

Abstract
This paper presents a case with dentinogenesis imperfecta (DI) associated with osteogenesis imperfecta. Systemic and dental manifestations of OI and its medical and dental treatments are discussed in this paper. A 5-year-old child with the diagnosis of OI was referred to the Dental School of Shaid Beheshti University of Medical Sciences. On clinical examination yellow/brown discoloration of primary teeth with the attrition of the exposed dentin and class III malocclusion was observed. Enamel of first permanent molars was hypoplastic. Radiographic examinations confirmed the diagnosis of DI. A histological study was performed on one of the exfoliating teeth, which showed abnormal dentin. Primary teeth with DI were more severely affected compared to permanent teeth; enamel disintegration occurred in teeth with DI, demonstrating the need for restricts recalls for these patients.
Keywords: Brittle bone disease, dental anomalies, dentinogenesis imperfecta, osteogenesis imperfecta
INTRODUCTION
Osteogenesis imperfecta (OI) or brittle bone disease considered as a genetically heterogeneous connective tissue disorder which is characterized by bone fragility and thus repeated bone fractures.[1,2] Consequently skeletal deformities may arise as a result of reduced bone mass and frequent bone fractures.[3] The incidence of OI varies between 6 and 20 in 100,000 newborns and its prevalence is 4-10 in 100,000 individuals.[4] According to Sillence,[5] OI is classified based on clinical, genetically, and radiographic features in four groups [Table 1]. Type I is a mild form of OI. Type II is the lethal form of OI, even during the prenatal and perinatal period. Type III patients show progressive limb deformation. Patients with type IV of OI are those who show moderate to severe phenotypes and do not fit into any of the first three categories. Some of them demonstrate heterogeneous features, which are not even according to Sillence[6] classification. There are three other new but uncommon types of OI. Hence, patients affected by these types do not demonstrate DI and blue sclera;[6] OI types V-VIII are called syndromes resembling OI.[7]
Table 1.

A reduction in collagen type I synthesis results in type I of OI, while quantitative or qualitative alterations in type I collagen leads to types II, III, and IV. Therefore all tissues rich in type I collagen may be affected in these patients as a result of impaired collagen synthesis. Some clinical signs and symptoms may arise including DI, blue sclera, hearing loss, growth deficiency, and joint laxity.[2,7,8]
Dentinogenesis imperfecta (DI) has been reported in more than 50% of patients suffering from OI.[9] DI is a hereditary disorder of dentin formation, which exhibits mostly an autosomal dominant (AD) trait.[10] Although DI type I is the oral manifestation of deficient collagen formation and is mainly associated with OI, DI types II and III are related to a mutation in the dentin sialophophosphoprotein (DSPP) gene.[10] Primary and permanent dentitions are both involved in DI type I, though primary teeth are more severely affected.[2] The most common clinical manifestations in teeth with DI are teeth discoloration (gray/opalescent, yellow/brown) and enamel fracture.[2,11] Radiographically, the crowns are bulbous as a result of significant cervical constriction, roots are short, and the dentin is defective although normal enamel in thickness and density exists.[2] All of the three types of DI demonstrate similar changes in dentin structure microscopically.[2] Normal mantle dentin, irregular circum pulpal dentin with abnormal dentinal tubules, and some atubular areas are evident in the histological study.[2]
Midface hypoplasia, CL III malocclusion, unilateral or bilateral crossbite, ectopic eruption of the first or second permanent molars and missing of second premolar are more prevalent in these patients. However, their dental development is age appropriate.[8]
According to Brustein[12] the mortality rate during infancy period was 70-80% and showed a reduction during puberty. However, women after menopause and men after the fifth decade of their life exhibit exacerbated symptoms.[13] Physical therapies will improve their muscle strength and functional skills and minimize deformation.[7] IV administration of pamidronate for these patients inhibits osteoclast activity by osteoclast apoptosis and preventing osteoclast formation.[7] This will lead to increased bone thickness and bone strength and finally decreased fracture incidence.[7]
Because of the numerous and severe skeletal and dental abnormalities occurring with OI, dental treatment is challenging for both patient and dentist. Being informed about the possible complexities encountering during the dental treatment for these children will help the dentist to manage their oral or dental problems. Therefore, the aim of this study was to describe the characteristics of OI, its oral and dental manifestations from a clinical, radiographic, and pathologic point of view by presenting a case.
CASE REPORT
A 5-year-old Iranian boy was referred to the Pediatric Department of Dental School, Shahid Beheshti University of Medical Sciences in 2010. According to the medical records he had experienced a vertebral fracture when he was just 20 days old. Taking skeletal radiography at that time revealed that also a femoral fracture occurred at birth. The patient's family history showed that his parents (mother, 27 and father 35 years old) were not related. Based on his medical and radiographic results, the diagnosis of OI was made, and intravenous pamidronate for every 4 months followed by daily use of calcium syrup was prescribed. Following numerous bone fractures due to poor muscle coordination at the toddler age and the bone fragility in these patients (two and eight times in the left and right femur, respectively) intramedulary rods were inserted within the right and left femurs at the age of 2.5 and 3.5 years accordingly. Figure 1 shows the left femur with the rod inside and the bowing in the right femur at the age 5, as intramedulary was removed due to the successful recovery.
Figure 1.
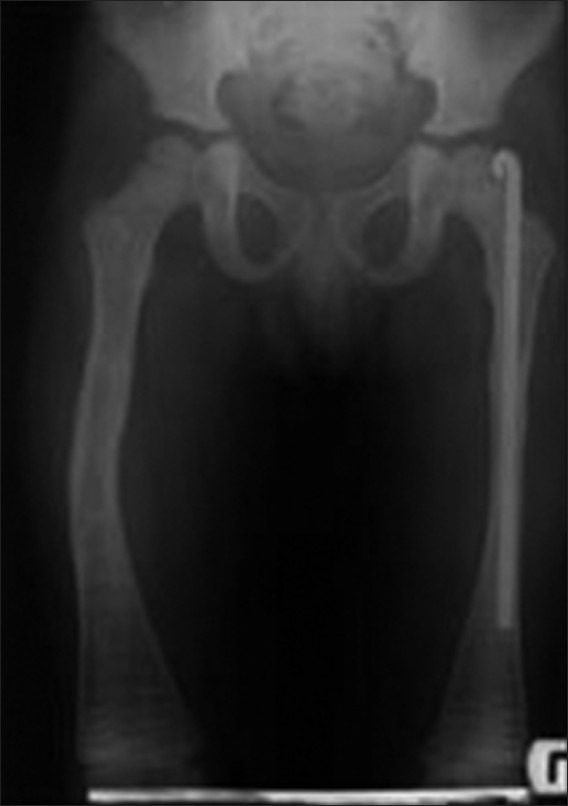
Bone deformity and the inserted rod in right and left femoral bone, respectively
Despite the abnormal physical development, the patient was mentally normal. Clinical examinations demonstrated limited mobility, short stature, bowing of the long bones, shortness of the neck, protuberance of the sternum, and relative macrocephaly. Due to these clinical signs and symptoms and the absence of hearing loss, according to OI classification [Table 1], the patient was diagnosed with type IV of OI. In the frontal view, the patient's face seemed symmetrical and hypoplasia of the maxilla and thus the CL III profile was evident [Figure 2]. Also there was posterior cross in the intraoral examination [Figure 3a].
Figure 2.
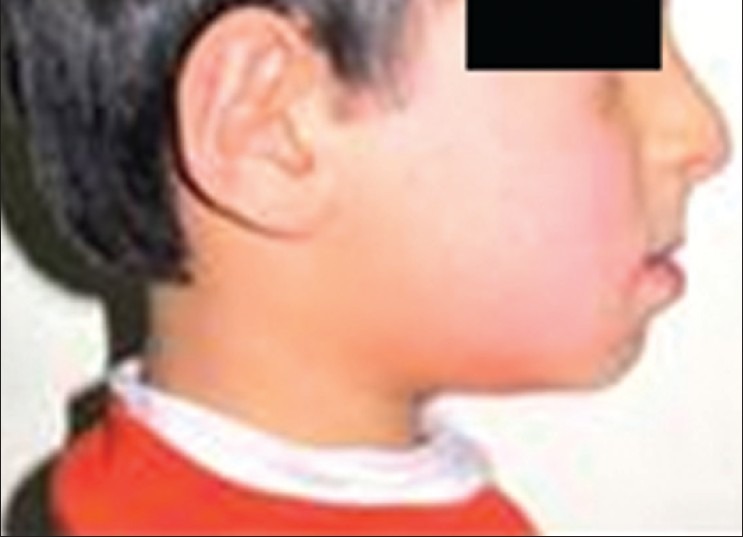
Sagittal view of patient's face
Figure 3.
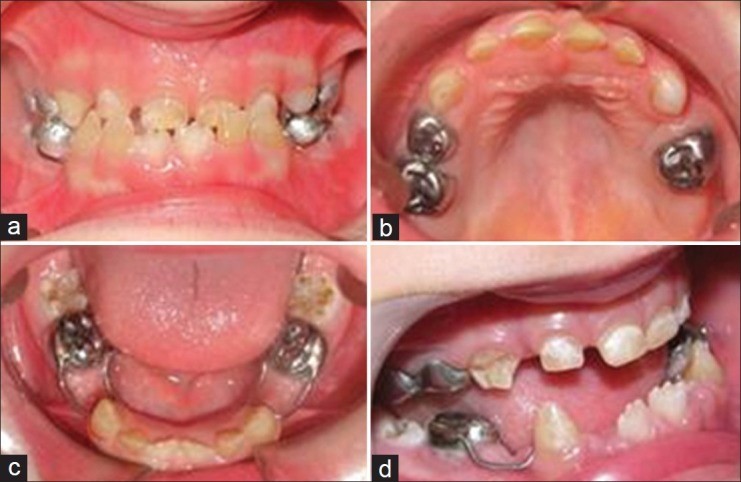
Intraoral view. (a) Frontal view of the occlusion. (b) Occlusal view of the upper arch. (c) Occlusal view of the lower arch after placement of space maintainers. (d) Enamel disintegration on the right upper canine (a lateral view)
Intraoral examinations showed yellow/brown discoloration in all primary teeth, with severe attrition on the lingual and vertical cracks on the labial side of the upper and lower anterior teeth. Moreover, enamel disintegration was evident on the buccal surface of the canines [Figure 3b–d].
Owing to the uncooperative behavior of the patient all the required dental treatments were performed under general anesthesia. The following treatment procedure was carried out prior to his attendance in our clinic: (a) Extraction of 64, 74, and 84. (b) Pulp therapy of 54, 55, 63, 65, 75, and 85 restored with stainless steel crowns or composite resins. (c) Composite resin restoration for 53, 63, and 73 [Figure 4]. Strict recommendations have been made by the operating team, for immediate space maintenance in the extraction areas. However, the patient was referred 4 months after the extensive dental treatment to the Pediatric Department of Shahid Beheshti University of Medical Sciences (Tehran, Iran). At the first visit a arch space available was evaluated and two band and loops were placed in the lower jaw to preserve the space of two extracted first primary molars. The upper jaw received a bonded facemask to stimulate the maxillary growth [Figure 5]. Radiographically, no pulp obliteration was observed and the bulbous appearance of the posterior teeth was masked by the full coverage of the crowns.
Figure 4.
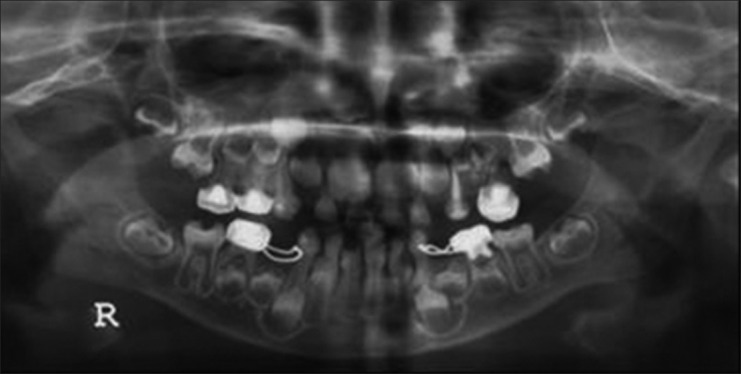
Panoramic radiography. Note the hypotrophic dentin and hypoplastic enamel on the first permanent molars
Figure 5.
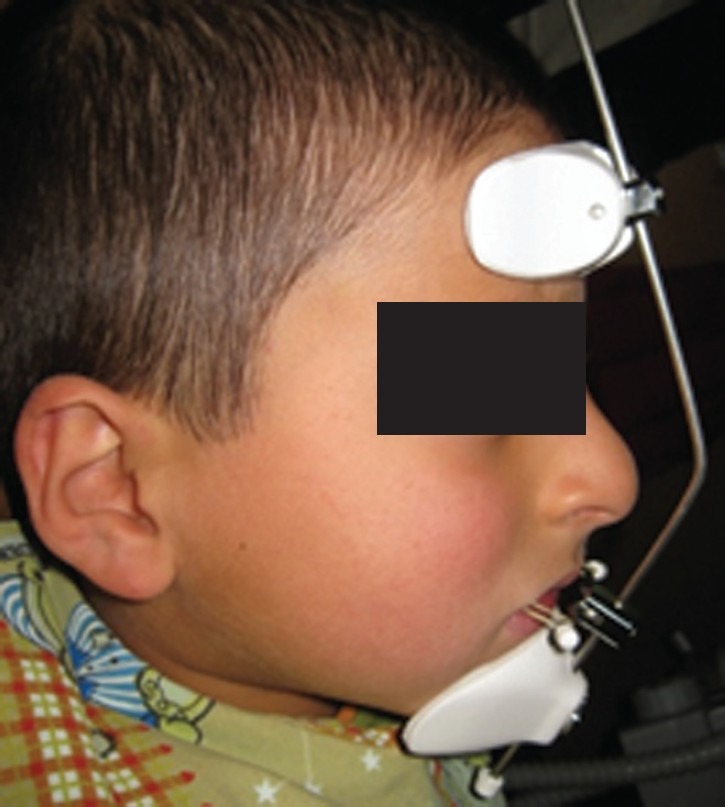
The patient received a face mask to stimulate the maxillary growth
By the age of 6, the mandibular central incisors and first permanent molars appeared in the mouth. Despite the lingual eruption of incisors, these teeth demonstrated no enamel or dentin defects or discolorations. However, enamel hypoplasia was evident in first permanent molars [Figure 3c], which was restored with composite resins immediately after eruption. The loose primary central incisors were removed and placed in a formalin 10% solution (Merck, Germany) for 24 hours. Then the teeth were prepared for histological examination under a steromicroscope (X400) (Nikon, USA): Following the decalcifying procedure by formic acid 10% for 6 weeks, the samples were sectioned, stained by Hematoxilin Eosin and observed by a light microscope.
Histologic examination showed an irregular periodic dentinogenesis, with numerous globular and interglobular dentin. Stereomicroscope evaluations exhibited a few disorganized tubular structures with an increased caliber, which was more prominent in circumpulpal dentin compared to the mantle dentin. The dentino-enamel junction (DEJ) mostly demonstrated an irregular scalloped appearance, although in some locations a straight smooth DEJ was also obvious. In certain regions in the middle third of the sectioned teeth the dentin was lost. Active odontoblasts in pulpal tissue were attributable to the normal physiologic exfoliative stage [Figure 6a–d].
Figure 6.

Microscopic view of the sectioned tooth, (a) Periodic irregular dentinogenesis, (b) Dentin detachment, (c) Dentinogenesis with a globular pattern, (d) Interglobular pattern
DISCUSSION
OI is a rare hereditary entity, diagnosed by disordered collagen formation and bone fragility. The disease is usually inherited as an AD trait, although recently mutations are also reported in some cases.[16] Our patient showed no family history of OI, so it may be assumed that it may be an AD trait with an incomplete penetrance or a new genetic mutation. However, because of the distinctive signs and symptoms, the early diagnosis of OI was made and further genetic assessments were unnecessary.
This patient showed repeated fractures in vertebral and femoral bones early in the life. Deformity and bowing in the long bones particularly in femur was obvious. According to Sillence,[5] these features are indicative for OI type IV. Although types IV and I of OI are similar in some ways, this patient did not experience prolonged bleeding, bone pain or heart problems, the less extraskeletal abnormalities, and more severe osseous involvements are investigative for type IV of OI.[14]
All patients suffering from OI display short statures as observed in this case. Relative macrocephaly due to the short size of the body and mid-face hypoplasia with a normal mandibular length resulting in posterior cross bite was similar to that described by O’Connell and Marini.[8] They described that the existing malocclusion in these patients is attributed to the disfigured dentoalveolar complex and the abnormal location, size, and weight of the head.[1] Schwartz and Tsipouras reported a prevalence of 75% for CL III malocclusion and 65% for posterior crossbite in OI patients.[1]
Dentiogenesis imperfecta (DI) has been noted in 80% of OI types III and IV.[8] According to DI classifications this patient was assigned to DI type I, as the other two types of DI merely demonstrate dental signs and do not accompany with any type of OI.[17] Intraoral examination of this boy revealed yellow/brown discoloration of the primary teeth and a normal appearance in permanent anterior teeth. This is consistent with the fact that primary dentition is usually more affected by DI than permanent dentition.[8] Moreover, yellow/brown discoloration of affected primary teeth is more prevalent than gray discoloration, stated by Majorana et al.[2] However, as extensive attrition and enamel fractures occurred more frequently in yellow/brown primary teeth than the gray discolored teeth, it is concluded that gray discoloration has a better prognosis.[2,8]
The patient showed vertical cracks and enamel detachments on the buccal or lingual side of the primary teeth. Children with DI experience vertical enamel fractures leading to exposure of the soft underneath dentin and consequently premature severe attrition.[4] Preserving the normal vertical dimension of these patients in primary and mixed dentition is only possible if the posterior teeth are completely covered by stainless steel crowns. As a result, all the existing posterior primary molars in this case were restored with stainless steel crowns.
The fully erupted first permanent molars in this patient exhibited hypoplastic enamel. Lindau et al.[18] reported an anomaly in the structure of the enamel of patients suffering from DI. They found a more irregular pattern of enamel in affected primary teeth compared to permanent dentition. This may be due to insufficient mesenchymal–ectodermal induction during the amelogenesis procedure which may lead to DEJ irregularity and hypomineralization. However, they stated that the extent of the dental involvement in primary dentition seems not to be a major predictor for the severity of DI in permanent teeth and there is no association between the enamel morphology and a certain type of OI.[18] Norgen and Malgren explained that permanent teeth of children with OI may display normal clinical and radiographic appearance but a histological defective dentin exists.[4] These cases like ours showed permanent mandibular incisors with normal appearance.
As all posterior primary teeth had received full coverage treatment, the bulbous shape of the crown was not discernible. First permanent molars exhibited hypotrophic enamel and broad, large root canals. According to Schwartz and Tsipouras the rate of pulpal obliteration appears to be different in various types of OI.[1] For instance, teeth in OI type III seem to show an accelerated pulp obliteration compared to other types.[1] Pulpal obliteration may merely occur subsequent to root completion (2-4 years after tooth eruption); therefore it is possible that the root canal of the first permanent teeth in this case will become obliterated in the future.
Other dental anomalies such as ectopic eruption and missing (10%) are more common in OI.[8] We found no missing or ectopic eruption in this case. However, at the time of evaluation the patient was too young to detect any ectopic eruption. Therefore, the patient was put on a regular recall.
Structural evaluations by means of a stereomicroscope revealed a periodic irregular dentinogenesis with abundant globular and interglobular calcifications throughout the dentin. A decline in the number, an increase in the diameter of dentinal tubules, and obvious dentin disorganization were observed. This was in line with the findings of Teixeira et al.[9] However, Shafer reported a lower number of dentinal tubules with larger calibers.[19] Rios et al. stated that while an absence of the tubules was present in all samples, the tubules diameters varied among different subjects with DI (either reduction or increase). This microscopic view was more prominent in circumpulpal dentin compared to mantle dentin.[11]
The DEJ appeared to have an irregular scalloping and in some areas a straight smooth pattern in the examined dental sections. The disarrangement in the DEJ may be considered as a secondary effect of defective predentin in adequate induction of preameloblasts.[18] Low wear resistance of affected teeth is related to the absence of DEJ scalloping described by Schwartz et al.[1] However, the abnormal dentinal structure has been suggested to be the main reason for enamel breakage.[20] Similarly, Levin noticed severe attrition in DI-affected teeth with a normal scalloping of DEJ.[21] They concluded that the enamel detachments principally occurred because of the defective molecular collagen or some impaired matrix molecules.[1] We observed numerous enamel disintegrations in anterior primary teeth, although a scalloping pattern of DEJ existed microscopically in several regions of these teeth.
CONCLUSION
OI is a heritable systemic connective tissue disorder with DI as its dental counterpart. It is essential for clinicians to be familiar with different medical and oral aspects of OI. Children with OI should be examined as soon as teeth are erupted to prevent loss of tooth structure and seen frequently to restore any new enamel fracture and maintain their oral health.
Footnotes
Source of Support: Nil.
Conflict of Interest: None declared.
REFERENCES
- 1.Schwartz S, Tsipouras P. Oral findings in osteogenesis imperfecta. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 1984;57:161–7. doi: 10.1016/0030-4220(84)90206-8. [DOI] [PubMed] [Google Scholar]
- 2.Majorana A, Bardellini E, Brunelli PC, Lacaita M, Cazzolla AP, Favia G. Dentinogenesis imperfect in children with osteogenesis imperfecta: A clinical and ultrastructural study. Int J Paediatr Dent. 2010;20:112–8. doi: 10.1111/j.1365-263X.2010.01033.x. [DOI] [PubMed] [Google Scholar]
- 3.Germain-Lee EL. A new culprit in osteogenesis imperfect. J Bone Miner Res. 2011;26:2795–7. doi: 10.1002/jbmr.540. [DOI] [PubMed] [Google Scholar]
- 4.Malmgren B, Norgren S. Dental aberrations in children and adolescents with osteogenesis imperfect. Acta Odontol Scand. 2002;60:65–71. doi: 10.1080/000163502753509446. [DOI] [PubMed] [Google Scholar]
- 5.Sillence DO, Senn A, Danks DM. Genetic heterogeneity in osteogenesis imperfect. J Med Genet. 1979;16:101–6. doi: 10.1136/jmg.16.2.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Glorieux FH, Rauch F, Plotkin H, Wart L, Travers R, Roughley P, et al. Type V osteogenesis imperfecta: a new form of brittle bone disease. J Bone Miner Res. 2000;15:1650–8. doi: 10.1359/jbmr.2000.15.9.1650. [DOI] [PubMed] [Google Scholar]
- 7.Huber MA. Osteogenesis imperfecta. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2007;103:314–20. doi: 10.1016/j.tripleo.2006.10.003. [DOI] [PubMed] [Google Scholar]
- 8.O’Connel AC, Marini JC. Evaluation of oral problems in an osteonesis imperfect population. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 1999;87:189–96. doi: 10.1016/s1079-2104(99)70272-6. [DOI] [PubMed] [Google Scholar]
- 9.Teixeira CS, Felippe MC, Felippe TF, Silva-Sousa YT, Sousa-Neto MD. The role of dentists in diagnosing osteogenesis imperfecta in patients with dentinogenesis imperfecta. J Am Dent Assoc. 2008;139:906–14. doi: 10.14219/jada.archive.2008.0277. [DOI] [PubMed] [Google Scholar]
- 10.Kim JW, Simmer JP. Hereditary dentin defects. J Dent Res. 2007;86:392–9. doi: 10.1177/154405910708600502. [DOI] [PubMed] [Google Scholar]
- 11.Rios D, Vieira AL, Tenuta LM, Machado MA. Osteogenesis imperfect and dentinogenesis imperfect: Associated disorder. Quintessence Int. 2005;36:695–701. [PubMed] [Google Scholar]
- 12.Brustein HC, Mautner RL. Osteogenesis imperfecta: Review of the medical and dental literature and report of a case. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 1976;42:42–52. doi: 10.1016/0030-4220(76)90030-x. [DOI] [PubMed] [Google Scholar]
- 13.Shapiro JR, McCarthy EF, Rossiter K, Ernest K, Gelman R, Fedarko N, et al. The effect of intravenous pamidronate on bone mineral density, bone histomorphometry, and parameters of bone turnover in adults with type IA osteogenesis imperfecta. Calcif Tissue Int. 2003;72:103–12. doi: 10.1007/s00223-001-1055-5. [DOI] [PubMed] [Google Scholar]
- 14.Niyibizi C, Wang S, Mi Z, Robbins PD. Gene therapy approaches for osteogenesis imperfect. Gene Ther. 2004;11:408–16. doi: 10.1038/sj.gt.3302199. [DOI] [PubMed] [Google Scholar]
- 15.Plotkin H. Syndromes with congenital brittle bones. BMC Pediatr. 2004;4(16):1–6. doi: 10.1186/1471-2431-4-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.McDonald RE, Avery DR. Dentistry for the child and adolescent. 9th ed. St Louis: Mosby Elsevier; 2011. p. 72. [Google Scholar]
- 17.Pinkham JR. Pediatric Dentistry. 4th ed. St Louis: Mosby Elsevier; 2005. p. 70. [Google Scholar]
- 18.Lindau BM, Dietz W, Hoyer I, Lundgreen T, Storhaug K, Norem JG. Morphology of dental enamel and dentin-enamel junction in osteogenesis imperfecta. Int J Paediat Dent. 1999;9:13–21. doi: 10.1046/j.1365-263x.1999.00101.x. [DOI] [PubMed] [Google Scholar]
- 19.Shafer WG, Hine MK, Levy BM. A text book of oral pathology. 4th ed. Philadelphia: W. B. Saunders Company; 1983. p. 60. [Google Scholar]
- 20.Sunderland EP, Smith CJ. The teeth in osteogenesis and dentinogenesis imperfect. Br Dent J. 1980;149:287–9. doi: 10.1038/sj.bdj.4804512. [DOI] [PubMed] [Google Scholar]
- 21.Levin LS, Brady JM, Melnick M. Scaning electron microscopy of teeth in dominant osteogenesis imperfect: support for genetic heterogeneity. Am J Med Genet. 1980;5:189–99. doi: 10.1002/ajmg.1320050213. [DOI] [PubMed] [Google Scholar]