

Abstract
Electron transfer is the simplest chemical reaction and constitutes the basis of a large variety of biological processes, such as photosynthesis and cellular respiration. Nature has evolved specific proteins and cofactors for these functions. The mechanisms optimizing biological electron transfer have been matter of intense debate, such as the role of the protein milieu between donor and acceptor sites. Here we propose a mechanism regulating long-range electron transfer in proteins. Specifically, we report a spectroscopic, electrochemical, and theoretical study on WT and single-mutant CuA redox centers from Thermus thermophilus, which shows that thermal fluctuations may populate two alternative ground-state electronic wave functions optimized for electron entry and exit, respectively, through two different and nearly perpendicular pathways. These findings suggest a unique role for alternative or “invisible” electronic ground states in directional electron transfer. Moreover, it is shown that this energy gap and, therefore, the equilibrium between ground states can be fine-tuned by minor perturbations, suggesting alternative ways through which protein–protein interactions and membrane potential may optimize and regulate electron–proton energy transduction.
Keywords: cytochrome oxidase, invisible states, paramagnetic proteins, NMR, spectroscopy
Long-range electron transfer (ET) reactions lie at the heart of bioenergetic pathways in all living organisms (1). Nature has evolved specialized proteins and cofactors performing ET with high efficiency over distances as long as 20 Å (2). The role of the protein milieu between the donor and acceptor sites and its fine-tuning has been matter of intense debate in the past two decades (3). Marcus theory predicts that, in the nonadiabatic regime, ET rates are determined by the product of two terms: a Franck–Condon factor that accounts for the ability of thermal fluctuations to induce energetic degeneracy of donor and acceptor, and the electronic matrix element or coupling 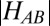 that represents the probability of electron tunneling between the degenerate states. Most structure-based ET coupling theories include parameters describing details of the donor and acceptor state electronic structure (4, 5). However, information needed to assess these parameters is unknown in most cases, and treatments often assume only one donor (and acceptor) electronic wave function involved, i.e., the so called redox-active molecular orbital, thereby emphasizing the role of the bridge. It has been shown that structural fluctuations of the bridge between donor and acceptor modulate
that represents the probability of electron tunneling between the degenerate states. Most structure-based ET coupling theories include parameters describing details of the donor and acceptor state electronic structure (4, 5). However, information needed to assess these parameters is unknown in most cases, and treatments often assume only one donor (and acceptor) electronic wave function involved, i.e., the so called redox-active molecular orbital, thereby emphasizing the role of the bridge. It has been shown that structural fluctuations of the bridge between donor and acceptor modulate 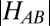 (6, 7), leading to different regimes depending on the time scale of the fluctuations with respect to the lifetime of the resonant donor-acceptor state. The structural features of the protein (and water) bridging donor and acceptor redox centers define the tunneling rates, even for large coupling fluctuations (8, 9). Prytkova et al. (10) have considered thermal averages over multiple ligand field states of Fe in heme proteins.
(6, 7), leading to different regimes depending on the time scale of the fluctuations with respect to the lifetime of the resonant donor-acceptor state. The structural features of the protein (and water) bridging donor and acceptor redox centers define the tunneling rates, even for large coupling fluctuations (8, 9). Prytkova et al. (10) have considered thermal averages over multiple ligand field states of Fe in heme proteins.
Here we report an example of a biological redox metal center (CuA) in which thermal fluctuations may populate two alternative ground-state electronic wave functions optimized for electron entry and exit, respectively, through two different and nearly perpendicular pathways. Moreover, we propose that biologically relevant perturbations may shift the equilibrium between both ground states, thus contributing to the directionality of ET reactions in CuA-containing enzymes.
CuA is a binuclear copper site that acts as an optimized hub for long-range ET in terminal oxidases and in N2O reductases (11). The two copper ions are bridged by two cysteine ligands, forming a nearly planar Cu2S2 diamond core characterized by a short Cu—Cu distance (2.4 Å; Fig. 1A). The coordination sphere of the metal site is completed by two terminal histidine residues and two weakly coordinated axial ligands provided by a methionine sulfur and a backbone carbonyl (Fig. 1A). Electrons shuttled by a soluble cytochrome c (Cyt) are delivered to the CuA site in subunit II of the oxidase and from there to the catalytic site embedded in subunit I, where O2 is reduced to water. These steps involve two long, nearly perpendicular pathways through the protein milieu. Despite the low driving forces, ET takes place with high rates along these two pathways (12–14). This efficiency has been attributed to the unique coordination features of the CuA site (Fig. 1A), which were suggested to yield both a low reorganization free energy (λ) and an electronic structure that enhances superexchange coupling by means of a high Cu—SCys covalency (15). In its oxidized form, CuA is a fully delocalized mixed-valence species (Cu1.5+Cu1.5+) whose electronic structure can be described by a double-well potential energy surface as a function of the Cu—Cu distance, with two ground states, a σu* and a πu level, with minima at ∼2.4 and ∼2.9 Å, respectively (Fig. 1B) (16, 17). NMR studies have revealed a fast interconversion between the σu* and πu ground states (18–20). However, the σu* ground state is considered as the only redox-active orbital in ET, based on (i) the low population of the πu ground state at physiological temperature, and (ii) the higher λ value predicted for the πu ground state, making its involvement in ET unlikely (16, 17).
Fig. 1.

(A) The CuA site from Thermus thermophilus ba3 oxidase (rendered from PDB ID 2CUA) (35). (B) Double-well potential describing the ground-state symmetry as a function of the Cu—Cu distance, including the calculated σ*u (blue) and πu (red) molecular orbitals. The large splitting between both states yields an adiabatic surface along the coordinate.
We recently reported that engineering of the weak axial ligand at position 160 of the CuA site of Thermus thermophilus (CuA hereafter) results in minor perturbations that tune the reduction potential (21) of the metal site. Here we show that these perturbations can also shift the fluctuating equilibrium between the σu* and πu ground states in the CuA site, favoring the population of the πu ground state, and, that far from being detrimental to the ET reaction, a large increase in the superexchange coupling through an alternative pathway compensates the larger λ measured for this ground state. Thus, we propose that thermally accessible ground states of different wave function symmetry can be switched, allowing for efficient directional long-range inter- and intraprotein ET with minor changes in the nuclear configuration at a low energy cost.
Results and Discussion
Effect of Axial Mutations on the Geometric and Electronic Structure of CuA Sites.
X-ray absorption spectra (XAS) were collected on samples of WT CuA and the M160H and M160Q mutants. Data analysis reveals that the metal–metal and metal–ligand distances in the Cu2S2 core are largely unaltered by the mutations (SI Appendix, Table S1 and Fig. S1). In addition, the structural differences between the oxidized and reduced forms are subtle in all cases. Addition of a S(Met) scatterer at 2.9 Å in the WT variants was consistent with the data, but did not improve significantly the fit. Instead, inclusion of an additional O/N scatterer definitely improved the fit for the mutants’ spectra, suggesting that the engineered axial ligands bind the copper center. Thus, introduction of axial ligands able to bind the copper ion do not give rise to dramatic structural changes in the Cu2S2 core.
The absorption and CD spectra of oxidized CuA centers are dominated by two intense SCys→Cu charge transfer (CT) bands at 21,270 and 18,700 cm−1, and NHis→Cu CT transitions at 28,000 and 25,800 cm−1 (Fig. 2B; SI Appendix, Fig. S4 and Table S3) (22). The low-energy feature at 12,650 cm−1 is an intervalence transition (ψ→ψ*) whose energy reflects the electronic coupling between the two copper ions, being sensitive to the ground-state identity (23). The absorption spectrum of M160Q closely resembles that of the WT protein, with a ψ→ψ* band at 12,750 cm−1, consistent with a σu* ground state for both protein variants (Fig. 2 B and C). The electronic spectrum of the M160H mutant, instead, presents some perturbations (Fig. 2B; SI Appendix, Fig. S4). To aid in the interpretation, we performed quantum mechanics (QM) calculations at the time-dependent density functional theory (TDDFT) level on this mutant, obtaining the spectra corresponding to both the σu* and πu ground state, shown in Fig. 2. The calculations show that the experimental UV-Vis spectral features result from contributions of both ground states. Specifically, for the πu ground state, the SCys→Cu CT bands blue-shift to ∼28,000 cm−1, partially overlapping with the NHis→Cu CT transitions, whereas the intervalence band appears at 12,100 cm−1, reflecting a decrease in the Cu—Cu interaction with respect to the WT protein. Thus, we can conclude that the perturbation of the weak axial ligand shifts the equilibrium toward the πu ground state, favoring its thermal accessibility (Fig. 2C; SI Appendix, Fig. S5). Resonance Raman (RR) spectra of the WT, M160Q, and M160H proteins recorded by exciting at 514 nm (19,455 cm−1) are almost identical (SI Appendix, Fig. S6 and Table S4), despite the fact that the absorption spectrum of M160H differs significantly from those of WT and M160Q. This finding is consistent with our QM calculations, which reveal that CuA sites with πu ground state do not present any significant absorption at the excitation wavelength and, thus, the similarity of the RR spectra reflects the structural identity of the three proteins in the σu* ground state. Coincidentally, the structural similarities between all studied species observed by extended X-ray absorption fine structure (EXAFS) is due to the accessibility of only the σu* state at the cryogenic temperatures used for these experiments (SI Appendix, Fig. S13). The electronic spectrum of M160H at room temperature is therefore consistent with the binding of His160 (as detected by XAS), with a larger impact in the electronic structure of the site compared with the M160Q mutation.
Fig. 2.

(A) 13C NMR spectra highlighting resonances from the cysteine residues in WT CuA and the M160H and M160Q mutants. (B) Experimental absorption spectra for WT CuA and the M160H and M160Q mutants. (C) Simulated spectra for σ*u and πu ground states on the WT geometry.
The electron spin density on 1H, 15N, and 13C nuclei on the metal ligands was determined by NMR spectroscopy on the oxidized, paramagnetic forms of the WT protein and the M160Q and M160H mutants. The high Cu—SCys covalency is preserved in all cases, as evidenced by the position of the β13C Cys resonances (Fig. 2A), which are sensitive probes of the unpaired spin density on the Cys S γ atoms. The NMR spectra of M160Q reveal specific perturbations in the signals from His114 (bound to the same copper ion as M160) and little effect on His157. On the contrary, in M160H the signals from both His114 and His157 are perturbed, with a reduced spin density in their imidazole rings. In this mutant, we identified a third spin system corresponding to an imidazole moiety with net unpaired spin density (SI Appendix, Figs. S2 and S3), which we attribute to His160. We conclude that His160 is tightly bound to the copper ion (supporting EXAFS data), and we attribute the unpaired electron spin density on His160 to the presence of dz2 character in the molecular orbital containing the unpaired electron. This phenomenon is unique for M160H, for the axial ligands in the WT protein (Met) and in M160Q (Gln) do not exhibit any unpaired spin density.
NMR allows to quantitate the populations of the σu* and πu ground states at room temperature. The chemical shifts of the β-1H of Cys153, and the α-13C from Cys149 are very sensitive to the σu*–πu energy gap because they present dramatic differences in their electron spin densities in the two possible ground states. These two nuclei display S-S-Cβ-Hβ /Cα dihedral angles of nearly 90°, resulting in minimal spin density delocalization in the σu* state, and a large spin delocalization in the πu ground state (Fig. 2A). These resonances show large differences between the studied proteins (Fig. 2; SI Appendix, Fig. S2). Analysis of the temperature dependences of their chemical shifts based on a Boltzmann distribution in a two-state model allowed us to determine the σu*–πu energy gaps of the WT, M160H, and M160Q proteins at 600, 200, and 900 cm−1, respectively (Table 1). These values imply that, at 25 °C, the population of the πu state is ∼30% in M160H and 1% in M160Q, compared with 5% for the WT protein. These results agree with the experimental and QM calculated absorption spectra, which indicate that the πu ground state is more accessible in M160H (Fig. 2B). Consistently, the sharper NMR lines in M160H can be accounted for by a more-accessible πu level, whereas the opposite holds for M160Q (Fig. 2).The electron paramagnetic resonance spectra of the different variants further confirm this trend (21). The g// values reflect the spin-orbit coupling of the σu* level with the πu excited state at the equilibrium geometry of the σu* ground state (∼2.5 Å; Fig. 1) and follow the trend M160Q > WT > M160H, consistent with the stabilization of the πu level provided by the axial His (Table 1). These data clearly demonstrate that the axial ligand tunes the energy gap between the σu* and the πu levels, and that the latter can be significantly populated upon this minor structural perturbation.
Table 1.
Summary of experimental and theoretical results for WT, M160Q, and M160H
| CuA variant | E(GSπu)–E(GSσu*), cm−1† | Populations at 25 °C, σu*:πu | E(ESπu)–E(GSσu*), cm−1‡ | Eψ→ψ*, cm−1§ | ε, mV¶ | kET, s−1|| | λexp, eV†† | λcalc, eV‡‡ | ΔG# = λexp/4, cm−1 | |
| σu* | πu | |||||||||
| M160H | 200 | 72:28 | 3,970 | 6,050 | 148 | 0.81 | 0.63 | 0.26 | 0.48 | 1,270 |
| WT | 600 | 95:5 | 4,690 | 6,325 | 293 | 2.03 | 0.46 | 0.32 | 0.63 | 928 |
| M160Q | 900 | 99:1 | 4,810 | 6,375 | 158 | 1.65 | 0.45 | 0.41 | 0.57 | 907 |
†Calculated from fits of the temperature dependences of the NMR signals. GS, ground state.
‡Calculated from the g// values reported by Ledesma et al. (21) as done by Gorelsky et al. (16). ES, excited state
§Calculated as half the energy of the intervalence band.
¶Reported by Ledesma et al. (21).
||Determined from electrochemical measurements (this work).
††Determined from the temperature dependences of kET.
‡‡Obtained from QM calculations.
Population of the πu Ground State May Be Achieved by Different Small Structural Fluctuations.
Mixed-valence model complexes reported in the literature are characterized by a πu ground state and a Cu—Cu distance of 2.9 Å, which is reflected in a red-shift of the intervalence band to 5,560 cm−1. This picture differs greatly from the one reported here, because the calculated and experimental position of the intervalence band for M160H exhibit much smaller red-shifts. The lack of evidence in favor of a long Cu—Cu distance prompted us to explore, through QM calculations, other reaction coordinates able to yield a πu ground state. We found that the σu*–πu energy gap is very sensitive to changes in the His114/His157 dihedral angle (with the highest occupied molecular orbital symmetry changing at an angle of ∼25°; SI Appendix, Fig. S10) and to deformations that disrupt the planarity of the Cu2S2 diamond core (SI Appendix, Fig. S11). None of these distortions is associated with a significant lengthening of the Cu—Cu distance, as depicted in Fig. 1. Moreover, the application of electric fields of biologically relevant magnitudes, which may act as an external perturbation, results in increased σu*–πu energy gaps along the Cu—Cu coordinate (SI Appendix, Fig. S12). We conclude that the alternative πu ground state may be populated through different reaction coordinates, all requiring approximately the same (low) energy and involving relatively minor geometric fluctuations that do not necessarily imply such a long Cu—Cu distance. Moreover, the energy gap (and the relative populations of each ground state) might be affected by perturbations such as mutations of the weak axial ligand and the presence of a biologically relevant electric field.
Protein Film Voltammetry.
The λ values of the WT CuA site and mutants were determined by cyclic voltammetry (CV) of protein samples adsorbed on biomimetic electrodes. Two types of independent experiments and data treatments were performed. In one case, CVs were recorded at constant temperature as a function of the scan rate, and λ values were obtained from simulations of the voltammograms in terms of the Marcus equation integrated to account for the density of states of the metal electrode (24). The second type of experiment consisted of CVs recorded as a function of temperature in a nonisothermal cell at variable scan rates. Rate constants in the latter case were obtained using Laviron’s formalism (24, 25), and λ was estimated from Arrhenius plots in a temperature range (15–40 °C) that, as shown in SI Appendix, Fig. S13, does not affect the relative population of both ground states. Despite the different uncertainties and sources of errors, both approaches yielded nearly identical results, with λ values of 0.46 (WT), 0.45 (M160Q), and 0.63 eV (M160H) (Table 1; SI Appendix, Figs. S7 and S8). The measured λ values for M160Q and WT CuA are expected to correspond to the σu* ground state, largely predominant in both variants in the temperature range of the experiments. This conclusion is confirmed by our QM calculations, which for WT CuA in σu* and πu ground-state configurations yield inner sphere λ values (λin) of 0.32 and 0.63 eV, respectively (Table 1). In contrast, the population of the πu ground state is 28% in the M160H mutant, implying that two species should be electrochemically distinguishable if both ground states were redox-active. However, only a single redox couple is detected over a large range of scan rates (10–10,000 mV⋅s−1) and temperatures, indicating that only one of the ground states is active in this mutant. The QM-calculated λin values for M160H are 0.26 eV and 0.48 eV for the σu* and πu ground states, respectively, suggesting that the measured λ value (0.63 eV) corresponds to the πu ground state. The similarity between calculated λin and experimental λ values and their sensitivity to the nature of the ground state suggests that in all cases, λout represents a small contribution.
The σu*–πu energy gaps for WT and M160Q are within the same range of the activation free energy (ΔG# ∼ λ/4) for ET of these variants, i.e., 0.12 eV (900 cm−1). Given that at room temperature the population of the σu* ground state is ≥95% in both cases, we conclude that the σu* state is the electrochemically detected species for WT and M160Q. Instead, for M160H ΔG# (0.16 eV) is 6.5× larger than the σu*–πu energy gap. This observation suggests that the crossing point between reactants and products can only be achieved for the πu state along the adiabatic surface, thereby supporting the conclusion that the πu state is the only redox-active species in this mutant (SI Appendix, Fig. S9 and accompanying text).
Of great significance, the measured ET rate for M160H is only 2.5-fold lower than in WT, thus revealing that the larger λ value (which would imply a sevenfold drop in kET), is compensated by an increase in |HAB|, as a consequence of populating the alternative πu ground state (SI Appendix, Fig. S7).
Interplay Between Electronic Coupling and Reorganization Free Energy.
The current picture for ET in terminal oxidases assumes that the protein framework forces the CuA site into the σu* ground state to avoid a detrimental increase of λ. However, here we have shown that the population of the πu state can be achieved through minor structural perturbations, and that this transition exerts a low impact on the value of λ. Previous pathways calculations considering only the σu* ground state weighted by metal–ligand covalencies suggested that the Cys ligands act as the electron access point (26). However, our pathways analysis in the ET complex between cytochrome c552 (Cyt c552) and the CuA domain of T. thermophilus (13) reveals that the axial Met160 ligand provides the favored electron entry point to the CuA site (SI Appendix, Tables S6 and S7). Even though the metal–ligand covalency is very low for the axial Cu—SMet bond in the σu* ground state (Fig. 3), our QM calculations predict a 10-fold increase in the πu ground state (SI Appendix, Table S8), which is supported by the finding of net electron spin density in the axial His ligand in M160H due to population of the πu level. Thus, a πu ground state provides an efficient electron entry pathway by means of an increased superexchange coupling along the Cu—SMet bond (Fig. 3). This effect results in an enhancement in the ET rate of up to two orders of magnitude, overriding the increase in λ when going from a σu* to a πu ground state. Met160 is not only in the preferred entry pathway, but it is also located next to the hydrophobic patch where Cyt c552 binds the CuA domain (Fig. 3) (13).
Fig. 3.

(Upper) Optimal ET pathway (red) from cytochrome c to CuA in the πu state, and (Lower) from CuA in the σu* state to the ba3 group of the oxidase. Rendered from the structure of T. thermophilus ba3 oxidase (PDB ID code 1XME) (41) and the model complex between the CuA domain and cytochrome c552 (PDB ID code 2FWL) (13).
The most efficient pathways for subsequent ET from reduced CuA to heme b involve His157 as the electron exit point. Our QM calculations on the reduced CuA site reveal that the covalency of the Cu—N(His157) bond is doubled in the σu* state compared with the πu state, suggesting that ET to heme b is favored through the σu* orbital. Thus, optimal interprotein ET from Cyt c552 to CuA involves a πu ground state as redox-active orbital, whereas subsequent ET from CuA is optimized for the σu* ground state of the reduced CuA (Fig. 3).
Implications for Biological ET.
Based on the present results, we propose that the availability of two alternative redox-active ground states enables efficient directional ET through the CuA site, which in the σu* and πu states is able to couple effectively with “upstream” and “downstream” redox partners, respectively. Although it cannot be completely ruled out for the in vivo process, such a pathway-switching mechanism does not necessarily imply a diode-like charge flow. However, the efficiency and directionality of the process depends on the σu*–πu equilibrium, which in turn might be regulated by minor perturbations. Specifically, transient binding of Cyt to CcO might induce structural perturbations populating the πu state, thus turning its associated pathway on. After the ET reaction takes place, the reduced state does not exhibit an energy minimum of πu symmetry, so the system relaxes toward the σu* ground state, which is optimized for subsequent intramolecular ET reactions. In line with this proposal, a recent study has provided evidence of a direct vibrational coupling between an ET metal site and the protein surface, suggesting that formation of a transient interprotein complex could be transmitted to the metal site (27). Moreover, transient binding of redox protein partners is known to modify the reduction potential of donor and acceptor sites (28) and to induce geometrical distortions of copper redox centers (29, 30). In fact, the study of the Cyt552—CuA complex reveals subtle structural perturbations at the CuA site in a redox state-dependent fashion (13). In addition, the prediction that moderate electric fields alter the barrier for σu*–πu interchange suggests that the local membrane potential could exert regulation on the electroprotonic energy transduction, as previously proposed based on different evidence (31, 32). Of utmost importance, however, even in the absence of external perturbations, the enhanced superexchange coupling of the πu state renders it suitable for participating in the ET reaction, even if poorly populated.
In summary, here we show that population of alternative electronic states can be favored by subtle perturbations, and suggest that their different covalencies could allow for directional ET. The involvement of transiently and lowly populated, or “invisible,” conformations has been recently recognized crucial for the function of a number of proteins (33, 34). In this respect, the finding that minor perturbations may favor the population of the otherwise invisible (but redox-active) πu state of CuA reveals a previously unidentified role for alternative electronic ground states in directional ET.
Materials and Methods
Protein Preparation.
Samples of WT and mutant CuA-soluble fragments from subunit II of the cytochrome ba3 from T. thermophilus were produced as described previously (18) and stored in 100 mM phosphate buffer (pH 6.0) with 100 mM KCl.
Resonance Raman.
RR spectra were recorded with 514 nm excitation (13 mW; Coherent Innova 70c) in backscattering geometry using a confocal microscope coupled to a single-stage spectrograph (JobinYvon XY 800) equipped with a CCD detector. Protein samples (1 mM) were placed into a rotating quartz cell for the measurements.
Electrochemical Determinations.
Cyclic voltammetry was performed with a Gamry REF600 potentiostat in a nonisothermal cell equipped with a polycrystalline gold bead working electrode, a Pt wire auxiliary electrode, and a Ag/AgCl reference electrode. Modification of the Au electrodes and measurement conditions are described in SI Appendix.
NMR Spectroscopy.
NMR experiments were carried out on a Bruker Avance II Spectrometer (600.13 MHz 1H frequency) as described in SI Appendix and in our previous work on WT CuA (18).
X-Ray Absorption Spectroscopy.
Cu K-edge (8.9 keV) EXAFS and X-ray absorption near-edge structure data were collected at the Stanford Synchrotron Radiation Laboratory operating at 3 GeV with currents between 100 and 80 mA on beam line 9-3. See SI Appendix for further details.
Optical Spectroscopy.
Absorption and circular dichroism spectra were acquired with Jasco V-670 and J-810 equipment, respectively, using thermostatized chambers and quartz cells.
Computational Methods.
Models were built from PDB ID code 2CUA (35) by replacing Met160 to generate the mutant structures, which were relaxed with AMBER 11 (36). QM and QM/molecular mechanics calculations were performed with Gaussian09 (37) and SIESTA/Hybrid (38, 39). Donor–acceptor electronic couplings were computed with the Pathways (40) plug-in for the Visual Molecular Dynamics program. See SI Appendix for further details.
Supplementary Material
Acknowledgments
We are grateful for the financial support from Agencia Nacional de Promoción Científica y Tecnológica (ANPCyT) Grants PICT 2007-314 and 2010-070; Universidad de Buenos Aires Grant 20020090100094; and National Institutes of Health Grant R01 GM054803 (to N.J.B.). This research was done using resources provided by the Open Science Grid, which is supported by the National Science Foundation and the US Department of Energy's Office of Science. A.J.V. is a Howard Hughes Medical Institute International Research Scholar; L.A.A. and D.Á.-P received fellowships from the Consejo Nacional de Investigaciones Científicas y Técnicas (CONICET); and G.N.L., A.J.V., and D.H.M. are staff members of CONICET. The NMR spectrometer was purchased with funds from ANPCyT and CONICET.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1204251109/-/DCSupplemental.
References
- 1.Marcus RA, Sutin N. Electron transfers in chemistry and biology. Biochim Biophys Acta. 1985;811:265–322. [Google Scholar]
- 2.Moser CC, Keske JM, Warncke K, Farid RS, Dutton PL. Nature of biological electron transfer. Nature. 1992;355(6363):796–802. doi: 10.1038/355796a0. [DOI] [PubMed] [Google Scholar]
- 3.Gray HB, Winkler JR. Long-range electron transfer. Proc Natl Acad Sci USA. 2005;102(10):3534–3539. doi: 10.1073/pnas.0408029102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hopfield JJ. Electron transfer between biological molecules by thermally activated tunneling. Proc Natl Acad Sci USA. 1974;71(9):3640–3644. doi: 10.1073/pnas.71.9.3640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Betts JN, Beratan DN, Onuchic JN. Mapping electron tunneling pathways: An algorithm that finds the ”minimum length”/maximum coupling pathway between electron donors and acceptors in proteins. J Am Chem Soc. 1992;114:4043–4046. [Google Scholar]
- 6.Skourtis SS, Balabin IA, Kawatsu T, Beratan DN. Protein dynamics and electron transfer: Electronic decoherence and non-Condon effects. Proc Natl Acad Sci USA. 2005;102(10):3552–3557. doi: 10.1073/pnas.0409047102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Skourtis SS, Waldeck DH, Beratan DN. Fluctuations in biological and bioinspired electron-transfer reactions. Annu Rev Phys Chem. 2010;61:461–485. doi: 10.1146/annurev.physchem.012809.103436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Balabin IA, Beratan DN, Skourtis SS. Persistence of structure over fluctuations in biological electron-transfer reactions. Phys Rev Lett. 2008;101(15):158102. doi: 10.1103/PhysRevLett.101.158102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Beratan DN, Betts JN, Onuchic JN. Protein electron transfer rates set by the bridging secondary and tertiary structure. Science. 1991;252(5010):1285–1288. doi: 10.1126/science.1656523. [DOI] [PubMed] [Google Scholar]
- 10.Prytkova TR, Kurnikov IV, Beratan DN. Ab initio based calculations of electron-transfer rates in metalloproteins. J Phys Chem B. 2005;109(4):1618–1625. doi: 10.1021/jp0457491. [DOI] [PubMed] [Google Scholar]
- 11.Ramirez BE, Malmström BG, Winkler JR, Gray HB. The currents of life: The terminal electron-transfer complex of respiration. Proc Natl Acad Sci USA. 1995;92(26):11949–11951. doi: 10.1073/pnas.92.26.11949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Farver O, Chen Y, Fee JA, Pecht I. Electron transfer among the CuA-, heme b- and a3-centers of Thermus thermophilus cytochrome ba3. FEBS Lett. 2006;580(14):3417–3421. doi: 10.1016/j.febslet.2006.05.013. [DOI] [PubMed] [Google Scholar]
- 13.Muresanu L, et al. The electron transfer complex between cytochrome c552 and the CuA domain of the Thermus thermophilus ba3 oxidase. A combined NMR and computational approach. J Biol Chem. 2006;281(20):14503–14513. doi: 10.1074/jbc.M601108200. [DOI] [PubMed] [Google Scholar]
- 14.Brzezinski P. Internal electron-transfer reactions in cytochrome c oxidase. Biochemistry. 1996;35(18):5611–5615. doi: 10.1021/bi960260m. [DOI] [PubMed] [Google Scholar]
- 15.Randall DW, Gamelin DR, LaCroix LB, Solomon EI. Electronic structure contributions to electron transfer in blue Cu and Cu(A) J Biol Inorg Chem. 2000;5(1):16–29. doi: 10.1007/s007750050003. [DOI] [PubMed] [Google Scholar]
- 16.Gorelsky SI, Xie X, Chen Y, Fee JA, Solomon EI. The two-state issue in the mixed-valence binuclear CuA center in cytochrome C oxidase and N2O reductase. J Am Chem Soc. 2006;128(51):16452–16453. doi: 10.1021/ja067583i. [DOI] [PubMed] [Google Scholar]
- 17.Olsson MHM, Ryde U. Geometry, reduction potential, and reorganization energy of the binuclear Cu(A) site, studied by density functional theory. J Am Chem Soc. 2001;123(32):7866–7876. doi: 10.1021/ja010315u. [DOI] [PubMed] [Google Scholar]
- 18.Abriata LA, Ledesma GN, Pierattelli R, Vila AJ. Electronic structure of the ground and excited states of the Cu(A) site by NMR spectroscopy. J Am Chem Soc. 2009;131(5):1939–1946. doi: 10.1021/ja8079669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Salgado J, Warmerdam G, Bubacco L, Canters GW. Understanding the electronic properties of the CuA site from the soluble domain of cytochrome c oxidase through paramagnetic 1H NMR. Biochemistry. 1998;37(20):7378–7389. doi: 10.1021/bi9728598. [DOI] [PubMed] [Google Scholar]
- 20.Bertini I, et al. The CuA center of a soluble domain from Thermus cytochrome ba3. An NMR investigation of the paramagnetic protein. J Am Chem Soc. 1996;118:11658–11659. [Google Scholar]
- 21.Ledesma GN, et al. The met axial ligand determines the redox potential in Cu(A) sites. J Am Chem Soc. 2007;129(39):11884–11885. doi: 10.1021/ja0731221. [DOI] [PubMed] [Google Scholar]
- 22.Xie X, et al. Perturbations to the geometric and electronic structure of the CuA site: Factors that influence delocalization and their contributions to electron transfer. J Am Chem Soc. 2008;130(15):5194–5205. doi: 10.1021/ja7102668. [DOI] [PubMed] [Google Scholar]
- 23.Gamelin DR, et al. Spectroscopy of mixed-valence CuA-type centers: Ligand-field control of ground-state properties related to electron transfer. J Am Chem Soc. 1998;120:5246–5263. [Google Scholar]
- 24.Honeychurch MJ. Effect of electron-transfer rate and reorganization energy on the cyclic voltammetric response of redox adsorbates. Langmuir. 1999;15:5158–5163. [Google Scholar]
- 25.Laviron E. General expression of the linear potential sweep voltammogram in the case of diffusionless electrochemical systems. J Electroanal Chem Interfac. 1979;101:19–28. [Google Scholar]
- 26.DeBeer George S, et al. A quantitative description of the ground-state wave function of Cu(A) by X-ray absorption spectroscopy: Comparison to plastocyanin and relevance to electron transfer. J Am Chem Soc. 2001;123(24):5757–5767. doi: 10.1021/ja004109i. [DOI] [PubMed] [Google Scholar]
- 27.Galinato MGI, et al. Heme-protein vibrational couplings in cytochrome c provide a dynamic link that connects the heme-iron and the protein surface. Proc Natl Acad Sci USA. 2012;109(23):8896–8900. doi: 10.1073/pnas.1200345109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gray KA, Davidson VL, Knaff DB. Complex formation between methylamine dehydrogenase and amicyanin from Paracoccus denitrificans. J Biol Chem. 1988;263(28):13987–13990. [PubMed] [Google Scholar]
- 29.Giudici-Orticoni MT, Guerlesquin F, Bruschi M, Nitschke W. Interaction-induced redox switch in the electron transfer complex rusticyanin-cytochrome c(4) J Biol Chem. 1999;274(43):30365–30369. doi: 10.1074/jbc.274.43.30365. [DOI] [PubMed] [Google Scholar]
- 30.Díaz-Moreno I, Díaz-Quintana A, Díaz-Moreno S, Subías G, De la Rosa MA. Transient binding of plastocyanin to its physiological redox partners modifies the copper site geometry. FEBS Lett. 2006;580(26):6187–6194. doi: 10.1016/j.febslet.2006.10.020. [DOI] [PubMed] [Google Scholar]
- 31.Kranich A, Ly HK, Hildebrandt P, Murgida DH. Direct observation of the gating step in protein electron transfer: Electric-field-controlled protein dynamics. J Am Chem Soc. 2008;130(30):9844–9848. doi: 10.1021/ja8016895. [DOI] [PubMed] [Google Scholar]
- 32.Alvarez-Paggi D, et al. Molecular basis of coupled protein and electron transfer dynamics of cytochrome c in biomimetic complexes. J Am Chem Soc. 2010;132(16):5769–5778. doi: 10.1021/ja910707r. [DOI] [PubMed] [Google Scholar]
- 33.Korzhnev DM, Kay LE. Probing invisible, low-populated states of protein molecules by relaxation dispersion NMR spectroscopy: An application to protein folding. Acc Chem Res. 2008;41(3):442–451. doi: 10.1021/ar700189y. [DOI] [PubMed] [Google Scholar]
- 34.Mittermaier A, Kay LE. New tools provide new insights in NMR studies of protein dynamics. Science. 2006;312(5771):224–228. doi: 10.1126/science.1124964. [DOI] [PubMed] [Google Scholar]
- 35.Williams PA, et al. The CuA domain of Thermus thermophilusba3-type cytochrome c oxidase at 1.6 Ǻ resolution. Nat Struct Mol Biol. 1999;6:509–516. doi: 10.1038/9274. [DOI] [PubMed] [Google Scholar]
- 36.Case DA, et al. 2010. AMBER 11 (Univ of California, San Francisco)
- 37.Frisch MJ, et al. 2009. Gaussian 09, revision A.02 (Gaussian, Inc., Wallingford, CT)
- 38.Izquierdo J, et al. Systematic ab initio study of the electronic and magnetic properties of different pure and mixed iron systems. Phys Rev B. 2000;61:13639–13646. [Google Scholar]
- 39.Crespo A, et al. A DFT-based QM-MM approach designed for the treatment of large molecular systems: Application to chorismate mutase. J Phys Chem B. 2003;107:13728–13736. [Google Scholar]
- 40.Beratan DN, Betts JN, Onuchic JN. Protein electron transfer rates set by the bridging secondary and tertiary structure. Science. 1991;252(5010):1285–1288. doi: 10.1126/science.1656523. [DOI] [PubMed] [Google Scholar]
- 41.Hunsicker-Wang LM, Pacoma RL, Chen Y, Fee JA, Stout CD. A novel cryoprotection scheme for enhancing the diffraction of crystals of recombinant cytochrome ba3 oxidase from Thermus thermophilus. Acta Crystallogr D Biol Crystallogr. 2005;61(Pt 3):340–343. doi: 10.1107/S0907444904033906. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.