17] Floating-Harbor syndrome is characterized by low birth weight, facial dysmorphism, short stature with delayed bone age, and developmental delay. Facial features include a triangular-shaped face, a large bulbous nose with a broad nasal bridge, a wide columella, deep-set eyes with long eyelashes, a wide mouth with thin lips, and a short smooth philtrum. Thumbs are short and broad, or bifid.[18] The majority of reported cases are sporadic, but a few familial cases, with predominantly autosomal dominant inheritance, have been reported.[18] Tricho-rhino-phalangeal syndrome (TRPS) is autosomal dominant malformation syndrome characterized by craniofacial and skeletal abnormalities. TRPS is characterized by sparse scalp hair, a bulbous tip of the nose, a long flat philtrum, a thin upper vermilion border, and protruding ears. Skeletal abnormalities include cone-shaped epiphyses at the phalanges, hip malformations, short stature, and shortening of all phalanges and metacarpals.[19] None of the present cases had hypotonia, anal anomalies, bifid thumbs, or cone-shaped epiphysis of phalanges. The diagnosis in present cases was made by appropriate database search including online databases.
Most cases of RSTS are sporadic, and families with more than 1 affected child are extremely rare.[20] The known genetic causes of RSTS are point mutations or microdeletions of CREB-binding protein gene (CREBBP) located on chromosome 16p 13.3 (in 50-60% affected patients), and of the EP300 gene encoding E1A binding protein p300 localized on 22q13.2 (in 5% patients).[6,8,21] The mutations causing RSTS are almost always de novo occurring in autosomal dominant fashion. Sharma et al. reported 13 patients of RSTS, and 10 of these had mutation in CREBBP gene, which is the first report on the molecular investigations in RSTS patients in the Indian population.[21] Adequate counseling of parents of a child with RSTS involves providing complete and comprehensive information on the syndrome itself, information regarding recurrence risks, and prenatal diagnosis. The empiric recurrence risk for a couple with a previous child with RSTS is as low as 0.1%.[4] Proper cytogenetic investigations including FISH studies can be done in selected cases, followed by proper molecular studies if needed. In married patients, the recurrence risk could be as high as 50% for offspring. If a cytogenetic or molecular abnormality has been detected, reliable prenatal diagnosis can be possible.[2]
An early diagnosis of RSTS is crucial, both for adequate information and for treatment of medical problems. In the first year of life, specific attention will be paid to the feeding problems, constipation, lacrimal duct stenosis, congenital heart defects, and glaucoma. In males, undescended testes can be surgically corrected if needed. If surgery or anesthetics are required, one should be aware that they are susceptible to tracheal collapse after muscle relaxants, which may cause intubation problems. Later in life, the constipation should not be ignored, and weight gain can occur around puberty. The combination of a narrow palate, micrognathia, hypotonia, obesity, and easy collapsibility of the laryngeal walls give rise to an extreme snoring and obstructive sleep apneas. Recurrent upper respiratory tract infections can lead to hearing loss. The abnormal talon cusp shape of teeth causes an increased risk for caries. Some patients have a tendency to develop keloids on upper chest and arms, sometimes after trauma, sometimes spontaneously, and treatment is disappointing.[2] Specific attention for the first symptoms of malignancy will allow an early recognition and thus increase the chances of successful intervention.[2] The delayed motor and cognitive development in RSTS needs continuous attention. Almost all patients will be best stimulated if they attend special schools for children with learning disabilities. In general, these children are friendly, happy, and are easy going.[12]
Conclusion
RSTS is regarded as one of the archetypical syndrome in clinical genetics in which multiple congenital malformations and intellectual impairment is caused by a generalized dysregulation of gene expression.[9] All of our patients had features consistent with RSTS as documented in literature, but webbing of neck, microphthalmia, and pachygyria were additional findings, which were seldom reported in associated with RSTS. Identifying these multiple malformations by an early diagnosis is important because multidisciplinary approach is required in the initial evaluation, treatment, and follow-up.
Footnotes
Source of Support: Nil
Conflict of Interest: No.
References
- 1.Rubinstein JH, Taybi H. Broad thumbs and toes and facial abnormalities.A possible mental retardation syndrome. Am J Dis Child. 1963;105:588–608. doi: 10.1001/archpedi.1963.02080040590010. [DOI] [PubMed] [Google Scholar]
- 2.Hennekam RC. Rubinstein-Taybi syndrome. Eur J Hum Genet. 2006;14:981–5. doi: 10.1038/sj.ejhg.5201594. [DOI] [PubMed] [Google Scholar]
- 3.Michail J, Matsoukas J, Theodorou S. Arched, clubbed thumb in strong abduction-extension & other concomitant symptoms. Rev Chir Orthop Reparatrice Appar Mot. 1957;43:142–6. [PubMed] [Google Scholar]
- 4.Hennekam RC, Stevens CA, Van de Kamp JJ. Etiology and recurrence risk in Rubinstein-Taybi syndrome. Am J Med Genet Suppl. 1990;6:56–64. doi: 10.1002/ajmg.1320370610. [DOI] [PubMed] [Google Scholar]
- 5.Kim SH, Lim BC, Chae JH, Kim KJ, Hwang YS. A case of Rubinstein-Taybi Syndrome with a CREB-binding protein gene mutation. Korean J Pediatr. 2010;53:718–21. doi: 10.3345/kjp.2010.53.6.718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.van Belzen M, Bartsch O, Lacombe D, Peters DJ, Hennekam RC. Rubinstein-Taybi syndrome (CREBBP, EP300) Eur J Hum Genet. 2011;19:118–20. doi: 10.1038/ejhg.2010.124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wiley S, Swayne S, Rubinstein JH, Lanphear NE, Stevens CA. Rubinstein-Taybi syndrome medical guidelines. Am J Med Genet A. 2003;119A:101–10. doi: 10.1002/ajmg.a.10009. [DOI] [PubMed] [Google Scholar]
- 8.Hosek J, Borkova A. The Rubinstein-Taybi syndrome or a broad thumb-hallux syndrome. Cas Lek Cesk. 2008;147:136–40. [PubMed] [Google Scholar]
- 9.Roelfsema JH, Peters DJ. Rubinstein-Taybi syndrome: Clinical and molecular overview. Expert Rev Mol Med. 2007;9:1–16. doi: 10.1017/S1462399407000415. [DOI] [PubMed] [Google Scholar]
- 10.Hennekam RC, Van Doorne JM. Oral aspects of Rubinstein-Taybi syndrome. Am J Med Genet Suppl. 1990;6:42–7. doi: 10.1002/ajmg.1320370607. [DOI] [PubMed] [Google Scholar]
- 11.Stevens CA, Hennekam RC, Blackburn BL. Growth in the Rubinstein-Taybi syndrome. Am J Med Genet Suppl. 1990;6:51–5. doi: 10.1002/ajmg.1320370609. [DOI] [PubMed] [Google Scholar]
- 12.Hennekam RC, Baselier AC, Beyaert E, Bos A, Blok JB, Jansma HB, et al. Psychological and speech studies in Rubinstein-Taybi syndrome. Am J Ment Retard. 1992;96:645–60. [PubMed] [Google Scholar]
- 13.Verhoeven WM, Tuinier S, Kuijpers HJ, Egger JI, Brunner HG. Psychiatric profile in rubinstein-taybi syndrome. A review and case report. Psychopathology. 2010;43:63–8. doi: 10.1159/000260045. [DOI] [PubMed] [Google Scholar]
- 14.Torres LC, de Lourdes Lopes Chauffaille M, Delboni TP, Okay TS, Carneiro-Sampaio M, Sugayama S. Rubinsteintaybi syndrome: A female patient with a de novo reciprocal translocation t(2; 16)(q36.3; p13.3) and dysgranulopoiesis. Clinics (Sao Paulo) 2010;65:107–9. doi: 10.1590/S1807-59322010000100016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Stef M, Simon D, Mardirossian B, Delrue MA, Burgelin I, Hubert C, et al. Spectrum of CREBBP gene dosage anomalies in Rubinstein-Taybi syndrome patients. Eur J Hum Genet. 2007;15:843–7. doi: 10.1038/sj.ejhg.5201847. [DOI] [PubMed] [Google Scholar]
- 16.Miller RW, Rubinstein JH. Tumors in Rubinstein-Taybi syndrome. Am J Med Genet. 1995;56:112–5. doi: 10.1002/ajmg.1320560125. [DOI] [PubMed] [Google Scholar]
- 17.Lyons MJ, Graham JM, Jr, Neri G, Hunter AG, Clark RD, Rogers RC, et al. Clinical experience in the evaluation of 30 patients with a prior diagnosis of FG syndrome. J Med Genet. 2009;46:9–13. doi: 10.1136/jmg.2008.060509. [DOI] [PubMed] [Google Scholar]
- 18.White SM, Morgan A, Da Costa A, Lacombe D, Knight SJ, Houlston R, et al. The phenotype of Floating-Harbor syndrome in 10 patients. Am J Med Genet A. 2010;152A:821–9. doi: 10.1002/ajmg.a.33294. [DOI] [PubMed] [Google Scholar]
- 19.Ludecke HJ, Schaper J, Meinecke P, Momeni P, Gross S, von Holtum D, et al. Genotypic and phenotypic spectrum in tricho-rhino-phalangeal syndrome types I and III. Am J Hum Genet. 2001;68:81–91. doi: 10.1086/316926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Akin MA, Gunes T, Akin L, Coban D, Oncu SK, Kiraz A, et al. Thyroid hypoplasia as a cause of congenital hypothyroidism in monozygotic twins concordant for Rubinstein-Taybi syndrome. J Clin Res Pediatr Endocrinol. 2011;3:32–5. doi: 10.4274/jcrpe.v3i1.07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sharma N, Mali AM, Bapat SA. Spectrum of CREBBP mutations in Indian patients with Rubinstein-Taybi syndrome. J Biosci. 2010;35:187–202. doi: 10.1007/s12038-010-0023-5. [DOI] [PubMed] [Google Scholar]