

Abstract
Gene expression occurs either as an episodic process, characterized by pulsatile bursts, or as a constitutive process, characterized by a Poisson-like accumulation of gene products. It is not clear which mode of gene expression (constitutive versus bursty) predominates across a genome or how transcriptional dynamics are influenced by genomic position and promoter sequence. Here, we use time-lapse fluorescence microscopy to analyze 8,000 individual human genomic loci and find that at virtually all loci, episodic bursting—as opposed to constitutive expression—is the predominant mode of expression. Quantitative analysis of the expression dynamics at these 8,000 loci indicates that both the frequency and size of the transcriptional bursts varies equally across the human genome, independent of promoter sequence. Strikingly, weaker expression loci modulate burst frequency to increase activity, whereas stronger expression loci modulate burst size to increase activity. Transcriptional activators such as trichostatin A (TSA) and tumor necrosis factor α (TNF) only modulate burst size and frequency along a constrained trend line governed by the promoter. In summary, transcriptional bursting dominates across the human genome, both burst frequency and burst size vary by chromosomal location, and transcriptional activators alter burst frequency and burst size, depending on the expression level of the locus.
Keywords: stochastic noise, automated single-cell imaging, human immunodeficiency virus, long terminal repeat promoter
There exists conflicting evidence over the predominant mode of gene expression in both prokaryotes and eukaryotes. The classical view of gene expression as a constitutive, Poisson-like accumulation of gene products (Fig. 1A) is supported by a comprehensive large-scale study in bacteria, demonstrating that > 400 genes appear to follow constitutive (or Poisson-like) gene expression (1). Constitutive expression has also been reported for subsets of human genes (2). Conversely, several elegant studies showed that specific promoters in bacteria and yeast express gene products in an episodic process (Fig. 1B), characterized by pulsatile bursts in transcription (3–9). Given this conflicting evidence, it remains unclear if episodic bursting is the predominant mode of gene expression across a genome or just a highlighted exception. If bursting is predominant, it is not clear if or how it depends on genomic location.
Fig. 1.

Fluctuations in gene expression to differentiate between alternate models of transcription across the genome. (A and B) Schematics of the constitutive, Poisson-expression model and the episodic, bursty gene-expression model, together with three expression trajectories from hypothetical genomic loci. Sites that exhibit constitutive (i.e., Poisson) expression exhibit small and relatively fast fluctuations in gene products over time. Alternatively, loci that exhibit episodic expression bursts generate large, slow fluctuations in gene expression. (C) The principle of noise space. The three-dimensional noise space consists of noise magnitude, noise autocorrelation, and mean expression level. Small, fast fluctuations have a small noise magnitude and short autocorrelation times and thus cluster (after normalization) at the origin of the noise magnitude-autocorrelation plane (gray region, Lower Left). Large, slow (i.e., bursty) fluctuations have expanded noise magnitude and extended autocorrelation times (red ovals). The three-dimensional space can be decomposed into two additional two-dimensional projections of noise magnitude and noise autocorrelation versus mean expression level (Lower Center and Lower Right). For episodic-bursty expression, a trajectory’s noise-space coordinates are invariably shifted away from the constitutive model into the burst model space depending on changes to their transcriptional parameters (10).
To globally determine if constitutive Poisson-like expression or episodic bursty expression dominates throughout the human genome, we capitalize on a recently proposed theoretical framework (10) for extracting the details of gene regulation from the time-resolved structure of fluctuations (i.e., noise) in gene expression. This analysis quantifies time-lapse expression trajectories to obtain three orthogonal measures of expression: the average expression level, the magnitude of expression fluctuations (as measured by the coefficient of variation squared, CV2), and the autocorrelation time of expression fluctuations (as measured by the noise autocorrelation time at half of its initial value, τ1/2) (11, 12) (Fig. 1C). Although this three-dimensional noise space is impractical to analyze directly, different two-dimensional projections of noise space allow the quantification of rate parameters in gene-regulatory models and provide a convenient method to differentiate between underlying gene-expression mechanisms, such as constitutive versus bursty transcription (Fig. 1C). For example, transcriptional bursting increases both noise magnitude and noise autocorrelation time and shifts points in the CV2 versus τ1/2 plane to the upper right quadrant relative to a constitutive expression model (Fig. 1C, Bottom Left). Conversely, translational bursting shifts noise magnitude, but not the autocorrelation time (10, 13).
Importantly, analysis of the τ1/2 axis is critical to fully parameterize two-state transcriptional bursting models (Fig. 1C), which always include at least three unknown parameters: the rate of transition to a transcriptionally active state (kon), the rate of transitioning to a transcriptionally inactive state (koff), and the rate of transcription once in the active state (km) (13, 14). Analyses of a single two-dimensional plane (e.g., CV2 versus expression level) cannot fully determine these three rate parameters. Conversely, analyses of CV2 versus expression level and τ1/2 versus expression level allow the determination of these three parameters, and analysis of CV2-versus-τ1/2 facilitates direct comparisons of data containing widely varying expression levels, because it removes the reciprocal dependence of noise magnitude on expression level (10).
The ability to accurately quantify these transcriptional rate parameters is essential for answering basic questions about the mechanisms that regulate transcription. Previous studies elegantly applied flow cytometry (6, 7) and time-lapse microscopy (1, 15, 16) to analyze gene-expression noise in large subsets of genes. However, a tedious experimental bottleneck of subcloning and expansion of isogenic populations necessarily limits the throughput of these noise-analysis approaches. Here, we circumvent this subcloning requirement to globally apply the analytical framework of noise space across the human genome and quantify these transcriptional rate parameters. The analysis addresses two specific questions regarding transcriptional regulation in human cells. (i) Does constitutive (Poisson-like) expression or episodic (bursty) expression dominate throughout the human genome? (ii) Does genomic location influence either mode of expression? For example, if bursting is operant, does genomic location influence burst size or burst frequency, and which is predominantly influenced? Recent studies (17–19) have tackled these questions for specific genes, but no clear and broad consensus has yet emerged.
Results
To globally apply the analytical framework of noise space to screen for constitutive versus bursty expression across the human genome, we capitalized on the semirandom pattern of integration exhibited by the HIV-1 lentivirus, where the majority of integrations (approximately 69%) occur within transcriptionally active regions (20, 21). Jurkat T lymphocytes were infected with HIV-based lentiviral vectors encoding a short-lived, two-hour half-life version of green fluorescent protein (referred to as d2GFP), to generate a library of cells in which the vector is integrated at a different genomic position in each individual cell (i.e., a polyclonal library) (Fig. 2A). To focus on measuring the intrinsic fluctuation dynamics of the genomic region surrounding the vector-integration site, we utilized a vector encoding the HIV-1 LTR promoter, which is relatively weak and heavily influenced by the expression dynamics of the local chromatin environment (22).
Fig. 2.

Extracting transcriptional parameters from the noise space. In individual isoclones, burst dynamics vary with genomic location. (A) Cells are infected with a lentiviral vector expressing a 2-h half-life GFP reporter (d2GFP) at a low multiplicity of infection (moi) to ensure a single semirandom integration in each cell. Individual single cells are isolated, grown (creating isoclone populations), and imaged by time-lapse fluorescence microscopy. (B and C) Single-cells are tracked for 12–18 h, and an individual cell’s mean expression level, variance (σ2), and autocorrelation time (τ1/2) are extracted from the time trace (e.g., the green circle represents a single cell’s noise space coordinate). A constitutive model of gene expression that displays abundance dependence (bold red arrows from black model lines) was used to normalize each cell’s noise magnitude (CV2) and autocorrelation (τ1/2). The normalized noise magnitudes and autocorrelations are plotted in a Δ log CV2 - Δ log τ1/2 noise map (Left). (D) Consistent shifts to the Upper Right quadrant in Δ log CV2 - Δ log τ1/2 space observed for three LTR isoclones (clones 3, 4, and 5), are indicative of transcriptional bursting relative to the least bursty isoclones (clones 1 and 2). Bursting dynamics varies between different clones as evidenced by shifts in both noise autocorrelation and magnitude. The isoclonal signature is taken from 18 h trajectories of 400 cells.
Initially, the analytical framework for distinguishing bursty expression was applied to five isoclonal populations: Each population was grown from a single parent cell, and all daughter cells, therefore, share the same LTR genomic integration site (Fig. 2
A–D). Cell fluorescence was then imaged for 18 h, and the resulting fluorescence intensity trajectories were used to construct a three-dimensional noise space with three axes: noise magnitude as measured by the coefficient of variation squared (CV2), noise autocorrelation represented by the half-autocorrelation time (τ1/2), and mean expression level (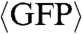 ) (Fig. 2
B–C and SI Appendix). Analysis of these trajectories in the noise space allows comparisons between isoclonal populations and differentiation between isoclones that exhibit constitutive transcription versus episodic bursts of transcription (Fig. 2D). To generate an initial baseline origin for noise-space analysis of the single-cell data, we identified isoclones that were the most Poissonian in their expression fluctuation profiles from a library of isoclonal populations (17) (SI Appendix). Two isoclonal populations exhibiting the fastest fluctuation autocorrelation decays (i.e., shortest autocorrelation times) were selected as the most Poissonian and used to establish an origin of the CV2-versus-τ1/2 noise map (Fig. 2D, clones 1 and 2). The isoclone heat map represents the probability of where a randomly chosen single cell would land on the noise map (Fig. 2D).
) (Fig. 2
B–C and SI Appendix). Analysis of these trajectories in the noise space allows comparisons between isoclonal populations and differentiation between isoclones that exhibit constitutive transcription versus episodic bursts of transcription (Fig. 2D). To generate an initial baseline origin for noise-space analysis of the single-cell data, we identified isoclones that were the most Poissonian in their expression fluctuation profiles from a library of isoclonal populations (17) (SI Appendix). Two isoclonal populations exhibiting the fastest fluctuation autocorrelation decays (i.e., shortest autocorrelation times) were selected as the most Poissonian and used to establish an origin of the CV2-versus-τ1/2 noise map (Fig. 2D, clones 1 and 2). The isoclone heat map represents the probability of where a randomly chosen single cell would land on the noise map (Fig. 2D).
Importantly, nontranscriptional phenomena that influence noise behavior (e.g., protein and mRNA lifetimes, GFP maturation, extrinsic noise) are already embedded in the noise of these isoclonal populations, specifically the most Poissonian reference clones. Thus, comparison between these necessarily precludes the possibility that noise-map shifts are due to these nontranscriptional phenomena. In addition, the high-frequency noise-processing technique minimizes extrinsic noise effects (SI Appendix) (12). Because it is likely that these two isoclones are somewhat bursty in their expression (SI Appendix), comparisons to these isoclones represent a highly conservative assay for bursty expression. Nevertheless, the results show that even in a small panel of five clones, there are marked changes in both noise magnitude and autocorrelation time of approximately 1.5-fold in normalized space (Fig. 2D). Clones 4 and 5 show significant changes in autocorrelation time with smaller differences in noise magnitude. These differences in noise magnitude were validated against conventional flow cytometry measurements (SI Appendix, Fig. S1). The agreement between data from the 12–18 h microscopy experiments and flow cytometry demonstrates that this CV2-versus-τ1/2 analysis has the fidelity to differentiate transcriptional dynamics between different isoclones.
We next extended the analysis to image polyclonal populations—consisting of thousands of integration sites (Fig. 3A)—to globally apply the analytical framework of noise space to screen for constitutive versus bursty expression across the human genome. We analyzed more than 8,000 distinct genomic loci with three different promoters integrated throughout the genome by imaging cells for 12–18 h (Fig. 3B). The constitutive expression origin of the CV2-versus-τ1/2 noise map previously determined for the most-Poissonian isoclones (Fig. 2D and SI Appendix) was compared to the 8,000 loci. To control for LTR-specific or vector-specific artifacts, we also tested self-inactivating lentiviral vectors that encode either promoters for human elongation factor 1α (EF1A) or human ubiquitin C (UBC) that in turn drive d2GFP. UBC and EF1A are promoters that drive essential cellular housekeeping genes: UBC promotes the ubiquitinization cascade by marking proteins for proteosomal degradation, and EF1A promotes the GTP-dependent binding of an aminoacyl-tRNA to ribosomes. UBC and EF1A are among the most abundant proteins in eukaryotic cells, and their promoters exhibit robust high-level expression across integration sites in different cell types (23, 24). Virtually all examined genomic loci for all promoters exhibit noise-map shifts to the upper right (Fig. 3B), indicating significant bursting in gene expression at virtually all genomic loci. Similar widespread bursting is also observed in the THP-1 human monocyte cell line and compared to a constitutive simulation (SI Appendix, Fig. S2). Both synchronized and unsynchronized cells exhibit similar shifts in noise space (SI Appendix, Fig. S3), indicating that transcriptional bursts appear to be common throughout the cell cycle, as reported (25).
Fig. 3.

Episodic-bursty expression dominates across the human genome. (A) To create the polyclonal population, cells are infected with a lentiviral vector expressing d2GFP so that each cell represents a unique clone harboring a single semirandom integration of reporter. (B) Resultant noise maps for over 8,000 individual cell trajectories for the HIV-1 LTR promoter, EF1A promoter, and UBC promoter. The constitutive origin is derived from Fig. 2D (18 h).
The UBC and EF1A promoters showed markedly lower CV2 values than the LTR promoter (Fig. 3B and SI Appendix, Fig. S4), which is consistent with our previous study (17) showing that the LTR promoter displays relatively higher levels of noise than other eukaryotic promoters in yeast (6, 7). This shift in noise magnitude is consistent with the well-known transcriptional elongation stall that characterizes LTR expression (26). This stall results in delayed switching to the transcriptional ON state in a two-state transcription model (17) and predicts that noise frequency (not only noise magnitude) (27) is modulated in different genomic or chromatin environments (SI Appendix). In agreement with this prediction, the distribution of points in the LTR noise map indicates significant differences from the UBC and EF1A noise maps. In further support, an alternate representation of the noise-map distribution that converts the distributions to centroids (with error bars) can be used to conveniently visualize these differences between promoters (SI Appendix, Fig. S4) (11).
Given conflicting reports on whether burst frequency varies with genomic location (17–19, 28), we next determined if transcriptional burst size, burst frequency, or both changed across the genome (Fig. 4A). As mentioned above, transcriptional bursting can be quantified by a two-state model of transcription (13, 14) in which switching between the two states occurs at rates kon and koff, and transcription only occurs in the ON state with a rate km (Eqs. 1–3 and Materials and Methods). The burst size, or number of mRNAs generated per activity pulse, is typically defined as km/koff and, in the limit of koff≫kon, the burst frequency is defined as kon (Fig. 4A) (17). To directly test if transcriptional burst frequency changes across genome location, we analyzed the polyclonal three-dimensional noise-space data to fit values for km, koff, and kon. LTR polyclonal trajectories are subclustered into groupings of approximately 60 cells, so that each subcluster represents cells in a specific range of gene-expression levels (SI Appendix, Fig. S5), and average noise autocorrelation is calculated for each subcluster by autocorrelation analysis (12). To validate this subclustering approach, we verified that CV2 values from subclustered polyclonal trajectories agree with CV2 from conventional flow cytometry data from isoclonal populations (Compare Fig. 4C to SI Appendix, Figs. S1 and S6) . Thus, each polyclonal subcluster corresponds to an isoclonal population in terms of average expression level (SI Appendix, Fig. S6). Strikingly, this genome-wide data demonstrate that autocorrelation time first increases with increasing expression and once an expression threshold is reached (gray line, Fig. 4B), autocorrelation time decreases as expression level increases (Fig. 4B). This pattern of concavity is inconsistent with constant burst frequency (i.e., constant kon) across genomic locations and provides a genome-wide measurement of kon and koff changes that is independent of km (10).
Fig. 4.

Transcriptional burst frequency and burst size vary equally across the genome and are strongly dependent on expression level. (A) Schematic of the two-state model of transcriptional bursting, where the promoter switches between ON and OFF states at rates kon and koff and transcribes at rate km in the ON state. Transcriptional dynamics are modulated through changes in burst size, burst frequency, or both. (B) Noise autocorrelation, noise magnitude (C), burst frequency (D), and burst size (E) versus abundance for polyclonal subclusters of 2,000 12-h Ld2G single-cell trajectories. Low and high abundance domains are separated by a solid gray threshold line which indicates the changes in the trends of noise autocorrelation, noise magnitude, and hence burst size and burst frequency is observed. (F and G) As a function of  , fold changes in burst size and frequency are comparable, with an initial increase of frequency in all promoters investigated.
, fold changes in burst size and frequency are comparable, with an initial increase of frequency in all promoters investigated.
Conventional approaches to quantify transcriptional burst kinetics analyze noise magnitude (CV2-versus- ) (6, 7) and the polyclonal data from Fig. 3 can also be analyzed in terms of noise magnitude on the CV2-versus-
) (6, 7) and the polyclonal data from Fig. 3 can also be analyzed in terms of noise magnitude on the CV2-versus- plane of noise space (Fig. 4C). This noise magnitude analysis shows a strong initial decrease in CV2 at low expression levels (Fig. 4C). Then when an expression threshold is reached (gray line, Fig. 4C) a leveling off of CV2 is observed for higher expression levels (Fig. 4C). However, noise magnitude is insufficient to uniquely parameterize the two-state model. Because burst size couples km and koff, it is only through the τ1/2 measurement, which is not influenced by km, that the two parameters can be differentiated from one another. Note, that the gray line in Fig. 4
B and C correspond to the same expression threshold.
plane of noise space (Fig. 4C). This noise magnitude analysis shows a strong initial decrease in CV2 at low expression levels (Fig. 4C). Then when an expression threshold is reached (gray line, Fig. 4C) a leveling off of CV2 is observed for higher expression levels (Fig. 4C). However, noise magnitude is insufficient to uniquely parameterize the two-state model. Because burst size couples km and koff, it is only through the τ1/2 measurement, which is not influenced by km, that the two parameters can be differentiated from one another. Note, that the gray line in Fig. 4
B and C correspond to the same expression threshold.
Fitting of the two-state model in the polyclonal three-dimensional noise map space shows a strong initial increase in burst frequency at low expression levels, whereas burst size remains almost constant (Fig. 4 D and E). Upon reaching a threshold expression level (gray vertical line in Fig. 4D), a switch in burst dynamics occurs, and burst size increases, whereas burst frequency remains constant (Fig. 4 D and E). The fold change in transcriptional burst size and burst frequency values reveals that both vary equally across genomic loci (Fig. 4F and SI Appendix, Fig. S6). In addition, the measured burst-size range predicts an average mRNA level of 110 molecules per cell, which is consistent with previous measurements that used single-molecule mRNA fluorescent in situ hybridization (29, 30). The success of fitting the three-dimensional noise space is reflected in the close agreement between a simulated autocorrelation curve and the experimental trend (Fig. 4B) with the fit model parameters. This fit shows that the assumed two-state model is sufficient to describe the measured system (SI Appendix). Much like LTR, UBC, and EF1A display similar fold changes in burst size and frequency and exhibit a similar pattern of increasing burst frequency, followed by increasing burst size (Fig. 4G). These data indicate that integration site influences burst kinetics, irrespective of promoter type (i.e., cis sequence). However, UBC and EF1A exhibit almost constant τ1/2 at the highest expression levels indicating increases in only km at these levels (SI Appendix, Fig. S7). Interestingly, these two strong promoters individually span the range of burst frequencies recently reported for a variety of mammalian genes (18) (SI Appendix, Fig. S7), whereas the LTR functions at much lower burst frequencies (Fig. 4).
To test how transcriptional activators influence burst dynamics (Fig. 4), transcription was perturbed with transcriptional activators, including the histone deacetylase inhibitor (HDAC) trichostatin A (TSA), and the cell-signaling molecule tumor necrosis factor α (TNF). TNF enhances expression by stimulating recruitment of a p50-RelA heterodimer to nuclear factor κB (NFκB) binding sites (31) and the HIV-1 LTR encodes multiple NFκB binding sites and is potently activated by TNF (22). We previously reported that TNF only changes burst frequency of the LTR while conserving burst size in a limited number of isoclones (17), and were interested to see how widespread this phenomena was across the genome. The τ1/2-versus- analysis of TNF stimulation shows a significant decrease in τ1/2 with increasing expression level (Fig. 5A). Here, τ1/2 decrease with increasing abundance is a direct indication of kinetic changes and demonstrates that increasing expression level cannot be explained solely by modulations of km (SI Appendix, Eq. S1) (10, 13).
analysis of TNF stimulation shows a significant decrease in τ1/2 with increasing expression level (Fig. 5A). Here, τ1/2 decrease with increasing abundance is a direct indication of kinetic changes and demonstrates that increasing expression level cannot be explained solely by modulations of km (SI Appendix, Eq. S1) (10, 13).
Fig. 5.

Transcriptional burst size and frequency are altered by transcriptional activators. (A–D) TNF addition (filled red circles) shifts the measured integration sites to the higher abundance and burst dynamic domain along the nondrug curve (empty circles). Large autocorrelation shifts implicate changes in burst kinetics. (E) Estimated residence times in the active (ON) and inactive (OFF) states.
Fitting of the different two-dimensional planes of three-dimensional noise space upon TNF induction (Fig. 5 A and B) demonstrates that both burst frequency and size significantly increase as expression levels increase, with burst frequency increasing at low expression levels and burst size increasing at higher expression levels (Fig. 5 C and D). Interestingly, there appears to be a threshold in expression level, above which kon plateaus to values observed before adding TNF, and koff appears to decrease. These data and analysis are corroborated by conventional flow cytometry measurements of 35 isoclonal populations (SI Appendix, Fig. S6). Overall, these results suggest that TNF induces expression from the LTR along existing burst trends (Fig. 5C and D), and the use of TSA, which induces expression through a different mechanism than TNF (32), corroborates this observation (SI Appendix). This observed decrease in koff with TNF induction leads to extended duration of bursts and is consistent with the reported inhibition of p50-HDAC1 repressive-complex formation at LTR NFκB sites by p50/RelA heterodimers (33)—the successful formation of HDAC1 leads to weakened recruitment of RNA polymerase II and weakened transcriptional initiation (34). The observed increases in kon are also consistent with increased recruitment of RNA polymerase II to the LTR promoter NFκB sites induced by TNF (35, 36). Fitted parameter estimates of LTR residency time in the presence of TNF were used to represent an average over the first 12 h of stimulation given the dynamic nonlinear nature of the NFκB response (37, 38). Collectively, these results enable estimation of LTR residency time in the transcriptional ON and OFF states and show that TNF extends duration in the ON state up to eightfold (Fig. 5E).
Discussion
The analysis of noise space presented here provides a high-throughput method to dynamically profile gene-regulatory mechanisms and the effects of perturbations on gene expression. A significant methodological advantage of analyzing three dimensions of noise space is the ability to more accurately constrain two-state transcriptional burst models and the polyclonal nature of the approach enables shotgun mapping of gene regulation dynamics on a genome-wide scale.
The resulting genome-wide data demonstrate that constitutive transcription is rare across the human genome. Instead, the overwhelming majority of human genomic loci appear to stochastically fire in episodic bursts. Analysis of noise space demonstrated that both transcriptional burst frequency and burst size vary in roughly equal degree across the human genome (Fig. 4 D–G). Intriguingly, there appears to be a threshold expression level below which integrations modulate only transcriptional burst frequency and above which only burst size is modulated (Fig. 4 B–G and SI Appendix, Figs. S6 and S7). This transition could result from recently reported refractory periods inherent to bursting kinetics (18, 39). Burst frequency can be increased at loci where transcriptional bursts are infrequent, but as frequency increases, the refractory period temporally precludes further increases in frequency. Therefore, once this frequency ceiling is reached, the only way to increase expression is to increase the transcription rate or extend the duration of each burst.
As proposed (40), widespread episodic bursting may allow limited transcriptional resources within the cell to be efficiently allocated to achieve high-level transcription across large numbers of loci. This efficient allocation of resources may be the biological analog of time-domain multiplexing approaches used to efficiently transmit data in signal processing applications.
Materials and Methods
Lentiviral Vectors.
Lentiviral vectors were cloned as described (41) and used to infect 5 × 105 Jurkat cells at a multiplicity of infection < 0.1, resulting in 25,000–50,000 infected cells each with a unique integration site. Cells were then sorted by FACS and fluorescently imaged on glass-bottom dishes in RPMI medium 1640 with 10% fetal calf serum and 1% penicillin-streptomycin.
Imaging.
Imaging was performed in humidified conditions at 37 °C and 5% CO2 for 12–24 h with a 40X (1.2 N.A.) oil-immersion objective on an Olympus DSU microscope equipped with an automated linear-encoded X-Y stage, as described in refs. (12) and (42). Image processing and cell tracking were performed in Matlab with an in-house algorithm (12) and a single 12-h experiment could generate up to 1,000 trajectories for analysis.
Calculations.
For each trajectory, noise autocorrelation (τ1/2) and noise magnitude (CV2) were calculated using an established noise-processing algorithm (11, 12). A reported theory (10, 13) of the two-state transcriptional bursting model yields analytical expressions for both the autocorrelation of the noise, τ1/2, and the noise magnitude, CV2(see SI Appendix, Materials and Methods). Detailed discussion of the noise-mapping approach, including analytical arguments and stochastic simulations (of Poissonian gene expression) by the Gillespie algorithm (43, 44), are described in detail in SI Appendix.
Transcriptional burst dynamics are quantified by deriving analytical expressions for burst size and burst frequency with formulations from previous analyses (13, 14, 17) and low promoter activity assumptions where koff≫kon, koff≫km, koff≫γp and km≫(γm + γp)):
 |
[1] |
 |
[2] |
 |
[3] |
where BS is the burst size, BF is the burst frequency, km is the transcription rate, kp is the translation rate, γm and γp are the mRNA and protein decay rates, respectively,  is the mean protein abundance, and b is the translational burst rate.
is the mean protein abundance, and b is the translational burst rate.  , or the mean number of GFP molecules in the measurements, is assumed to be directly proportional to
, or the mean number of GFP molecules in the measurements, is assumed to be directly proportional to  , the mean fluorescence intensity. Eqs. 2 and 3 reveal that measurements of CV2 and
, the mean fluorescence intensity. Eqs. 2 and 3 reveal that measurements of CV2 and  are sufficient to quantify burst size and burst frequency within a constant, which is only dependent on the translation rate and decay rates of mRNA and protein. Assuming these remain constant, while varying integration site or promoter sequence, an abundance-dependent burst size and frequency trend can be directly resolved.
are sufficient to quantify burst size and burst frequency within a constant, which is only dependent on the translation rate and decay rates of mRNA and protein. Assuming these remain constant, while varying integration site or promoter sequence, an abundance-dependent burst size and frequency trend can be directly resolved.
Supplementary Material
ACKNOWLEDGMENTS.
We thank Hana El-Samad, Ido Golding, Jim Kadonaga, Laurie Boyer, Alex Hoffmann, John Cooke, David Karig, and members of the Weinberger and Simpson labs for helpful comments. B.S.R. was supported by National Science Foundation Graduate Research Fellowship Grant 1144247 and by National Institutes of Health (NIH) Molecular Biophysics Training Grant GM08326. L.S.W. acknowledges support from the Pew Scholars Program in the Biomedical Sciences and the Alfred P. Sloan Research Fellowship Program. This work was supported by the NIH Director’s New Innovator Award (OD006677) (to L.S.W.), the in-house research program at the Center for Nanophase Materials Sciences at Oak Ridge National Laboratory (sponsored by the Office of Basic Energy Sciences, US Department of Energy) (to R.D.D. and M.L.S.), and by the National Institute of General Medical Sciences National Systems Biology Centers at University of California at San Diego (P50 GM085764) and University of California, San Francisco (P50 GM081879).
Note.
Full decomposition of koff, kon, and kmcan only be performed through the use of the full 3D noise space.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1213530109/-/DCSupplemental.
References
- 1.Taniguchi Y, et al. Quantifying E. coli proteome and transcriptome with single-molecule sensitivity in single cells. Science. 2010;329:533–538. doi: 10.1126/science.1188308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yunger S, Rosenfeld L, Garini Y, Shav-Tal Y. Single-allele analysis of transcription kinetics in living mammalian cells. Nat Methods. 2010;7:631–633. doi: 10.1038/nmeth.1482. [DOI] [PubMed] [Google Scholar]
- 3.Golding I, Paulsson J, Zawilski SM, Cox EC. Real-time kinetics of gene activity in individual bacteria. Cell. 2005;123:1025–1036. doi: 10.1016/j.cell.2005.09.031. [DOI] [PubMed] [Google Scholar]
- 4.Pedraza JM, Paulsson J. Effects of molecular memory and bursting on fluctuations in gene expression. Science. 2008;319:339–343. doi: 10.1126/science.1144331. [DOI] [PubMed] [Google Scholar]
- 5.Cai L, Friedman N, Xie XS. Stochastic protein expression in individual cells at the single molecule level. Nature. 2006;440:358–362. doi: 10.1038/nature04599. [DOI] [PubMed] [Google Scholar]
- 6.Newman JR, et al. Single-cell proteomic analysis of S. cerevisiae reveals the architecture of biological noise. Nature. 2006;441:840–846. doi: 10.1038/nature04785. [DOI] [PubMed] [Google Scholar]
- 7.Bar-Even A, et al. Noise in protein expression scales with natural protein abundance. Nat Genet. 2006;38:636–643. doi: 10.1038/ng1807. [DOI] [PubMed] [Google Scholar]
- 8.So LH, et al. General properties of transcriptional time series in Escherichia coli. Nat Genet. 2011;43:554–560. doi: 10.1038/ng.821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Blake WJ, M KÆrn, Cantor CR, Collins JJ. Noise in eukaryotic gene expression. Nature. 2003;422:633–637. doi: 10.1038/nature01546. [DOI] [PubMed] [Google Scholar]
- 10.Cox CD, McCollum JM, Allen MS, Dar RD, Simpson ML. Using noise to probe and characterize gene circuits. Proc Natl Acad Sci USA. 2008;105:10809–10814. doi: 10.1073/pnas.0804829105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Austin DW, et al. Gene network shaping of inherent noise spectra. Nature. 2006;439:608–611. doi: 10.1038/nature04194. [DOI] [PubMed] [Google Scholar]
- 12.Weinberger LS, Dar RD, Simpson ML. Transient-mediated fate determination in a transcriptional circuit of HIV. Nat Genet. 2008;40:466–470. doi: 10.1038/ng.116. [DOI] [PubMed] [Google Scholar]
- 13.Simpson ML, Cox CD, Sayler GS. Frequency domain chemical Langevin analysis of stochasticity in gene transcriptional regulation. J Theor Biol. 2004;229:383–394. doi: 10.1016/j.jtbi.2004.04.017. [DOI] [PubMed] [Google Scholar]
- 14.Kepler TB, Elston TC. Stochasticity in transcriptional regulation: Origins, consequences, and mathematical representations. Biophys J. 2001;81:3116–3136. doi: 10.1016/S0006-3495(01)75949-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sigal A, et al. Variability and memory of protein levels in human cells. Nature. 2006;444:643–646. doi: 10.1038/nature05316. [DOI] [PubMed] [Google Scholar]
- 16.Cohen AA, et al. Dynamic proteomics of individual cancer cells in response to a drug. Science. 2008;322:1511–1516. doi: 10.1126/science.1160165. [DOI] [PubMed] [Google Scholar]
- 17.Singh A, Razooky B, Cox CD, Simpson ML, Weinberger LS. Transcriptional bursting from the HIV-1 promoter is a significant source of stochastic noise in HIV-1 gene expression. Biophys J. 2010;98:L32–L34. doi: 10.1016/j.bpj.2010.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Suter DM, et al. Mammalian genes are transcribed with widely different bursting kinetics. Science. 2011;332:472–474. doi: 10.1126/science.1198817. [DOI] [PubMed] [Google Scholar]
- 19.Skupsky R, Burnett JC, Foley JE, Schaffer DV, Arkin AP. HIV promoter integration site primarily modulates transcriptional burst size rather than frequency. PLoS Comput Biol. 2010;6:e1000952. doi: 10.1371/journal.pcbi.1000952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mitchell RS, et al. Retroviral DNA integration: ASLV, HIV, and MLV show distinct target site preferences. PLoS Biol. 2004;2:1127–1137. doi: 10.1371/journal.pbio.0020234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schroder AR, et al. HIV-1 integration in the human genome favors active genes and local hotspots. Cell. 2002;110:521–529. doi: 10.1016/s0092-8674(02)00864-4. [DOI] [PubMed] [Google Scholar]
- 22.Jordan A, Defechereux P, Verdin E. The site of HIV-1 integration in the human genome determines basal transcriptional activity and response to Tat transactivation. EMBO J. 2001;20:1726–1738. doi: 10.1093/emboj/20.7.1726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kim DW, Uetsuki T, Kaziro Y, Yamaguchi N, Sugano S. Use of the human elongation factor 1 alpha promoter as a versatile and efficient expression system. Gene. 1990;91:217–223. doi: 10.1016/0378-1119(90)90091-5. [DOI] [PubMed] [Google Scholar]
- 24.Ramezani A, Hawley TS, Hawley RG. Lentiviral vectors for enhanced gene expression in human hematopoietic cells. Mol Ther. 2000;2:458–469. doi: 10.1006/mthe.2000.0190. [DOI] [PubMed] [Google Scholar]
- 25.Harper CV, et al. Dynamic analysis of stochastic transcription cycles. PLoS Biol. 2011;9:e1000607. doi: 10.1371/journal.pbio.1000607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kao SY, Calman AF, Luciw PA, Peterlin BM. Anti-termination of transcription within the long terminal repeat of HIV-1 by tat gene product. Nature. 1987;330:489–493. doi: 10.1038/330489a0. [DOI] [PubMed] [Google Scholar]
- 27.Sakane N, et al. Activation of HIV transcription by the viral Tat protein requires a demethylation step mediated by lysine-specific demethylase 1 (LSD1/KDM1) PLoS Pathog. 2011;7:e1002184. doi: 10.1371/journal.ppat.1002184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lo MY, Rival-Gervier S, Pasceri P, Ellis J. Rapid transcriptional pulsing dynamics of high expressing retroviral transgenes in embryonic stem cells. PLoS One. 2012;7:e37130. doi: 10.1371/journal.pone.0037130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Raj A, Peskin CS, Tranchina D, Vargas DY, Tyagi S. Stochastic mRNA synthesis in mammalian cells. PLoS Biol. 2006;4:1707–1719. doi: 10.1371/journal.pbio.0040309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Singh A, Razooky B, Dar RD, Weinberger LS. Dynamics of protein noise can distinguish between alternative sources of gene-expression variability. Mol Syst Biol. 2012;8:1–9. doi: 10.1038/msb.2012.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Vallabhapurapu S, Karin M. Regulation and function of NF-kappaB transcription factors in the immune system. Annu Rev Immunol. 2009;27:693–733. doi: 10.1146/annurev.immunol.021908.132641. [DOI] [PubMed] [Google Scholar]
- 32.VanLint C, Emiliani S, Ott M, Verdin E. Transcriptional activation and chromatin remodeling of the HIV-1 promoter in response to histone acetylation. EMBO J. 1996;15:1112–1120. [PMC free article] [PubMed] [Google Scholar]
- 33.Hayden MS, Ghosh S. Signaling to NF-kappaB. Genes Dev. 2004;18:2195–2224. doi: 10.1101/gad.1228704. [DOI] [PubMed] [Google Scholar]
- 34.Williams SA, et al. NF-kappaB p50 promotes HIV latency through HDAC recruitment and repression of transcriptional initiation. EMBO J. 2006;25:139–149. doi: 10.1038/sj.emboj.7600900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.West MJ, Lowe AD, Karn J. Activation of human immunodeficiency virus transcription in T cells revisited: NF-kappaB p65 stimulates transcriptional elongation. J Virol. 2001;75:8524–8537. doi: 10.1128/JVI.75.18.8524-8537.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Barboric M, Nissen RM, Kanazawa S, Jabrane-Ferrat N, Peterlin BM. NF-kappaB binds P-TEFb to stimulate transcriptional elongation by RNA polymerase II. Mol Cell. 2001;8:327–337. doi: 10.1016/s1097-2765(01)00314-8. [DOI] [PubMed] [Google Scholar]
- 37.Williams SA, Kwon H, Chen LF, Greene WC. Sustained induction of NF-kappa B is required for efficient expression of latent human immunodeficiency virus type 1. J Virol. 2007;81:6043–6056. doi: 10.1128/JVI.02074-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hoffmann A, Levchenko A, Scott ML, Baltimore D. The IkappaB-NF-kappaB signaling module: Temporal control and selective gene activation. Science. 2002;298:1241–1245. doi: 10.1126/science.1071914. [DOI] [PubMed] [Google Scholar]
- 39.Suter DM, Molina N, Naef F, Schibler U. Origins and consequences of transcriptional discontinuity. Curr Opin Cell Biol. 2011;23:657–662. doi: 10.1016/j.ceb.2011.09.004. [DOI] [PubMed] [Google Scholar]
- 40.Cai L, Dalal CK, Elowitz MB. Frequency-modulated nuclear localization bursts coordinate gene regulation. Nature. 2008;455:485–490. doi: 10.1038/nature07292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Weinberger LS, Burnett JC, Toettcher JE, Arkin AP, Schaffer DV. Stochastic gene expression in a lentiviral positive-feedback loop: HIV-1 Tat fluctuations drive phenotypic diversity. Cell. 2005;122:169–182. doi: 10.1016/j.cell.2005.06.006. [DOI] [PubMed] [Google Scholar]
- 42.Weinberger LS, Shenk T. An HIV feedback resistor: Auto-regulatory circuit deactivator and noise buffer. PLoS Biol. 2007;5:67–81. doi: 10.1371/journal.pbio.0050009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gillespie DT. General method for numerically simulating stochastic time evolution of coupled chemical-reactions. J Comput Phys. 1976;22:403–434. [Google Scholar]
- 44.McCollum JM, Peterson GD, Cox CD, Simpson ML, Samatova NF. The sorting direct method for stochastic simulation of biochemical systems with varying reaction execution behavior. Comput Biol Chem. 2006;30:39–49. doi: 10.1016/j.compbiolchem.2005.10.007. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.