

Abstract
Kartagener's syndrome is a very rare congenital malformation comprising of a classic triad of sinusitis, situs inversus and bronchiectasis. Primary ciliary dyskinesia is a genetic disorder with manifestations present from early life and this distinguishes it from acquired mucociliary disorders. Approximately one half of patients with primary ciliary dyskinesia have situs inversus and, thus are having Kartagener syndrome. We present a case of 12 year old boy with sinusitis, situs inversus and bronchiectasis. The correct diagnosis of this rare congenital autosomal recessive disorder in early life is important in the overall prognosis of the syndrome, as many of the complications can be prevented if timely management is instituted, as was done in this in this case.
Keywords: Bronchiectasis, primary ciliary dyskinesia, Kartagener's syndrome, sinusitis, situs inversus
Introduction
Siewert first described the combination of situs inversus, chronic sinusitis, and bronchiectasis[1] in 1904. However, Manes Kartagener[2] first recognized this clinical triad as a distinct congenital syndrome in 1933. Because Kartagener described this syndrome in detail, it bears his name. Kartagener's syndrome (KS) is inherited via an autosomal recessive pattern. Its incidence is about 1 in 30,000 live births. Male patients with this syndrome are almost invariably infertile because of immotile spermatozoa. The immotility is due to variety of ultrastructural defects in respiratory cilia and sperm tail.[3] Afzelius[4] was the first to recognize the relationship between KS and male infertility when he observed lack of dynein arms in the sperms and cilia of four subjects, three with KS and a fourth one, brother of one of the three subjects. However, our patient was 12 years old; hence, we did not have spermatozoa for microscopic diagnosis.
Case Report
A twelve year-old boy came to our institute with complaints of recurrent sore throat and cough with yellowish expectoration intermittently for past three to four years with episodes of fever; weight loss of about two to three kg over past one year; chronic wheezing; and chronic use of antibiotics for past two to three years with occasional steroids.
Examination
The patient was a moderately built fair young boy, who was dyspneic, coughing, and short of breath. He was febrile, conscious, and oriented. No pallor and no icterus. Clubbing grade 2 was present. JVP was not raised and no edema feet. A few submental lymph nodes were palpable, which were mildly tender. Auscultation of the chest revealed diffuse bilateral ronchi with scattered crepitations over both infrascapular regions. Chest expansion was reduced. Surprisingly, we could not hear his heart sounds on the left side, but they were appreciable over right side and those were normal cardiac sounds. Apex beat was palpable over the fifth intercostal space on the right side of chest. Rest of the physical and systemic examination were normal.
Investigations
We did an electrocardiogram (ECG) on him with both right and left-sided chest leads, which revealed inverted “P” waves in L1 and AVL [Figure 1]. The ECG was normal on right-sided chest leads [Figure 2].
Figure 1.
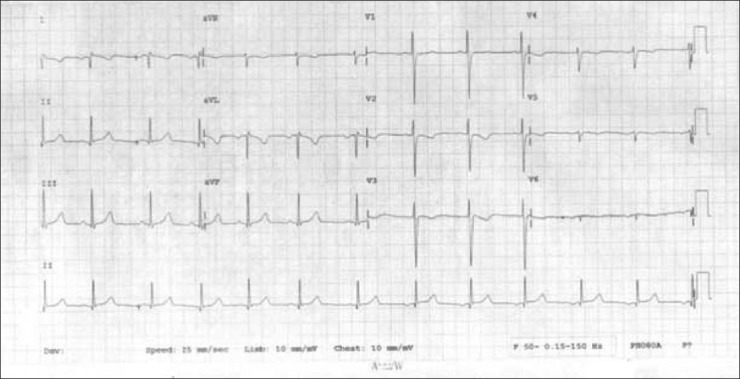
ECG with left sided chest leads
Figure 2.
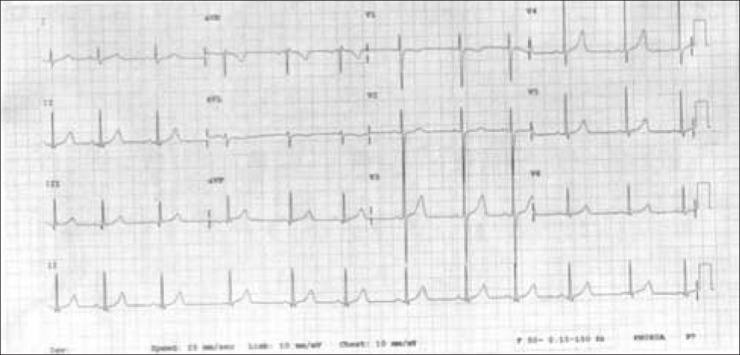
ECG with right sided chest leads
His chest X-ray PA view revealed the cardiac shadow and apex on the right side. Therefore, at this moment, he was a case of dextrocardia [Figure 3].
Figure 3.
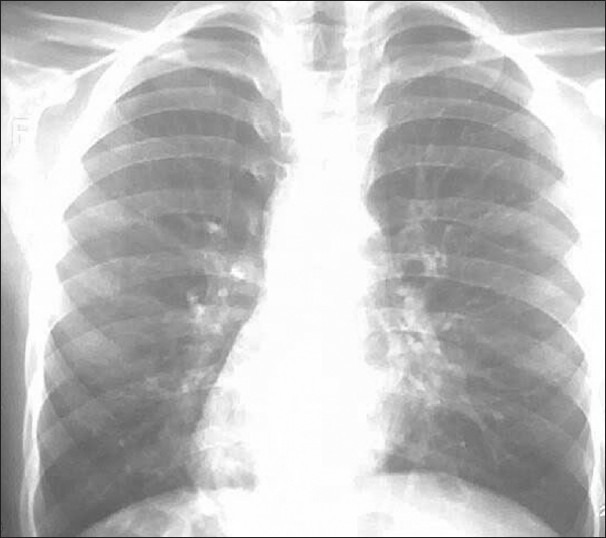
Chest X- Ray PA View shoing dextrocardias
The chest X-ray revealed increased bronchovesicular markings and suspicious bronchiectatic features. At this moment, we suspected him to be a case of situs inversus with KS.
The ultrasonography of abdomen revealed situs inversus, but rest of the scan was normal. His high resolution computed tomography (HRCT) chest revealed bronchiectatic changes [Figure 4] and CT abdomen again confirmed situs inversus [Figure 5]. X-ray and CT para nasal sinus revealed pansinusitis [Figure 6].
Figure 4.
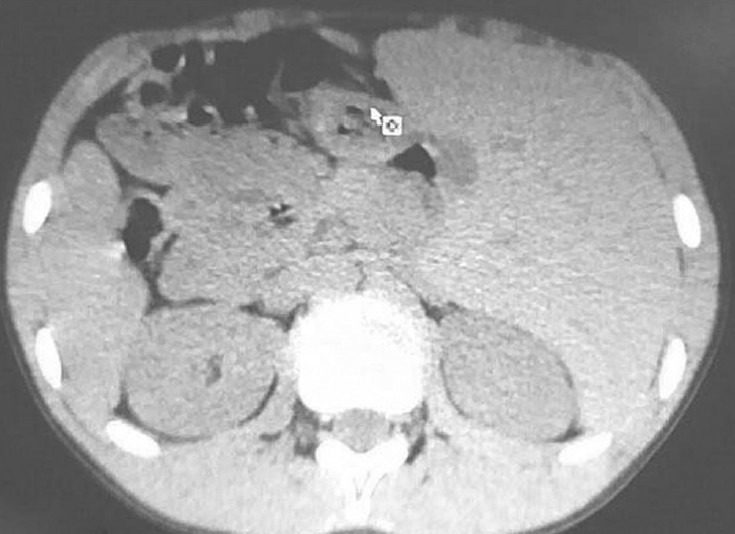
CT abdomen showing situs inversus
Figure 5.
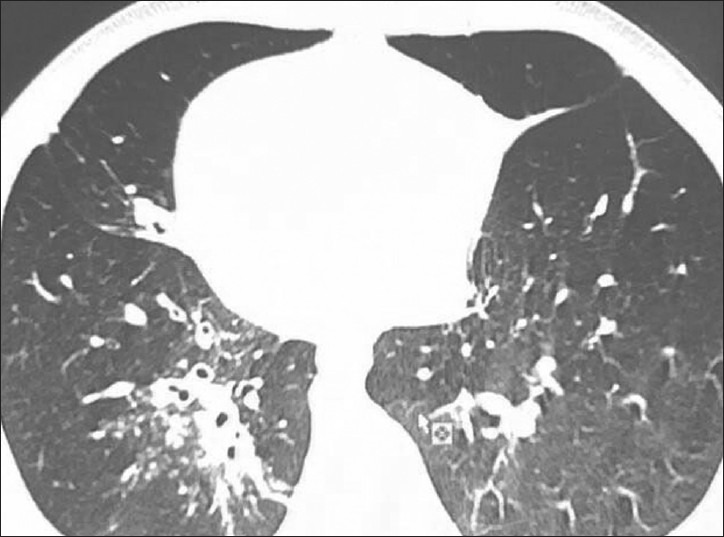
CT Chest showing bronchiectasis
Figure 6.
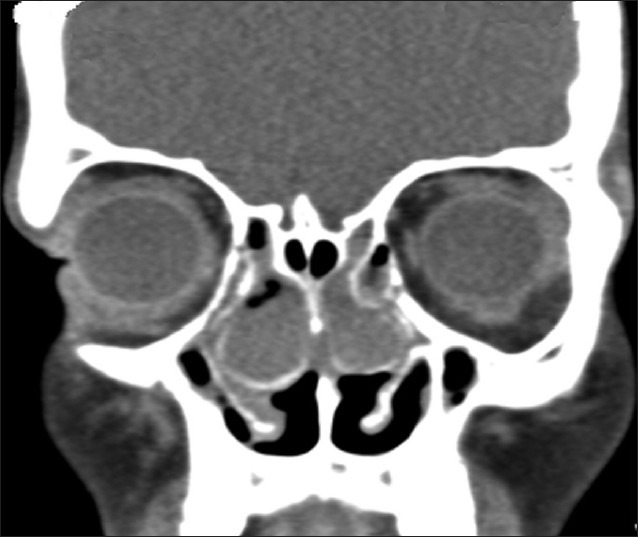
CT PNS showing chronic sinusitis
2D echo was consistent with dextrocardia; all four chambers were normal. Doppler study confirmed situs inversus of aorta and inferior vena cava [Figure 7].
Figure 7.
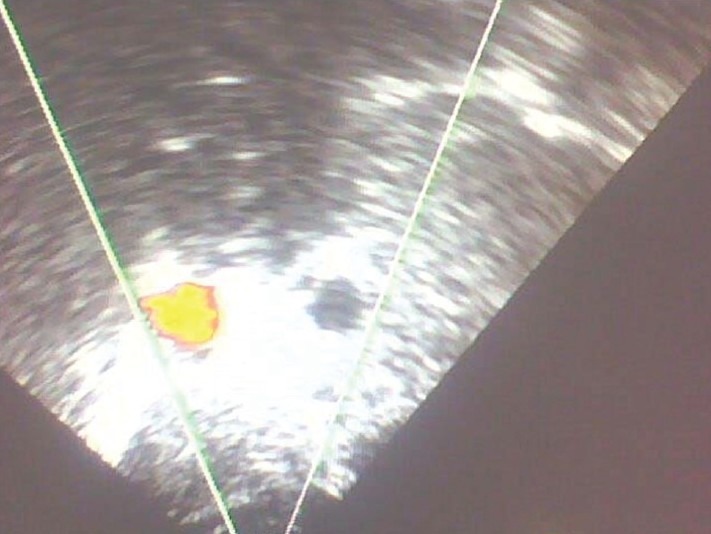
Color Doppler showing transposition of Aorta and Inferior Vena Cava
His hemogram revealed a total leucocyte count of 14700/cu.mm. erythrocyte sedimentation rate was 35 mm after 1st hour. Liver and renal functions were normal as was the urine examination.
Anti nuclear antibody, anti nutrophilic cytoplasmic antibody, and human immunodeficiency virus serolgy were negative. Serum angiotensin converting enzyme levels and Serum Immunoglobulin E (IgE) were normal. Tuberculosis (TB) gold Quantiferon was negative. Sputum for acid fast bacilli did not reveal the tuberculosis bacilii. Gram stain of the sputum revealed numerous gram positive cocci in pairs and gram negative bacilli. Sputum culture was sterile.
Course in the hospital
He was operated for sinusitis (functional endoscopic sinus surgery) at the same institute by our ENT team. Bilateral middle turbinates were fused with the uncinate process. Fused portion was opened using microdibrider and frank pus was found in both bulla ethmoidalis. Bulla was opened and pus was removed. Bilateral inferior meatal antrostomy was done. Subsequently, he was advised vaccination for influenza and pneumococcal vaccine, inhaled steroid and bronchodilator, a mucolytic, and antibiotic course with regular follow-up in ENT, Pulmonology, and Internal Medicine OPD. After 2 months of follow-up, the patient is doing well, although he continues to have intermittent cough of mild intensity.
Discussion
KS is a rare disorder. Sinusitis, bronchiectasis, situs inversus and male infertility occurring in this condition are attributed to abnormal ciliary motility. Cilia may be immotile or may show uncoordinated and inefficient movement patterns. Camner[5] and coworkers first suggested ciliary dyskinesia as the cause of KS in 1975. They described two patients with KS who had immotile cilia and immotile spermatozoa. These patients had poor mucociliary clearance because the cilia that lined their upper airways were not functioning.
The lower respiratory tract contains ciliated epithelium from the trachea to the respiratory bronchioles. Each ciliated cell gives rise to approximately 200 cilia that vary in length from 5 to 6 μm and decrease in size as the airway becomes smaller. Patients with primary ciliary dyskinesia exhibit a wide range of defects in ciliary ultrastructure and motility, which ultimately impairs ciliary beating and mucociliary clearance. The most common defect, first described by Afzelius, is a reduction in the number of dynein arms, which decreases the ciliary beat frequency.[4] Sturgess et al.[6] described how the radial spoke, which serves to translate outer microtubular sliding into cilial bending, was absent in some patients with primary ciliary dyskinesia. Cilia in other patients lacked central tubules; however, instead of the central tubules, an outer microtubular doublet transposed to the cell of the axoneme was present that displayed an abnormal 8+1 doublet-to-tubule pattern. Both the radial spoke and the transposed doublet defects impaired mucociliary clearance. The diagnostic criteria recommended for this syndrome are history of chronic bronchial infection and rhinitis from early childhood, combined with one or more of following features: (a) situs inversus or dextrocardia in a patient or a sibling, (b) living but immotile spermatozoa, (c) tracheobronchial clearance, which is absent or nearly so.[7] Our case had all these features; however, due to age, immotile spermatozoa could not be demonstrated.
Treatment of this rare congenital disorder includes antibiotics, intravenous or oral, intermittent or continuous, and are used to treat upper and lower airway infections. Hemophilus influenzae and Staphylococcus aureus are the most common organisms.[8] Long-term low-dose prophylactic antibiotics may be necessary in children. Obstructive lung disease/bronchiectasis should be treated with inhaled bronchodilators, mucolytics, and chest physiotherapy. The evidence base is largely anecdotal, but there may also be a role for inhaled antibiotics, inhaled and oral corticosteroids, and recombinant DNAs. Influenza and pneumococcal vaccination should be encouraged.
Surgical care involves tympanostomy tubes that will reduce recurrent infections and conductive hearing loss. Repeated insertions may be required and chronic otitis media can be an annoying complication. Endoscopic sinus surgery and the formation of a nasal antral window underneath the inferior turbinate may afford a transient improvement in upper and lower respiratory tract symptoms. Taiana and associates[9] reported on successful pulmonary operations in a 25-year-old man, consisting of left lower lobectomy, lingulectomy, and anterior segmentectomy of the left upper lobe. Lung transplantation and heart-lung transplantation have occasionally been tried in severe cases with some success.[10,11]
On follow-up, our case improved significantly after endoscopic sinus surgery and the incidence of recurrent infections reduced drastically, which emphasizes the fact that early diagnosis and treatment of associated complications of this rare syndrome significantly improves the quality of life and prognosis of the patients. Late diagnosis with established bronchiectasis worsens the overall the prognosis, even with the best of treatment modalities. A high degree of suspicion about KS among pediatricians around the globe is the need of the hour.
Footnotes
Source of Support: Nil
Conflict of Interest: None declared.
References
- 1.Kartagener M. Zur Pathogenese der Bronchiektasien. Bronchiektasien bei Situs inversus viscerum. Beitr Klin Tuberk. 1933;83:489–501. [Google Scholar]
- 2.Kartagener M, Horlacher A. Bronchiektasien bei Situs Viscerum inversus. Schweiz Med Wocshenschr. 1935;65:782–4. [Google Scholar]
- 3.Samuel I. Kartagener's syndrome with normal spermatozoa. JAMA. 1987;258:1329–30. [PubMed] [Google Scholar]
- 4.Afzelius BA. A human syndrome caused by immotile cilia. Science. 1976;193:317–9. doi: 10.1126/science.1084576. [DOI] [PubMed] [Google Scholar]
- 5.Camner P, Mossberg B, Afzelius BA. Evidence of congenitally nonfunctioning cilia in the tracheobronchial tract in two subjects. Am Rev Respir Dis. 1975;112:807–9. doi: 10.1164/arrd.1975.112.6.807. [DOI] [PubMed] [Google Scholar]
- 6.Sturgess JM, Chao J, Wong J, Aspin N, Turner JA. Cilia with defective radial spokes: A cause of human respiratory disease. N Engl J Med. 1979;300:53–6. doi: 10.1056/NEJM197901113000201. [DOI] [PubMed] [Google Scholar]
- 7.Bent J., III Kartagener syndrome. e medicine. 2009;69:39–41. [Google Scholar]
- 8.Rosen MJ. Chronic cough due to bronchiectasis: ACCP evidence-based clinical practice guidelines. Chest. 2006;129:122S–31S. doi: 10.1378/chest.129.1_suppl.122S. [DOI] [PubMed] [Google Scholar]
- 9.Taiana JA, Villegas AH, Schieppati E. Kartagener's syndrome: Report of a case treated by pulmonary resection; review of the literature. J Thorac Surg. 1955;30:34–43. [PubMed] [Google Scholar]
- 10.Otgün I, Karnak I, Tanyel FC, Senocak ME, Büyükpamukçu N. Surgical treatment of bronchiectasis in children. J Pediatr Surg. 2004;39:1532–6. doi: 10.1016/j.jpedsurg.2004.06.009. [DOI] [PubMed] [Google Scholar]
- 11.Alvarez A, Algar FJ, Santos F, Lama R, Baamonde C, Cerezo F, et al. Pediatric lung transplantation. Transplant Proc. 2005;37:1519–22. doi: 10.1016/j.transproceed.2005.02.005. [DOI] [PubMed] [Google Scholar]