

Abstract
Although the pimeloyl moiety was long known to be a biotin precursor, the mechanism of assembly of this C7 α,ω-dicarboxylic acid was only recently elucidated. In Escherichia coli, pimelate is made by bypassing the strict specificity of the fatty acid synthetic pathway. BioC methylates the free carboxyl of a malonyl thioester, which replaces the usual acetyl thioester primer. This atypical primer is transformed to pimeloyl-acyl carrier protein (ACP) methyl ester by two cycles of fatty acid synthesis. The question is, what stops this product from undergoing further elongation? Although BioH readily cleaves this product in vitro, the enzyme is nonspecific, which made assignment of its physiological substrate problematical, especially because another enzyme, BioF, could also perform this gatekeeping function. We report the 2.05-Å resolution cocrystal structure of a complex of BioH with pimeloyl-ACP methyl ester and use the structure to demonstrate that BioH is the gatekeeper and its physiological substrate is pimeloyl-ACP methyl ester.
Keywords: cofactor biosynthesis, esterase, protein–protein interaction
Recent work delineated the assembly pathway of the enigmatic pimeloyl moiety of biotin. Labeling studies in Escherichia coli had shown that pimelate, a C7 α,ω-dicarboxylic acid, is made by head-to-tail incorporation of three intact acetate units with one of the carboxyl groups being derived from CO2 (1, 2). The differing origins of the carboxyl groups indicated that free pimelate was not a synthetic intermediate. The acetate incorporation pattern was consistent with use of the synthetic pathway that produces the usual monocarboxylic fatty acids. However, synthesis of a dicarboxylic acid using the fatty acid synthetic pathway appeared precluded by the strongly hydrophobic active sites of the fatty acid synthetic enzymes (3), which seemed unlikely to tolerate the charged carboxyl group in place of the usual terminal methyl group. The solution to this conundrum was provided by the characterization of two enzymes, BioC and BioH, which do not directly catalyze pimelate synthesis but instead allow fatty acid synthesis to assemble the pimelate moiety (4). Such circumvention of the specificity of normal fatty acid synthesis begins by BioC (an O-methyltransferase) conversion of the ω-carboxyl group of malonyl-acyl carrier protein (ACP) to a methyl ester using S-adenosyl-l-methionine (SAM) as a methyl donor (Fig. 1). Conversion to a methyl ester neutralizes the negative charge and provides a methyl carbon that mimics the methyl ends of normal fatty acyl chains. The malonyl-ACP methyl ester can now enter the fatty acid synthetic pathway where it is condensed with malonyl-ACP by a 3-oxoacyl-ACP synthase in a decarboxylating Claisen reaction to give 3-oxoglutaryl-ACP methyl ester. The methyl ester shielding allows the 3-oxo group to be processed to a methylene group by the standard fatty acid reductase-dehydratase-reductase reaction sequence. The resulting glutaryl-ACP methyl ester would then be elongated to the C7 species, and another round of the reductase-dehydratase-reductase cycle would give pimeloyl-ACP methyl ester (Me-pimeloyl-ACP). Once synthesis of the pimelate chain is complete, the ester is no longer required and must be removed, because the freed carboxyl group will eventually be used to attach biotin to essential metabolic enzymes.
Fig. 1.

Proposed E. coli biotin synthetic pathway showing the catalytic role of BioH. Biotin biosynthesis is initiated by BioC methylation of ACP (or malonyl-CoA) to give malonyl-ACP methyl ester, which, after two successive cycles of fatty acid synthesis (FAS), becomes pimeloyl-ACP methyl ester. Once pimelate is synthesized, BioH hydrolyzes the methyl ester bond to expose the ω-carboxyl group and terminate chain elongation. The ACP moiety is subsequently released in the BioF reaction, the first step of biotin ring assembly.
Prior experiments demonstrated that BioH is responsible for cleavage of the methyl ester, but the physiological substrate of the enzyme was unclear. Chemical logic argues that the most attractive substrate would be Me-pimeloyl-ACP because its cleavage would act as a gatekeeper to prevent further chain elongation to azelayl-ACP methyl ester, a physiologically useless product. However, because the methyl ester introduced by BioC is at the far end of the pimeloyl moiety from the carbon atom incorporated to initiate the biotin ureido ring, the ester could remain until later in the pathway. In such a case, the gatekeeper function could be provided by BioF, the 7-keto-8-aminopelargonic acid (KAPA) synthase that begins ring formation, because the reaction consumes the thioester required for further fatty acid chain elongation. Hence, the problem distills down to the question of whether BioH acts before or after BioF (Fig. S1A). This issue is further complicated by the findings that BioH is known to be a rather promiscuous hydrolase. This substrate promiscuity may reflect recent recruitment of E. coli BioH for biotin synthesis and the fact that the bioH gene has not been fully integrated into the biotin synthetic pathway. E. coli bioH differs from the other genes of the pathway in that it is neither located within the bio operon nor regulated by the BirA repressor/biotin protein ligase (5, 6), which is in contrast to other bacteria where bioH resides within the biotin operon (7). Thus, the E. coli bioH gene may encode a less-specific protein than those encoded by the integrated bioH genes.
In this paper we report experiments indicating that BioH acts before BioF. However, the promiscuity of both enzymes made this experiment less than compelling, and thus we obtained the 2.05-Å resolution cocrystal structure of a complex of BioH with Me-pimeloyl-ACP as an independent test of our interpretation (Fig. 2). The BioH–ACP interface contacts identified in the structure (Fig. 3) were demonstrated to be required for biotin synthesis in vivo, indicating that Me-pimeloyl-ACP is the physiological substrate of BioH, and that BioH is the gatekeeper.
Fig. 2.
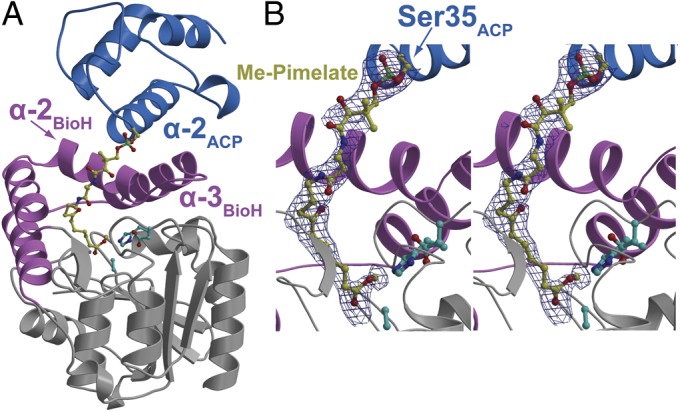
Overall structure of Me-pimeloyl-ACP in complex with BioH S82A. (A) The α/β core domain of BioH is colored gray, with the capping helices colored pink. Helices 2 and 3 of the capping domain are labeled. The BioH catalytic triad residues are shown in stick-ball representation with carbon atoms in cyan. The ACP molecule is colored blue with the helix 2 labeled. The phosphopantetheine-linked pimeloyl methyl ester is shown in stick-ball representation with carbon atoms colored yellow. (B) Zoomed-in stereoview of the phosphopantetheinylated pimeloyl methyl ester. Superimposed is a difference Fourier electron density map (contoured at 2.3σ over background in blue and 8.0σ over background in red) calculated with coefficients |Fobs| − |Fcalc| and phases from the final refined model with the coordinates of the ligand deleted before one round of refinement.
Fig. 3.

Ionic interactions between BioH and ACP are mediated by residues lying on the BioH capping helices and ACP helix2. Structural elements are represented and colored as in Fig. 2. (A) Ionic interaction at the amino terminus of ACP-α2 comprised of main-chain amide carbonyl oxygen and side-chain carboxylate oxygen atoms from ACP (carbon atoms colored gray), making salt bridge interactions (shown as black dashes) with BioH residues R138 and R142 (carbon atoms colored green). (B) Ionic interactions at the carboxyl terminus of ACP-α2 constitute the side-chain carboxylate oxygen atoms from aspartate and glutamate residues of ACP and the basic BioH residues R155 and R159.
Results
Evidence That BioH Acts Before BioF.
The order of action of the genes in a microbial metabolic pathway can often be deduced by cross-feeding experiments among mutant strains. However, in pimelate synthesis this approach fails because the protein-bound intermediates cannot diffuse. To counter this problem, the previously reported in vitro cell-free extract system (4) that converted malonyl-CoA to dethiobiotin (DTB) was simplified to a coupled system where four enzymes convert Me-pimeloyl-ACP to DTB. The design was to add either BioH or BioF to an in vitro system containing the substrates required for their reactions, and then remove the enzyme by use of an affinity tag (Fig. S1A). The second enzyme (either BioF or BioH) is then added with BioA and BioD and their substrates. Following further incubation, DTB synthesis is assayed. The experiment with addition of BioH or BioF in the opposite order would be done in parallel and the results of the two orders of addition compared. Assuming strict enzyme specificity addition of the enzymes in the correct order would give DTB, whereas the incorrect order of addition would not. However, we expected less than “black or white” results due to the known substrate promiscuity of BioH (8–10). The results of these experiments indicated that addition of BioH before BioF gave about twice as much DTB synthesis as the reverse order of addition in all four independent trials of the experiments, one of which is shown in Fig. S1B. Reaction 7 established that BioH cleaves the ω-methyl ester of KAPA methyl ester and that BioF accepts Me-pimeloyl-ACP in place of pimeloyl-ACP, although both off-target reactions are slower than the physiological reactions (reaction 8). Given these off-target reactions, the results we obtained were perhaps as good as might be expected. Another possible source of background would be inefficient removal of the first enzyme. To deal with this possibility, we (rather perversely) used the tag system having the highest known affinity, biotin-streptavidin. The BioH and BioC proteins were expressed as fusions to an affinity tag that becomes biotinylated by the E. coli biotin-protein ligase, BirA. The biotinylated proteins were purified on a monomeric avidin column before use. Control experiments showed that neither protein preparation contained free biotin (which would have scored as DTB; Fig. S1B). Note that the bead-conjugated streptavidin was added in large molar excess.
Given that the order-of-addition experiments gave the less-than-ideal results, we sought an independent means to test whether BioH is the gatekeeper. The promiscuity of E. coli BioH raised the question of how a nonspecific enzyme can catalyze a specific reaction. In the absence of other factors, BioH might derail biotin synthesis by hydrolyzing intermediates before Me-pimeloyl-ACP, and thereby abort the pathway. Previously we proposed that the structure of the ACP moiety of the substrate could bestow the needed specificity (4). Early work on acyl-ACPs demonstrated that the protein sequesters the first 6–8 carbon atoms of the acyl chain from solvent (11), a picture borne out by X-ray structures and molecular dynamics simulations (12–14). The protected acyl chain carbon atoms reside within a hydrophobic tunnel formed by a bundle of four helices. Hence, the bundle would largely or completely protect the ester groups of the shorter intermediates from BioH. Only upon chain elongation to the C7 species would the ester group become fully exposed to BioH such that it could enter the active site. In this scenario, the slow hydrolysis of glutaryl-ACP methyl ester would be due to the dynamic nature of ACP structure that would leave the methyl ester transiently unprotected. To test this model of the putative gatekeeper reaction, we solved the cocrystal structure of a complex of Me-pimeloyl-ACP with BioH S82A, which lacks the catalytic serine nucleophile.
BioH:Me-Pimeloyl-ACP Structure: Crystal Packing and Overall Structure.
The binary complex (both proteins were those of E. coli K-12) was crystallized and X-ray diffraction data collected to a limiting resolution of 2.05 Å (relevant data collection and refinement statistics are presented in Table S1). The crystals occupy space group P1. Despite attempts using numerous strategies, the data could not be scaled in higher symmetry settings. The crystallographic asymmetric unit consists of two monomers of BioH and two ACP molecules. The arrangement within the asymmetric unit consists of two productive complexes between the BioH holo enzyme and the ACP substrate (Fig. S2). The average thermal (B) factors are 40.8 Å2 for the BioH chains and 65.9 Å2 for the ACP chains, suggesting some degree of flexibility for the ACPs.
BioH belongs to the α/β hydrolase superfamily of enzymes. The tertiary structure consists of a core domain of a seven-member central β-sheet flanked on either side by α helices and a four-helical capping domain (K121 through T185) present at the top of the loops emanating from the central β-sheet. The canonical S82-H235-D207 catalytic triad of BioH residues lies at the interface of the core and capping domains (9) (Fig. 2). The S82 to alanine mutation places the Cβ atom of A82 side chain at a distance of 3.1 Å from the pimeloyl carboxylate carbon atom, which would be the site of nucleophilic addition (Fig. S3). The ACP molecule adopts the well-described four-helix bundle structure with a hydrophobic core described in prior studies of ACP (8–10). The second helix of the ACP helical bundle (ACP-α2) lies directly above the BioH capping-domain helices and provides most of the residues that interact with BioH. The ACP serine residue that anchors the phosphopantetheine arm, S35, is positioned at the amino terminus of the ACP-α2 helix.
Ionic Contacts Rigidly Position ACP-α2 Relative to BioH.
The contacts between BioH and the ACP molecule are mediated solely by the ACP-α2 helix and the second and third α helices of the BioH capping domain (Fig. S4). The ACP-α2 is nearly perpendicular to the BioH capping-domain helices, which in turn are antiparallel to each other. Two sites of ionic interactions are predicated on the density of negative charge on the ACP surface, which are engaged by corresponding interactions with basic amino acid side chains in BioH. The first site of interaction occurs between R138 and R142 in BioH and the main chain carbonyl of Q13 and the side chain carboxylates of D34 and D37 in ACP (Fig. 3A). Slight variability is observed in the interactions of R138 within different BioH-ACP surfaces. The second ionic interaction site engages the guanidinium side chains of R155 and R159 of BioH with the carboxylate side chains of E46 and D55, together with the main chain of I53 in ACP (Fig. 3B). These two sites lie at either end of the ACP-α2 helix, and are thus postulated to rigidly hold the ACP-α2 in the required position relative to BioH to direct the ACP-S35–conjugated phosphopantetheinylated substrate into the enzyme active site. Additional hydrophobic interactions occur with the side chains of ACP-α2 residues L36, V39, and M43, which project into a completely solvent-occluded cavity generated by BioH residues A145, L146, and M149.
Hydrophobic Interactions Direct the Substrate for Catalysis.
Clear and continuous electron density in the difference Fourier maps allows for unambiguous assignment of all atoms of the phosphopantetheine-linked pimeloyl methyl ester (Fig. 2B), which is covalently attached to Ser35 located in the amino terminus of the ACP-α2 helix. The dipole moment generated by the 14-residue ACP-α2 helix is postulated to stabilize the negative charge of the phosphate group. The phosphopantetheine arm makes minimal contacts with BioH. A water molecule, found only in one of the two complexes, interacts with an amide carbonyl oxygen of the pantetheine arm through a long hydrogen bond (3.4 Å), and is also bonded (3.3 Å) to the K121 main chain amide nitrogen. No other appropriately positioned water molecules can be identified at the interaction site. The phosphopantetheine arm is juxtaposed against a BioH surface lined with hydrophobic amino acid side chains I120, V124, F128, L146, M149, and ACP-L36 (Fig. 4 and Fig. S5A).
Fig. 4.

A hydrophobic channel directs the methyl pimeloyl moiety toward the active site for catalysis. The structural elements are colored as in Fig. 2. The phosphopantetheine-linked pimeloyl methyl ester is shown in stick-ball representation with carbon atoms colored yellow. (A) Internal cavity around the methyl-pimeloyl thioester at a distance of 4.5 Å is shown as a partially transparent surface colored green. The pantetheine sulfur atom is marked by an arrow, and the seven-carbon pimeloyl chain is traced by numerals against carbon atoms. (B) Side chains of all residues at a maximum distance of 4.5 Å from the phosphopantetheine-linked pimeloyl methyl ester are shown with their side-chain atoms in stick-ball representation with the carbon atoms colored green.
The five pimeloyl carbon chain methylenes are similarly buttressed by several hydrophobic residues of BioH. These residues construct a very narrow hydrophobic channel that acts as an approach tunnel to the enzyme active site (Fig. 4 and Fig. S5B). The hydrophobic channel begins at the site where the pantetheine sulfur atom is positioned, and terminates at the catalytic triad. All residues that form the hydrophobic channel are positioned on the capping domain helices and loops emanating from the central β-sheet of BioH.
Pimeloyl Methyl Ester Engagement in the BioH Active Site.
The quality of the electron density maps allows for the unequivocal assignment of the carbonyl oxygen and the alcohol substituents at the pimeloyl ester C7 carbon atom. The carboxylate oxygen atom of the pimeloyl moiety bearing methyl ester is stabilized by a lone interaction with the imidazole side chain of H235 (2.9 Å). The other carboxylate oxygen is stabilized by interactions with the main-chain amide nitrogen atoms of L83 (2.8 Å) and W22 (2.8 Å). These two interactions constitute the “oxyanion hole” for stabilization of the tetrahedral hemiacetal intermediate generated during the protease/esterase reaction coordinate (15) (Fig. S3).
The methyl carbon atom of the ester moiety points into a hydrophobic cage defined by the side chains of L24, W81, and F143. The hydrophobic cage for BioH likely causes the destabilization of the carboxylate negative charge for the product pimeloyl acid moiety, and thus contributes to product expulsion from the active site.
Effects of BioH Surface-Residue Substitutions on Esterase Activity in Vitro.
To test the enzymological and physiological relevance of the ionic interactions between BioH and ACP-α2 helix, we replaced these BioH residues (R138, R142, R155, and R159) with Ala and assembled a panel of single, double, triple, and quadruple mutants. We assayed the esterase activity of these BioH mutants in vitro using the cognate substrate Me-pimeloyl-ACP. The production of pimeloyl-ACP, which migrates more slowly than the substrate in a destabilizing urea-PAGE system, was monitored over 20 min (Fig. 5). The wild-type enzyme and the single Arg-to-Ala substitution mutants had comparable activities, whereas the S82A mutant was completely inactive. With these BioH proteins, production of pimeloyl-ACP could be seen in as little as 30 s, and the reaction approached completion in ∼10 min. The activities of proteins containing two Ala substitutions were significantly reduced. The earliest sign of pimeloyl-ACP occurred at 2 min, and at 20 min the reactions remained incomplete. The activities of triple and quadruple Ala substitution mutants were greatly reduced, producing significantly less pimeloyl-ACP over the same time period. This result indicated that at least three Arg residues were needed to maintain a wild-type level of activity. Given the crystal structure, interpretation of these data in terms of the binding affinity between BioH and the ACP moiety of Me-pimeloyl-ACP seems straightforward and was confirmed by an electrophoretic mobility shift assay in which binding of the highly acidic Me-pimeloyl-ACP to S82A BioH increased the mobility of BioH in nondenaturing gels (Fig. S6A). S82A derivatives of the BioH R-to-A substitution mutants were found to have decreased affinities for the substrate that paralleled the activity assays. The double, triple, and quadruple Ala substitution mutants showed no detectable binding of Me-pimeloyl-ACP, whereas the protein retaining all four Arg residues had a Kd of 3.1 μM. Finally, we assayed the ability of each of the mutant proteins to cleave 4-nitrophenyl acetate, a commonly used colorimetric esterase substrate. Each of the mutant proteins had essentially wild-type activity (2 mmol min−1⋅mg−1 BioH) on this model substrate, indicating that active site function remained intact in the mutant proteins.
Fig. 5.

Substitution of alanine for the BioH arginine residues at positions 138, 142, 155, and 156 reduced in vitro cleavage of pimeloyl-ACP methyl ester. The product is the slower-migrating pimeloyl-ACP, which can be resolved in a destabilizing urea-PAGE system (SI Materials and Methods). Activity was assayed in vitro with 200 μM Me-pimeloyl-ACP and 5 nM enzyme for 20 min. Wild-type enzyme and single Ala substitution mutant proteins demonstrated similar catalytic rates, whereas the S82A protein was completely inactive. Double mutants showed significant reductions in activity, whereas the activities of the triple and quadruple mutants were nearly abolished. Ppant, 4′-phosphopantetheine.
Effects of BioH Surface-Residue Substitutions on Biotin Synthesis.
If the interactions seen in the crystal structure are of physiological relevance, decreased in vitro activities of the BioH proteins carrying Ala substitutions should result in decreased abilities to support biotin synthesis; this was tested using an E. coli strain in which both the bioH and pcnB genes had been deleted and that also carried pMS421, a lacIq plasmid. The various BioH proteins were expressed in plasmid pET28b, a vector often used to express proteins from its LacI-controlled phage T7 promoter. In this case, however, no phage T7 RNA polymerase was present, and bioH transcription was due to use of the T7 promoter by E. coli RNA polymerase (16). The ∆pcnB mutation was included to decrease the copy number of the BioH plasmids by ∼10-fold (17). Replication of pMS421, which provided additional LacI to allow better control of BioH expression, is unaffected by the pcnB deletion. These manipulations were intended to allow detection of BioH mutants having significant, but incomplete, losses of esterase activity, which could be masked by high-level expression. Successful complementation absolutely required the catalytic S82 residue and the single R-to-A substitutions only modestly affected complementation activity (Fig. S7A), although a brief lag was observed before exponential growth ensued. This growth lag was further extended in the double mutants and became much more prominent in the triple and quadruple mutants (Fig. S7A). Because biotin is not consumed (as in most growth assays) but allows catalysis, the lag is explained by the time required to accumulate protein-bound biotin to the threshold level required for fatty acid synthesis. As growth proceeds, the levels of protein-bound biotin may fall below the threshold, but because E. coli can grow through several generations on accumulated lipids before growth cessation, the growth curve reaches stationary phase. In contrast, colony formation requires a much greater number of generations and thus provides a more stringent assay, as shown by the inability of the triple and quadruple BioH mutants to form colonies (Fig. S7B). The effects of the alanine substitutions on growth were completely reversed by addition of biotin to the medium.
Discussion
Our 2.05-Å resolution cocrystal structure of BioH S82A in complex with its Me-pimeloyl-ACP substrate revealed salt-bridge interactions between the BioH capping domain and residues positioned close to the termini of ACP α2 helix (Fig. 3 and Fig. S5). This mode of ACP binding is also seen in the P450BioI–acyl ACP complexes (13), the castor desaturase–apo ACP complex (18), and the EntE adenylation domain complex with the peptidyl carrier protein EntB (19). Binding both ends of ACP helix 2 by BioH correctly positions the catalytically tethered serine residue (which lies at the amino terminus of the helix) of the carrier protein relative to BioH, to form a catalytically productive complex.
The cocrystal structure also provided an independent means to test the fate of the esterified pimelate moiety in the biotin biosynthetic pathway, namely through the disruption of interactions that mediate complex formation between BioH and ACP. Substitution of Ala for four Arg BioH residues, designed to disrupt interactions between BioH and the ACP moiety of Me-pimeloyl-ACP, inhibited ester hydrolysis in vitro and compromised the ability of plasmids encoding the altered enzymes to complement growth of the bioH deletion strain. When two or more BioH Arg residues were replaced, the enzymes lost activity in both the in vivo and in vitro assays and were completely inactivated with three or four replacements. Indeed, the effects of the various mutations on in vitro enzymatic activity and in vivo biotin synthetic ability were strikingly similar. Moreover, electrophoretic shift assays of binding of Me-pimeloyl-ACP to BioH S82A versions of the Arg substitution proteins showed progressive loss of binding as the number of substitutions increased (Fig. S6A). Finally, all of the BioH mutant proteins retained a fully functional active site, as assayed by the ability to cleave a model esterase substrate. These data demonstrate that Me-pimeloyl-ACP is the physiological substrate of BioH, and that BioH functions as the gatekeeper in pimeloyl moiety synthesis by terminating elongation of the pimeloyl chain by the fatty acid synthetic pathway.
Though the enzyme–carrier protein interactions are conserved in BioH, BioI, and the castor desaturase, variability is found in the interactions of the substrate phosphopantetheine arm. In BioH, minimal interactions hold the phosphopantetheine arm within the active site, with one surface of the arm placed near hydrophobic residues and the other side of the arm being solvent-exposed (Fig. S5A). In contrast, extensive water-mediated contacts were observed for the phosphopantetheine arm in the P450BioI-acyl ACP crystal structures (13) (in the castor desaturase-apo ACP crystal structure, the phosphopantetheine arm is truncated due to lack of electron density) (18). Although the complete functional implications for the lack of direct or water-mediated interactions between BioH and the phosphopantetheine arm of acyl-ACP are not clear, it provides the basis for postulating that efficient BioH hydrolysis of the Me-pimeloyl moiety requires tethering to an ACP molecule.
The structure reported argues that substrate specificity for ACP-bound substrates has been engineered into BioH, such that only a pimeloyl methyl ester chain can be accepted. Although BioH does indeed accept glutaryl (C5), adipyl (C6), suberyl (C8), and azelayl (C9) methyl esters conjugated to ACP as substrates in addition to the physiological substrate pimeloyl (C7) methyl ester, the hydrolysis of these unnatural substrates was slower than that of the pimeloyl substrate (Fig. S6B). An inspection of the BioH hydrophobic substrate channel, and the manner in which the phosphopantetheinylated fatty acid is directed into the enzyme active site, provides a rationale for these observations. The length of the hydrophobic channel cavity is organized to allow the pimeloyl moiety to efficiently reach the BioH catalytic triad, whereas the shorter glutaryl chain has difficulty in traversing this cavity, and longer chains would have steric clashes with the side chains of the BioH hydrophobic cavity, which would mitigate binding. The C6 (adipyl) species was a fair BioH substrate (Fig. S6B) but is not germane to the pathway. The C5 substrate bound BioH (Fig. S6A), but catalysis was slow (Fig. S6B), and the C9 substrate bound BioH very poorly (Fig. S6A). Hence, the dimensions of this hydrophobic cavity generate the specificity for BioH. This model for BioH specificity is supported by the structure-based primary sequence alignment of the BioH proteins of diverse bacteria (Fig. S8), which shows a strict conservation of the hydrophobic residues implicated in generating the hydrophobic tunnel and for hydrophobic interactions with the pantetheine arm. The known ability of BioH to cleave short-chain (but not long-chain) 4-nitrophenol esters (8, 9) and the methyl ester of dimethylbutyryl-S-methyl mercaptopropionate (10) are consistent with the hydrophobicity and narrow aperture of the BioH channel, although accommodation of the bulky 4-nitrophenol group argues that the bottom of the channel can expand. Expansion would also explain the observed hydrolysis of the ethyl, propyl, and butyl esters of pimeloyl-ACP (4).
The complex of BioH with Me-pimeloyl-ACP is formed without significant structural changes in either the enzyme or the ACP moiety. Indeed, it seems that the only structural change required is partition of the acyl chain from the ACP hydrophobic core into the hydrophobic active site channel of BioH (vide infra). Following completion of the reaction, the newly formed carboxyl group would cause expulsion of the pimeloyl-pantetheine moiety from the BioH hydrophobic channel and return of the methylene chain to the ACP hydrophobic core with the carboxyl group protruding from the helical bundle and exposed to solvent.
We had previously postulated that BioC, the SAM-dependent methyl transferase, which methylates malonyl-ACP (or malonyl-CoA) to shunt a small portion of the flux of the fatty acid synthetic pathway toward biotin synthesis (Fig. 1), may be an intrinsically poor catalyst (4). This lack of catalytic efficiency is also characteristic of the downstream enzymes of the pathway (20–23), and has been attributed to the traces of biotin required for cellular growth (24). To verify this premise, we sought to determine the kinetic constants for the esterase reaction mediated by BioH and attempted several in vitro assays, all of which were unsuccessful. However, an estimate for the maximal catalytic rate for the enzyme can be obtained from the gel-shift assay (Fig. 5). Wild-type BioH routinely hydrolyzed a 40,000-fold molar excess of Me-pimeloyl-ACP in less than 10 min. Assuming the substrate/enzyme molar ratio used in our in vitro experiments to be saturating for the enzyme, the kcat value for the ester hydrolysis by BioH can be minimally estimated to be 60 s−1. This value is orders of magnitude greater than those reported for the downstream enzymes of the pathway (20–23). This mismatch of BioH catalytic activity with the other enzymes of the pathway and with the physiological need for only extremely modest amounts of biotin may indicate that the enzyme has not yet been fully integrated into E. coli biotin synthesis. Other indications are that the E. coli bioH gene is not located within bio operon or regulated by BirA, the repressor that controls expression of the other bio genes. Catalytically relevant amino acids identified by the crystal structure presented here are found to be highly conserved between BioH sequences that lie outside or within the bio operon under BirA control (Fig. S8). Hence the mode of substrate engagement, and the model proposed for the generation of substrate specificity within the enzyme active site, seems to be independent of genetic positioning of the bioH gene.
Materials and Methods
Bacterial Media, Strains, Plasmids, and Oligonucleotides.
Protein Purifications and Substrate Synthesis.
See SI Materials and Methods for purification.
Crystallization, Phasing, and Structure Determination.
The complex of Me-pimeloyl-ACP with BioH S82A was crystallized by sparse matrix screening, followed by optimization of screening hits. See SI Materials and Methods for detailed procedures. The crystal structure of the complex of Me-pimeloyl-ACP with BioH S82A was determined by molecular replacement using apo wild-type BioH and E. coli ACP as the search models. See SI Materials and Methods for detailed procedures.
Assays.
BioH proteins were assayed for their carboxylesterase activity on Me-pimeloyl-ACP. Hydrolysis of the ester bond produces a slower-migrating pimeloyl-ACP that can be resolved by the conformationally sensitive electrophoretic mobility-shift assay described previously (4). See SI Materials and Methods for detailed procedures. See SI Materials and Methods for the in vivo complementation, BioH-ACP substrate interaction, and 4-nitrophenyl acetate hydrolysis assays.
Supplementary Material
Acknowledgments
We thank Drs. Keith Brister and Joseph Brunzelle, and the staff at Life Sciences Collaborative Access Team at Argonne National Laboratory for facilitating data collection. This work was supported by National Institutes of Health Grant AI15650 from the National Institute of Allergy and Infectious Diseases (to J.E.C.) and the National Institute of General Medical Sciences (S.K.N.).
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission. C.K. is a guest editor invited by the Editorial Board.
Data deposition: Atomic coordinates for the BioH S82A in complex with Me-pimeloyl-ACP have been deposited in the Protein Data Bank, www.pdb.org (PDB ID code 4ETW).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1207028109/-/DCSupplemental.
References
- 1.Ifuku O, et al. Origin of carbon atoms of biotin. 13C-NMR studies on biotin biosynthesis in Escherichia coli. Eur J Biochem. 1994;220:585–591. doi: 10.1111/j.1432-1033.1994.tb18659.x. [DOI] [PubMed] [Google Scholar]
- 2.Sanyal I, Lee S, Flint DH. Biosynthesis of pimeloyl-CoA, a biotin precursor in Escherichia coli, follows a modified fatty acid synthesis pathway: 13C-labeling studies. J Am Chem Soc. 1994;116:2637–2638. [Google Scholar]
- 3.White SW, Zheng J, Zhang YM, Rock The structural biology of type II fatty acid biosynthesis. Annu Rev Biochem. 2005;74:791–831. doi: 10.1146/annurev.biochem.74.082803.133524. [DOI] [PubMed] [Google Scholar]
- 4.Lin S, Hanson RE, Cronan JE. Biotin synthesis begins by hijacking the fatty acid synthetic pathway. Nat Chem Biol. 2010;6:682–688. doi: 10.1038/nchembio.420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Barker DF, Campbell AM. Use of bio-lac fusion strains to study regulation of biotin biosynthesis in Escherichia coli. J Bacteriol. 1980;143:789–800. doi: 10.1128/jb.143.2.789-800.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Koga N, Kishimoto J, Haze S, Ifuku O. Analysis of the bioH gene of Escherichia coli and its effect on biotin productivity. J Ferment Bioeng. 1996;81:482–487. [Google Scholar]
- 7.Rodionov DA, Mironov AA, Gelfand MS. Conservation of the biotin regulon and the BirA regulatory signal in Eubacteria and Archaea. Genome Res. 2002;12:1507–1516. doi: 10.1101/gr.314502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kwon MA, Kim HS, Oh JY, Song BK, Song JK. Gene cloning, expression, and characterization of a new carboxylesterase from Serratia sp. SES-01: Comparison with Escherichia coli BioHe enzyme. J Microbiol Biotechnol. 2009;19:147–154. doi: 10.4014/jmb.0804.287. [DOI] [PubMed] [Google Scholar]
- 9.Sanishvili R, et al. Integrating structure, bioinformatics, and enzymology to discover function: BioH, a new carboxylesterase from Escherichia coli. J Biol Chem. 2003;278:26039–26045. doi: 10.1074/jbc.M303867200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Xie X, Wong WW, Tang Y. Improving simvastatin bioconversion in Escherichia coli by deletion of bioH. Metab Eng. 2007;9:379–386. doi: 10.1016/j.ymben.2007.05.006. [DOI] [PubMed] [Google Scholar]
- 11.Cronan JE., Jr Molecular properties of short chain acyl thioesters of acyl carrier protein. J Biol Chem. 1982;257:5013–5017. [PubMed] [Google Scholar]
- 12.Chan DI, Stockner T, Tieleman DP, Vogel HJ. Molecular dynamics simulations of the Apo-, Holo-, and acyl-forms of Escherichia coli acyl carrier protein. J Biol Chem. 2008;283:33620–33629. doi: 10.1074/jbc.M805323200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cryle MJ, Schlichting I. Structural insights from a P450 carrier Pprotein complex reveal how specificity is achieved in the P450(BioI) ACP complex. Proc Natl Acad Sci USA. 2008;105:15696–15701. doi: 10.1073/pnas.0805983105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Roujeinikova A, et al. Structural studies of fatty acyl-(acyl carrier protein) thioesters reveal a hydrophobic binding cavity that can expand to fit longer substrates. J Mol Biol. 2007;365:135–145. doi: 10.1016/j.jmb.2006.09.049. [DOI] [PubMed] [Google Scholar]
- 15.Jaeger KE, Dijkstra BW, Reetz MT. Bacterial biocatalysts: Molecular biology, three-dimensional structures, and biotechnological applications of lipases. Annu Rev Microbiol. 1999;53:315–351. doi: 10.1146/annurev.micro.53.1.315. [DOI] [PubMed] [Google Scholar]
- 16.Somerville RL, Shieh TL, Hagewood B, Cui JS. Gene expression from multicopy T7 promoter vectors proceeds at single copy rates in the absence of T7 RNA polymerase. Biochem Biophys Res Commun. 1991;181:1056–1062. doi: 10.1016/0006-291x(91)92044-k. [DOI] [PubMed] [Google Scholar]
- 17.Masters M, et al. The pcnB gene of Escherichia coli, which is required for ColE1 copy number maintenance, is dispensable. J Bacteriol. 1993;175:4405–4413. doi: 10.1128/jb.175.14.4405-4413.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Guy JE, et al. Remote control of regioselectivity in acyl-acyl carrier protein-desaturases. Proc Natl Acad Sci USA. 2011;108:16594–16599. doi: 10.1073/pnas.1110221108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sundlov JA, Shi C, Wilson DJ, Aldrich CC, Gulick AM. Structural and functional investigation of the intermolecular interaction between NRPS adenylation and carrier protein domains. Chem Biol. 2012;19:188–198. doi: 10.1016/j.chembiol.2011.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Alexeev D, Baxter RL, Smekal O, Sawyer L. Substrate binding and carboxylation by dethiobiotin synthetase—a kinetic and X-ray study. Structure. 1995;3:1207–1215. doi: 10.1016/s0969-2126(01)00256-8. [DOI] [PubMed] [Google Scholar]
- 21.Eliot AC, Sandmark J, Schneider G, Kirsch JF. The dual-specific active site of 7,8-diaminopelargonic acid synthase and the effect of the R391A mutation. Biochemistry. 2002;41:12582–12589. doi: 10.1021/bi026339a. [DOI] [PubMed] [Google Scholar]
- 22.Farrar CE, Siu KK, Howell PL, Jarrett JT. Biotin synthase exhibits burst kinetics and multiple turnovers in the absence of inhibition by products and product-related biomolecules. Biochemistry. 2010;49:9985–9996. doi: 10.1021/bi101023c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Webster SP, et al. Mechanism of 8-amino-7-oxononanoate synthase: Spectroscopic, kinetic, and crystallographic studies. Biochemistry. 2000;39:516–528. doi: 10.1021/bi991620j. [DOI] [PubMed] [Google Scholar]
- 24.Cronan JE., Jr Interchangeable enzyme modules. Functional replacement of the essential linker of the biotinylated subunit of acetyl-CoA carboxylase with a linker from the lipoylated subunit of pyruvate dehydrogenase. J Biol Chem. 2002;277:22520–22527. doi: 10.1074/jbc.M201249200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.