

Abstract
The term neurofibromatosis (NF) is used for a group of genetic disorders that primarily affect the cell growth of neural tissues. Neurofibromatosis type 1 (NF1), also known as von Recklinghausen's disease, is the most common type of NF, and accounts for about 90% of all cases. It is one of the most frequent human genetic diseases, with a prevalence of one case in 3,000 births. The expressivity of NF1 is extremely variable, with manifestations ranging from mild lesions to several complications and functional impairment. Oral manifestations can be found in almost 72% of the NF1 patients. The aim of this article is to report the NF1 in a family with different manifestations and to review the literature.
Keywords: Genetic diseases, neurofibromatosis type 1, von Recklinghausen's disease
INTRODUCTION
The term neurofibromatosis (NF) is referred to a group of genetic disorders that primarily affect the cell growth of neural tissues. There are two forms of NF: Type 1 (NF1) and type 2 (NF2).[1–3] These two forms have few common features and are caused by mutations on different genes.[4,5]
Neurofibromatosis type 1, also known as von Recklinghausen's disease, is a neurodermal dysplasia. This disease was first described by Friederich Daniel Von Recklinghausen, the pathologist, in 1882.[4,6] The pathological alterations behind it begin in the embryonic period, prior to differentiation of the neural crest.[4,7] NF1 is the most common type of NF and is estimated to occur in 90% of all cases. It is one of the most frequent human genetic diseases, with a prevalence of one in 3,000 births.[1,8] There is no sex or racial predilection. NF1 is an autosomal dominant disease caused by a spectrum of mutations that affect the NF1 gene located at the 17q11.2 chromosome,. It has one of the highest rates of spontaneous mutation among genetic diseases in humans. Only 50% of the NF1 patients have a positive family history of the disease. The rest of the patients represent spontaneous mutations. The expressivity of the disease is extremely variable, with manifestations ranging from mild lesions to several complications and functional impairment. The penetrance, otherwise, is 100%.[2,8]
Café-au-lait spots, axillary and inguinal freckling, optic gliomas, Lisch nodules (pigmented hamartomas of the iris), spinal and peripheral nerve neurofibromas, neurological or cognitive impairment, scoliosis, abnormalities in the oral and maxillofacial region, malignant tumors of the nerve sheath, pheochromocytoma, vasculopathy, and specific bone lesions are common clinical features of NF1.[2,5] Oral manifestations can be found in almost 72% of NF1 patients.[9] Café-au lait spots are also seen in some other diseases [R] We report on a family with NF1, who have different manifestations of the disease.
CASE REPORTS
Five members of a family, a mother and her four offsprings, referred to the Department of Oral and Maxillofacial Medicine to be diagnosed and received oral and dental treatments. During clinical examination, we found clinical manifestations of neurofibromatosis in all of them. We have described all of them from older to younger in the following text:
Case 1
The mother, who was 50 years old, had several soft tissue cutaneous nodules on the body (neurofibroma), including the head and neck, and multiple hyperpigmented macules (Café-au-lait pigmentation) [Figure 1]. She did not have any systemic problems in the organs of her body or any oral manifestation. All her offsprings had skin neurofibroma and Café-au-lait pigmentations in variable sizes and frequencies [Figure 2]. She noted that her first child died because of severity of manifestations. She also mentioned that her mother and aunt had Café-au-lait spots and neurofibroma on their skin.
Figure 1.
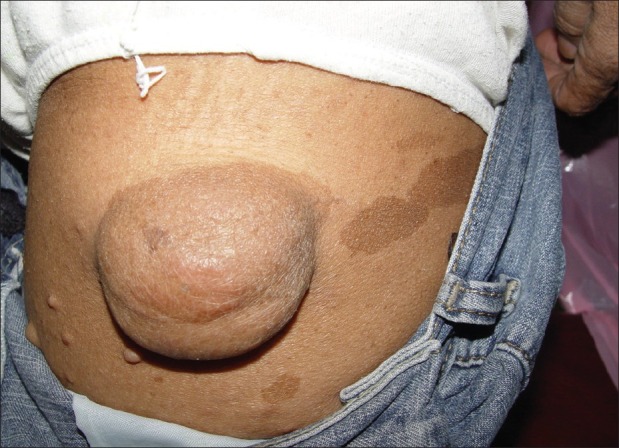
Pelexiform neurofibroma and café-au-lait spots in the mother
Figure 2.
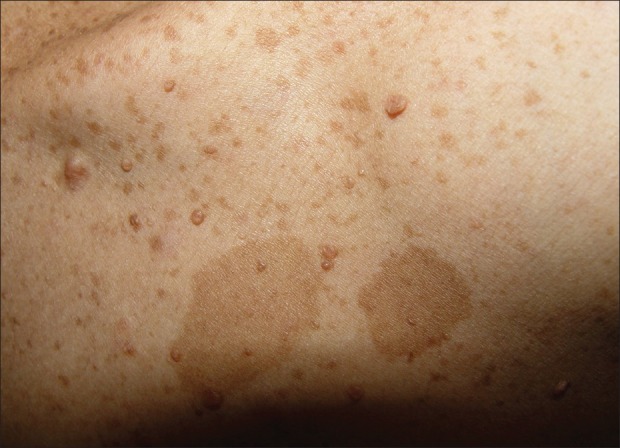
Café-au-lait spots and small neurofibromas in the third offspring
Case 2
The first offspring, a female, 25 years old, reported eight surgeries on her face because of the involvement of her left facial nerve; thus, she had facial asymmetry and a lowered left ear. She had several neurofibromas and Café-au-lait pigmentations on her body. She was the only member of her family who had oral manifestation of NF1. During oral examination, we saw three new formations of neurofibromatic masses on the left side of her palate and sublingual area [Figures 3–5].
Figure 3.
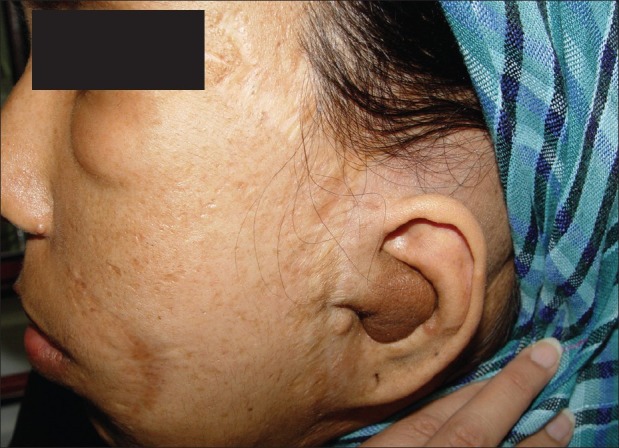
Lateral view of the face in the first offspring. Facial asymmetry and ear involvement are present
Figure 5.
First offspring — palatal neurofibroma
Figure 4.
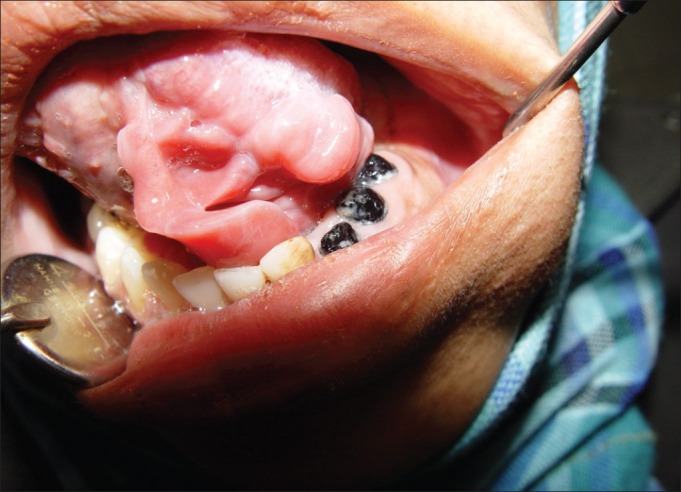
First offspring — sublingual neurofibroma
Case 3
The second offspring was a 23-year-old female. She had lost her left eyesight because of an optic nerve tumor. She had received radiotherapy and the development of the tumor had been stopped. She also had neurofibroma and Café-au-lait pigmentations on her body, especially on the back with no oral manifestation.
Case 4
The third offspring, a male, was 20 years old. He had a history of seizure in his childhood and an arteriovenous shunt, due to operation on his brain. He had experienced headaches. He did not have any oral manifestation, but he had skin lesions similar to those of the other family members.
Case 5
The last offspring was a 17-year-old female. She was with scoliosis and because of this problem had a pathological fracture on the vertebral column [Figure 6]. She did not have any oral manifestation. As a symptom of the disease, she had very few limited pigmentations on her trunk and over the scoliosis area.
Figure 6.
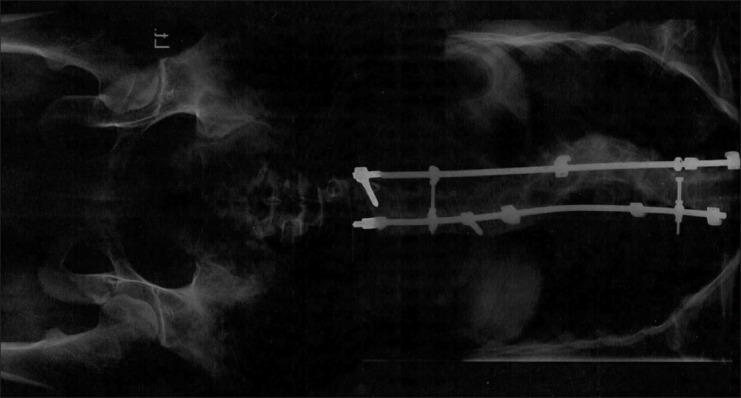
Scoliosis in the last offspring
DISCUSSION
NF-1 is caused by an alteration of the NF-1 gene. This gene is a tumor suppressor located on the long arm of chromosome 17 (17q11.2).[3,7] Loss of this gene's function due to a mutation leads to an increase in cell proliferation and development of tumors.[3]
The NF1 gene encodes neurofibromin, a cytoplasmic protein that is predominantly expressed in neurons, Schwann cells, oligodendrocytes, and leukocytes. It is a multidomain molecule with the ability to regulate several intracellular processes, including the RAS-cyclic AMP pathway, ERK / MAP kinase cascade, adenylyl cyclase, and the cytoskeletal assembly.[10]
This disease is a slowly evolving neurodermic dysplasia. Troubles begin at the embryonic stage before differentiation of the neural crests. After birth, the disease evolves in bursts, especially during growth, puberty, and pregnancy. This is a dominant autosomal hereditary disease with total penetrance and variable expression; about 50% of cases, however, are sporadic. Von Recklinghausen's neurofibromatosis has the highest rate of spontaneous mutation in all human genetic diseases. Its incidence is 1 to 2 in 2000 to 3000 of the population as a whole, but reaches to 1 in 200 of mentally retarded individuals. There is neither racial nor sexual dominance.[4,11]
Pigmented lesions are a common manifestation in NF-1. These lesions usually appear during the first years of life or are present at birth, either as Café-au-lait spots or freckles.[3–5,7] Café-au-lait spots are hyperpigmented maculae that may vary in color from light to dark brown. Their borders may be smooth or irregular. They may appear anywhere on the skin, but they are less common on the face. Inguinal and axillary freckles (Crowe's sign) are frequently present. In some patients with NF1, freckling may occur diffusely over the trunk, extremities, upper eyelids, and base of the neck.[3] This family exhibited more than six Café-au-lait spots and bilateral axillary and inguinal freckles. Their mother reported that all her children had Café-au-lait spots at birth, which had become more dominant.
Neurofibromas are benign complex tumors. They arise from peripheral nerve sheaths and constitute one of the main manifestations of NF1. A solitary neurofibroma may occur in an individual who does not have NF1, but multiple neurofibromas tend to develop in a person with NF1. Clinical observations suggest that there are at least two major types of neurofibroma which may differ widely in their natural history: ‘Discrete’ or ‘localized’ and ‘plexiform’ neurofibroma.[3,5]
A localized neurofibroma arises from a single site along a peripheral nerve and presents as a focal mass with well-defined margins. It can occur superficially or may involve deeper peripheral nerves. A localized neurofibroma is the most common type of neurofibroma occurring in NF1 patients. They are rarely, if ever, present at birth and usually appear in late childhood or early adolescence.[3] The number of localized neurofibromas tends to increase with age, which varies widely from person to person. NF1 patients may have few, hundreds, or even thousands of localized neurofibromas. Neurofibromas are found mostly on the skin. Nevertheless, many organs may be involved, including the stomach, intestines, kidney, bladder, larynx, and heart. In the head and neck region, the most commonly affected sites are the scalp, cheek, neck, and oral cavity.[1] Several soft tissue lesions, comparable to the localized neurofibroma, could be seen all over the mother's body. Her offsprings also had these lesions, but to a lesser extent.
Plexiform neurofibroma spreads along the peripheral nerve and may affect some nervous rami. The cranial nerves most involved are the fifth, ninth, and tenth.[3,5]
This is a poorly-circumscribed and locally invasive tumor. About 21% of the patients with NF-1 have plexiform neurofibromas.[7] The morbidity of plexiform neurofibromas in NF-1 is high, as they tend to grow up to a great size, producing disfigurement. Moreover, the risk of malignization is between 2 and 5%.[3] Plexiform neurofibroma on the face can also cause facial asymmetry.[12] The older offspring was with facial asymmetry because of plexiform neurofibroma and paralysis of the facial nerve on the left side of her face.
Skeletal involvement is present in almost 40% of the patients with NF-1. Scoliosis is the most common skeletal pathology.[4,6,7] In this family the last offspring had a pathological fracture of her vertebral column because of scoliosis.
Various neurological pathologies can also be found, such as, hamartomas of the iris, neurinomas of the acoustic nerve, tumors of the central nervous system (gliomas, glioblastomas), macrocephalies, and mental retardation (in 40% of cases).[4,5] The third offspring had a history of seizure in his childhood and had an arteriovenus shunt due to operation on his brain. There was no mass or tumor in his CT scan report, but diagnosis was intraventricular obstructive hydrocephaly.
Optic pathway tumors (OPT) are a frequent finding. OPT is usually localized in the prechiasmal, chiasmal, and postchiasmal regions. Massive involvement of the optic system can damage the optic nerves and result in blindness.[13] The second daughter of this family had lost the sight in her left eye because of optic nerve involvement. She was treated with radiotherapy; but in some reports radiotherapy is associated with an important morbidity.[13]
Oral manifestations were found in 72% of the patients with NF1, in a study performed by Shapiro et al.[12] According to a survey performed by D’Ambrosio et al., 66% of his NF1 patients had at least one intraoral manifestation of the disease and 58% presented with manifestations in the maxilla and the mandible, which were detected on panoramic radiographs.[9]
Neurofibromas may appear in any tissue, soft or hard, in the oral cavity. The most commonly affected site is the tongue.[3,4,7,14]
Shapiro et al. stated that gingival involvement is 5%.[12]
Localized oral neurofibromas usually appear as asymptomatic nodules covered by normally colored mucosa.[3,4] However, when they are adjacent to the cranial nerves, they can impair motor function of the facial or hypoglossal nerves or the sensitivity of the trigeminal nerve.[3,12] The first offspring had three localized neurofibromas in her mouth. One on the left side of palate and two in the sublingual area.
Radiological changes with oral neurofibromas are widening of the mandibular channel, mandibular foramen, and mental foramen.[3,4,15] We found these characteristics in the first offspring [Figure 7].
Figure 7.
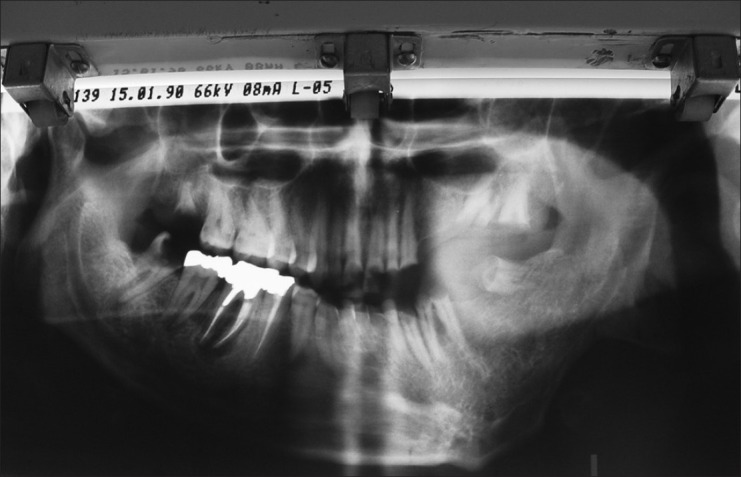
Panoramic view of the first offspring
Histologically, neurofibromas are composed of a mixture of Schwann cells, perineurial cells, and endoneurial fibroblasts, which are not capsulated.[16] Schwann cells account for about 36 to 80% of the lesional cells. These constitute the predominant cellular type and they usually have widened nuclei with an undulated shape and sharp corners. In an electron microscope image, Schwann cells can be seen embracing the axons. These can be highlighted by silver or acetylcholinesterase staining or immunohistochemical techniques when using the optic microscope. It is estimated that between 0,7 and 31% of the cells are perineurial cells. In very few cases can these types of cells predominate.[16]
Neurofibromatous lesions usually evolve slowly, without pain, but during growth, puberty, or pregnancy, their evolution may be accelerated.[4]
Multiorgan occurrence of NF1 requires a multidisciplinary approach. As there is no medical treatment for NF1, the management must be toward prevention and control of the complications. Although the rate of malignant transformation of NF1 is low (3 – 5%), these neoplasms can cause other clinical problems, including esthetic and functional compromising. Surgical treatment, therefore, is not always satisfactory, as the complete removal of large and also multiple lesions is very difficult. Surgical intervention is indicated when the patient's function is impaired. Risk, possible complication, and expected benefit gained by such procedures should be considered.[17] In this family, the first offspring had had eight operations before, and because of the extent of lesions, she had facial paralysis and facial disfigurement; but the lesions recurred and even extended to her ear, sublingual area and palate. She also had several neurofibromas on her body. She had several consultations with many specialists and because of the proximity of the lesions to the nerves, all of them believed that the lesions should not be removed. On account of the proximity to the facial and hypoglossal nerves, we could not excise all her oral neurofibromas. However, because of the risk of malignant transformation, we decided to perform an incisional biopsy from the sublingual lesion and one skin neurofibroma of the first offspring. Fortunately, malignant transformation was not seen and the diagnosis was neurofibroma. Histology of the skin and the oral lesion showed a non-capsulated tumor, composed of cells having spindle wavy nuclei [Figure 8]. Subsequently, we removed all the remaining palatal neurofibroma.
Figure 8.
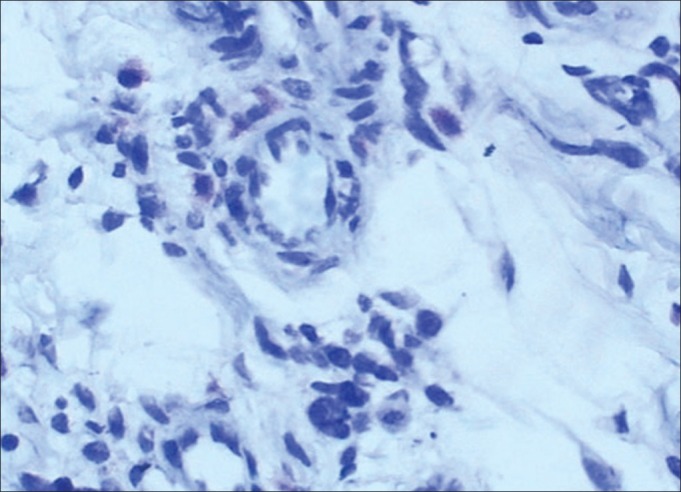
Immunohistochemical feature of the oral neurofibroma in the first offspring
The third offspring had an arteriovenus shunt because of a former operation on his brain, during childhood, to treat the seizures. Radiotherapy or chemotherapy was not recommended.[4] When he referred to our department, he had several untreated teeth and dental roots. We extracted all the remaining roots and referred him to the Department of Prosthodontics.
The NF1 patients may receive genetic consulting.[18] These patients should be advised that the disorder is autosomal dominant and that the inheritability is 50% in both sexes. As NF1 is one of the most common genetic diseases, and oral manifestations can be found in almost 72% of the cases, dentists should be aware of the characteristics of this disease. It is important to conduct a long-term follow-up, because of local complications and the risk of malignant transformation. In cases with a rapid increase in size of the neurofibroma and presence of pain, the probability of malignant transformation must be considered.[19] Incisional biopsy should be performed for histopathological evaluation.
CONCLUSIONS
On account of the fact that neurofibromatosis is an autosomal dominant inherited disease, genetic consulting is necessary before marriage and before becoming pregnant, as well, The patients with this disorder have special problems because of their disease and many of the dentists do not treat their dental problems. Therefore, it is our duty, as oral and maxillofacial medicine specialists, to manage their dental problems such as dental extractions, endodontic therapy, restorative therapy, and so on.
Cafe-au-lait pigmentation is seen in many diseases such as neurofibromatosis type 1 and 2, McCune-Albright syndrome, tuberous sclerosis, fanconi anemia, and so on.
Footnotes
Source of Support: Nil.
Conflict of Interest: None declared.
REFERENCES
- 1.Gorlin RJ, Cohen MM, Levin LF. Syndromes of the head and neck. Oxford: Oxford University Press; 1990. pp. 353–416. [Google Scholar]
- 2.Friedman JM, Gutmann DH, MacCollin M, Riccardi Y. Phenotype, natural history and pathogenesis. Baltimore: The Johns Hopkins University Press; 1999. Neurofibromatosis. [Google Scholar]
- 3.Cunha KS, Barboza EP, Dias EP, Oliveira FM. Neurofibromatosis type I with periodontal manifestation. A case report and literature review. Br Dent J. 2004;196:457–60. doi: 10.1038/sj.bdj.4811175. [DOI] [PubMed] [Google Scholar]
- 4.Bekisz O, Darimont F, Rompen EH. Diffuse but unilateral gingival enlargement associated with von Recklinghausen neurofibromatosis: A case report. J Clin Periodontol. 2000;27:361–5. doi: 10.1034/j.1600-051x.2000.027005361.x. [DOI] [PubMed] [Google Scholar]
- 5.García-de Marcos JA, Dean-Ferrer A, Alamillos-Granados F, Ruiz- Masera JJ, García-de Marcos MJ, Vidal-Jiménez A, et al. Gingival neurofibroma in a neurofibromatosis type 1 patient. Med Oral Patol Oral Cir Bucal. 2007;12:E287–91. [PubMed] [Google Scholar]
- 6.Hillier JC, Moskovic E. The soft tissue manifestations of neurofibromatosis type 1. Clin Radiol. 2005;60:960–7. doi: 10.1016/j.crad.2005.02.008. [DOI] [PubMed] [Google Scholar]
- 7.Bongiorno MR, Pistone G, Aricò M. Manifestations of the tongue in Neurofibromatosis type 1. Oral Dis. 2006;12:125–9. doi: 10.1111/j.1601-0825.2005.01168.x. [DOI] [PubMed] [Google Scholar]
- 8.Cotran RS, Kumar V, Robbins SL. Robbins pathologic basis of disease. 8th ed. Philadelphia: Saunders Company; 2010. [Google Scholar]
- 9.D’Ambrosio JA, Langlais RP, Young RS. Jaw and skull changes in neurofibromatosis. Oral Surg Oral Med Oral Pathol. 1988;66:391–6. doi: 10.1016/0030-4220(88)90252-6. [DOI] [PubMed] [Google Scholar]
- 10.Trovo-Marqui AB, Tajara EH. Neurofibromin: A general outlook. Clin Genet. 2006;70:1–13. doi: 10.1111/j.1399-0004.2006.00639.x. [DOI] [PubMed] [Google Scholar]
- 11.Reynols RL, Pineda CA. Neurofibromatosis: Review and report of case. J Am Dent Assoc. 1988;117:735–7. doi: 10.14219/jada.archive.1988.0121. [DOI] [PubMed] [Google Scholar]
- 12.Shapiro SD, Abramovitch K, van Dis ML, Skoczylas LJ, Langlais RP, Jorgenson RJ, et al. Neurofibromatosis: Oral and radiographic manifestations. Oral Surg Oral Med Oral Pathol. 1984;58:493–8. doi: 10.1016/0030-4220(84)90350-5. [DOI] [PubMed] [Google Scholar]
- 13.Gucev Z, Krstevska-Konstantinova M, Tasic V, Jancevska A, Kirovski I, Pop-Jordanova N. Four generations in a family with neurofibromatosis 1: Precocious puberty and optic nerve tumor (OPT) Prilozi. 2010;31:253–9. [PubMed] [Google Scholar]
- 14.Apostolidis C, Anterriotis D, Rapidis AD, Angelopoulos AP. Solitary intraosseous neurofibroma of the inferior alveolar nerve: Report of a case. J Oral Maxillofac Surg. 2001;59:232–5. doi: 10.1053/joms.2001.20508. [DOI] [PubMed] [Google Scholar]
- 15.Ide F, Shimoyama T, Horie N, Kusama K. Comparative ultrastructural and immunohistochemical study of perineurioma and neurofibroma of the oral mucosa. Oral Oncol. 2004;40:948–53. doi: 10.1016/j.oraloncology.2004.04.015. [DOI] [PubMed] [Google Scholar]
- 16.Holtzman L. Radiographic manifestation and treatment consideration in a case of multiple neurofibromatosis. J Endod. 1998;24:442–3. doi: 10.1016/S0099-2399(98)80030-7. [DOI] [PubMed] [Google Scholar]
- 17.Maceri DR, Saxon KG. Neurofibromatosis of the head and neck. Head Neck Surg. 1984;6:842–50. doi: 10.1002/hed.2890060407. [DOI] [PubMed] [Google Scholar]
- 18.Vincent SD, Williams TP. Mandibular abnormalities in neurofibromatosis. Case report and literature review. Oral Surg Oral Med Oral Pathol. 1983;55:253–8. doi: 10.1016/0030-4220(83)90324-9. [DOI] [PubMed] [Google Scholar]
- 19.White SC, Pharoh MJ. Oral radiology principles and interpretation. 6th ed. Louis: Mosby com; 2009. [Google Scholar]