

Abstract
Oral tolerance is a promising approach to induce unresponsiveness to various antigens. The development of tolerogenic vaccines could be exploited in modulating the immune response in autoimmune disease and allograft rejection. In this study, we investigated a nonviral gene transfer strategy for inducing oral tolerance via antigen-encoding chitosan-DNA nanoparticles (NP). Oral application of ovalbumin (OVA)-encoding chitosan-DNA NP (OVA-NP) suppressed the OVA-specific delayed-type hypersensitivity (DTH) response and anti-OVA antibody formation, as well as spleen cell proliferation following OVA stimulation. Cytokine expression patterns following OVA stimulation in vitro showed a shift from a Th1 toward a Th2/Th3 response. The OVA-NP-induced tolerance was transferable from donor to naïve recipient mice via adoptive spleen cell transfer and was mediated by CD4+CD25+ T cells. These findings indicate that nonviral oral gene transfer can induce regulatory T cells for antigen-specific immune modulation.
INTRODUCTION
The intestinal mucosa is constantly challenged by numerous external antigens. The majority consist of food antigens and commensal bacteria against which the immune system usually reacts with systemic unresponsiveness. This phenomenon is known as oral tolerance (17). In recent years, various experimental models exploiting oral tolerance showed its potential in prevention and treatment of diseases such as encephalomyelitis, arthritis, uveitis, myasthenia gravis, type 1 diabetes, and allograft rejection (3, 16, 26, 34, 44, 46, 48). However, translation of oral tolerance into clinical studies proved to be difficult (7, 14, 24, 33, 39, 43). Possible explanations could be the required antigen dose, the purity of the antigen, modifications of the antigen during the gastrointestinal passage, and the ways in which the antigen is expressed and presented to the immune system of the gut. Furthermore, developing tolerogenic vaccines on a protein basis for oral tolerance requires selection and purification of the antigen. A potential alternative could be the use of DNA-encoded vaccines, applied via a nonviral gene delivery system, resulting in direct expression of the antigen in the gut.
Chitosan, a nontoxic biodegradable polycationic polymer with low immunogenicity, was shown to be a useful oral gene carrier (8, 27, 28). Chitosan has been complexed with plasmid DNA, forming chitosan-DNA nanoparticles (NP), which are stable during the gastrointestinal passage and will be phagocytized in the gut, resulting in gene expression (2). It was shown that feeding of factor VIII-encoding chitosan-DNA NP to hemophilia A mice resulted in increased factor VIII plasma levels (6, 15) and that oral application of erythropoietin-encoding chitosan-DNA NP led to a significant increase of hematocrit levels (8). In rodent models of diabetes, chitosan-DNA NP encoding insulin or glucagon-like peptide 1 were able to decrease blood glucose concentrations (23, 31, 32). In addition, there is potential for chitosan-DNA NP to be used for immune modulation. Intranasal vaccination with pneumococcal surface antigen A-encoding chitosan-DNA NP or pulmonary application of chitosan-DNA NP encoding T cell epitopes from Mycobacterium tuberculosis led to immune stimulation (4, 45). Roy et al. showed that oral administration of chitosan complexed with DNA encoding a dominant peanut allergen is effective in reducing murine anaphylactic responses to peanuts (35).
Although it has been shown that nonviral gene application for immune modulation offers a promising way to induce systemic tolerance for the prevention and treatment of autoimmune, allergic disease and allograft rejection, the underlying immunological mechanisms are less well understood. In this study, we directly compared the effectiveness of protein- and DNA-based tolerogenic vaccines to ovalbumin as a model antigen. In addition, we analyzed the potential of ovalbumin-encoding chitosan-DNA NP (OVA-NP) to induce oral tolerance to OVA and characterized the cellular mechanisms mediating this tolerance induction.
MATERIALS AND METHODS
Materials.
Chitosan (medium molecular weight [MMW]; degree of deacetylation [DD], 79%), ovalbumin (grade V), Freund's adjuvant (complete, i.e., containing 1 mg/ml killed M. tuberculosis), para-nitrophenyl-phosphate (pNPP), and an anti-mouse IgG (Fc-specific) antibody (alkaline phosphatase conjugated; produced in goat) were purchased from Sigma-Aldrich (Deisenhofen, Germany).
Plasmids.
pEGFP was purchased form Clontech (Germany). The enhanced green fluorescent protein (EGFP)-encoding region (764 bp) was cloned into pcDNA3.1 (Invitrogen, Karlsruhe, Germany) to obtain EGFP/pcDNA3.1. BamHI and EcoRI (Roche, Mannheim, Germany) were used as restriction enzymes. OVA/pcDNA3 was a kind gift from Martin Vabulos (Max-Plank-Institut for Biochemistry, Martinsried, Germany) (37). Plasmids were isolated using the Qiagen Plasmid Maxikit (Hilden, Germany) according to the manufacturer's protocol.
Nanoparticle formulation.
Nanoparticles were made by complex coacervation of chitosan and DNA as described before (35). Briefly, plasmid solution (200 μg/ml) was mixed 1:1 with sodium sulfate (90 mM) and heated to 55°C. MMW-chitosan was purified before use. Therefore, it was reprecipitated with ammonia from a chitosan solution (0.5% [wt/vol] chitosan in 1% acetic acid). The precipitate was washed two times in deionized water and vacuum dried. The purified chitosan (37 mg) was solubilized in 200 ml of acetic acid (25 mM), and the pH value was adjusted to 5.5 with NaOH. After sterile filtration of the chitosan solution, it was also heated to 55°C. The heated plasmid and chitosan solutions were mixed 1:1 and vortexed at high speed for 30 s. Particles were stored at room temperature.
Mice.
BALB/c mice (H-2d) were originally purchased from Charles River (Sulzfeld, Germany). All mice used in this study were aged between 6 and 12 weeks at the time of experimental use and were bred and maintained at the animal facility of the Department of Experimental Surgery at the University of Erlangen-Nürnberg under specific-pathogen-free conditions and treated in accordance with the institutional and state guidelines.
Gene expression analysis.
Mice were fed chitosan-DNA NP containing 50 μg plasmid encoding EGFP (EGFP-NP). At 0 h, 3 h, 6 h, 12 h, 24 h, and 48 h after oral application, mesenteric lymph nodes (MLN) and Peyer's patches were removed and RNA was isolated using the Qiagen RNeasy minikit (Hilden, Germany). After cDNA synthesis, quantitative real-time PCR (qRT-PCR) was performed on a StepOne Real-Time PCR system (Applied Biosystems, Darmstadt, Germany). The amplification protocol started with an initial holding at 50°C for 2 min and then at 95°C for 10 min. This was followed by a two-step PCR program consisting of 95°C for 15 s and 60°C for 1 min for 40 cycles. Each sample was analyzed in duplets. The obtained qRT-PCR values were normalized against GAPDH. The sequences of the primers and the hybridization probes were as follows: GAPDH forward, 5′-TTC ACC ACC ATG GAG AAG GC-3′; GAPDH reverse, 5′-GGC ATG GAC TGT GGT CAT GA-3′; probe, 5′-FAM-TGC ATC CTG CAC CAC CAA CTG CTT AG-TAMRA-3′; EGFP forward, ′-CGA CGG CAA CTA CAA GAC-3′; EGFP reverse, 5′-TAG TTG TAC TCC AGC TTG TGC-3′; probe, 5′-FAM-ACT TCA AGG AGG ACG GCA ACA TCC T-TAMRA-3′.
Immunization and antigen treatment.
To induce oral tolerance, mice were given an oral administration of 25 mg ovalbumin (OVA) in phosphate-buffered saline (PBS) on days 0 and 2 as the established treatment arm (1, 25, 41) or OVA-encoding chitosan-DNA NP (OVA-NP) containing 50 μg plasmid-DNA (OVA/pcDNA3) on days 0, 2, 5, 7, and 9. Four groups of control animals received either a corresponding amount of chitosan-DNA NP encoding the enhanced green fluorescent protein (EGFP/pcDNA3.1) or pure chitosan solution, or 50 μg naked OVA/pcDNA3-plasmid, or PBS. Mice were immunized by subcutaneous injection of 100 μg OVA in 100 μl of a 1:1 mixture of PBS and Freund's adjuvant (CFA) on day 12.
Measurement of DTH.
Seven days after immunization, mice were challenged by subcutaneous injection of 50 μg OVA in 30 μl PBS into the right ear pinna, while 30 μl PBS was injected into the left ear pinna as a control. Ear swelling was measured before and 24 h after injection with a digital micrometer (Kroeplin, Schlüchtern, Germany). The delayed-type hypersensitivity (DTH) response was calculated as follows: OVA-specific ear swelling = (right ear thickness − left ear thickness)24 h − (right ear thickness − left ear thickness)0 h.
Measurement of OVA-specific antibody production.
Immediately after measuring ear thickness, blood samples were collected and OVA-specific IgG-antibody production was measured via enzyme-linked immunosorbent assay (ELISA). Ninety-six-well microtiter plates were coated with OVA in PBS (100 μg/ml; 100 μl/well) and incubated overnight at 4°C. The wells were then washed with Tween/PBS (0.5 μl/ml; 200 μl/well) and blocked with 3% bovine serum albumin (BSA) in PBS (100 μl/well) at room temperature for 2 h. Serum samples were serially diluted 1:2 with 1% BSA in PBS, starting at an initial 1:250 dilution. After washing of the blocked wells, 100 μl of the diluted serum samples was added, and the samples were incubated for 1 h at room temperature. After a washing step, 100 μl of a 1:10,000 diluted alkaline phosphatase-conjugated anti-mouse IgG-antibody was pipetted into the wells. One hour later, the wells were washed before 100 μl of substrate solution (para-nitrophenyl-phosphate in 1 M diethanolamine buffer; 1 mg/ml) was added. After 30 min at room temperature, the reaction was stopped with 3 M NaOH (100 μl/well). The optical density at 405 nm (OD405) (reference OD, 490 nm) was measured with an ELISA reader (SLT; Tecan, Crailsheim, Germany). Results are shown as endpoint dilution titers. The cutoff values were determined for a 95% confidence interval based on four negative-control sera analyzed in parallel, as described before (18).
Determination of antigen-specific proliferation.
Fourteen days after the first immunization (day 26), mice received a second immunization. Briefly, on day 39, mice were sacrificed, and their spleen mononuclear cells (MNC) were isolated as described before (41). Aliquots (200 μl) of MNC suspensions (2.5 × 106 cells/ml) were cultivated with different OVA concentrations (0, 5, 25, and 50 μg/ml) in 96-well microtiter plates for 72 h. Afterwards, 10 μl [H3]thymidine was added (1 μCi/well). Eighteen hours later, cells were harvested and thymidine incorporation was measured. The results were expressed as counts per minute (cpm).
Cytokine measurement.
Eight days after immunization, mice were sacrificed, and their spleen MNC were isolated. Splenocytes (1.5 × 106) were cultured in 96-well round plates in a final volume of 200 μl in the presence of 20 μg/ml OVA in R10+ medium at 37°C with 5% CO2 and humidified atmosphere as described before (41). Supernatants were harvested after 48 h and stored at −20°C until assayed. Cytokine production was quantified using the mouse cytometric bead assay (BD, Germany) on a FACSCanto II flow cytometer (BD, Germany). Transforming growth factor β (TGF-β) concentration was analyzed using an ELISA kit (eBioscience, Germany).
Transfer of tolerance.
Mice were fed OVA-NP containing 50 μg plasmid DNA or PBS as control on days 0, 3, 5, 7, and 10. On day 11, mice were sacrificed and their spleen MNC were isolated. Naïve recipient mice were injected with 2 × 107 splenocytes in 200 μl PBS in the tail vein. The next day, recipient mice were immunized against OVA. Seven days after immunization, the OVA-specific DTH response was measured.
Beside the complete spleen cell fraction (3 × 106 cells in 200 μl PBS), purified CD4+ T cells (1 × 106 cells; purity, >76%) or a CD4+ T cell-depleted cell fraction (3 × 106 cells, depletion of CD4+ T cells, >90%) isolated from the spleens of OVA-NP fed mice was transferred. In further experiments, CD4+CD25+ T cells (2 × 105 cells; purity, >90%) or CD4+CD25− T cells (2 × 105 cells; depletion of CD4+CD25+ T cells, >95%) were adoptively transferred. The cell subsets were stained using the CD4+CD25+ regulatory T cell isolation kit (Miltenyi Biotec) and sorted by magnetically activated cell-sorting (MACS) columns as described by the manufacturer.
Statistical analysis.
Results are given as the means per group ± standard deviations (SD). A normal distribution of the data was confirmed. The data were analyzed using an unpaired two-tailed Student's t test. When more than two groups were compared, a one-way analysis of variance (ANOVA) test followed by Dunnett's multiple-comparison test was used. P values of <0.05 were considered significant. Statistical analysis was performed using GraphPad Prism version 5.03 for Windows (GraphPad Software, San Diego, CA).
RESULTS
Gene expression kinetics after oral application of chitosan-DNA NP.
To analyze gene expression kinetics after oral nanoparticle administration, mice received a single dose of antigen-encoding chitosan-DNA NP containing 50 μg plasmid DNA. Three hours after oral application, mRNA of the encoded antigen was already detected in the Peyer's patches (PP) and mesenteric lymph nodes (Fig. 1A and B). The maximum expression was reached after 6 h in both compartments, and the mRNA remained detectable for up to 48 h. To address whether systemic levels of the gene product can be measured, serum samples of mice receiving OVA-encoding chitosan-DNA NP were analyzed using an OVA-specific ELISA system. However, at none of the time points were systemic levels of OVA detectable (data not shown).
Fig 1.
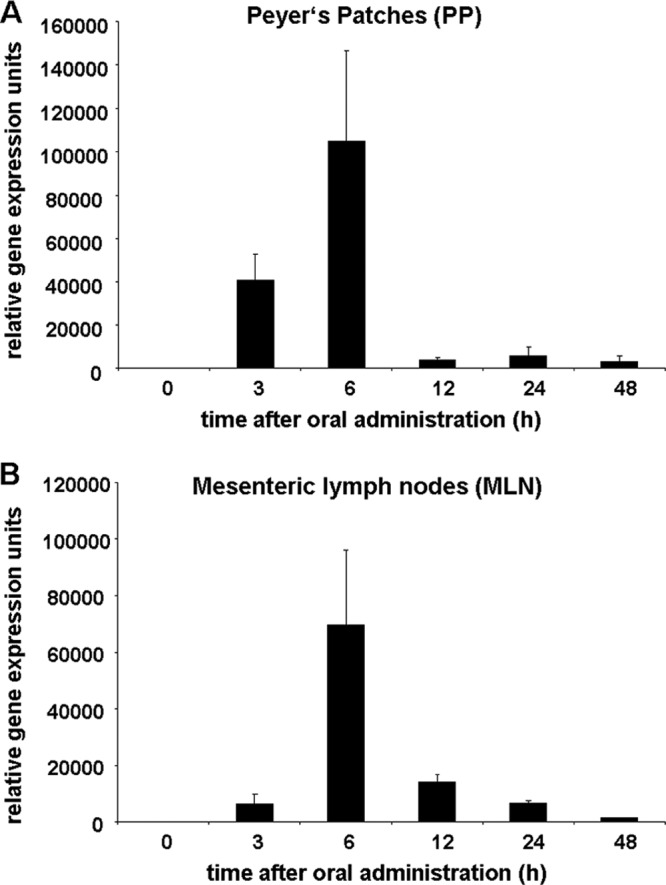
Gene expression kinetics after oral application of chitosan-DNA NP. Mice were fed a single dose of antigen-encoding chitosan-DNA NP containing 50 μg of plasmid DNA. At 0 h, 3 h, 6 h, 12 h, 24 h, or 48 h, Peyer's patches (A) and mesenteric lymph nodes (B) were removed and analyzed for gene expression by qRT-PCR. The results were normalized against GAPDH and are shown in relative expression units, representing the means of three independent experiments ± SDs.
OVA-NP suppress delayed hypersensitivity.
First, we investigated whether oral application of OVA-encoding chitosan-DNA NP (OVA-NP) could suppress the cell-mediated OVA-specific delayed-type hypersensitivity response. Mice were fed either OVA-NP or OVA protein before immunization with the antigen. The treatment regime for OVA-NP is shown in Fig. 2A. Feeding OVA-NP significantly suppressed the OVA-specific ear swelling, as effectively as OVA protein did.
Fig 2.
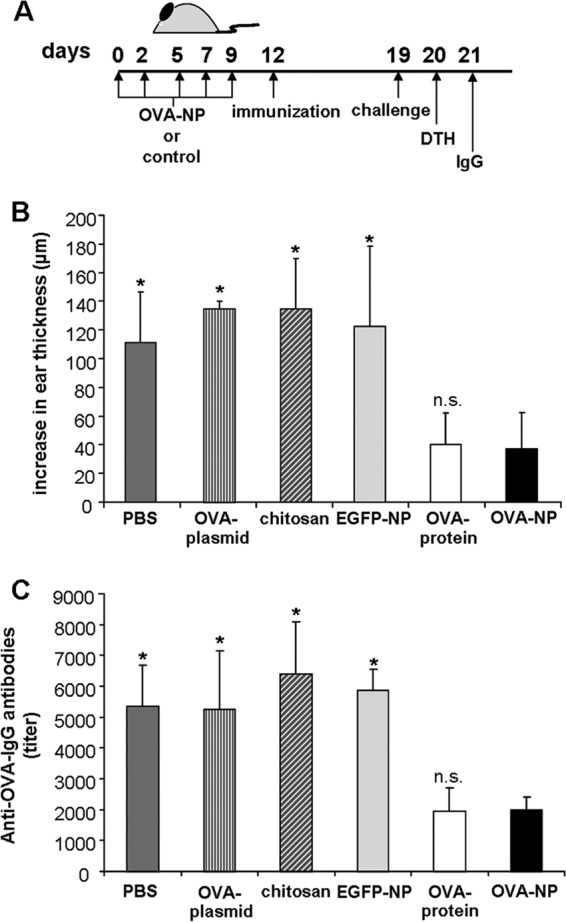
OVA-encoding chitosan-DNA nanoparticles suppress delayed hypersensitivity and OVA-specific antibody formation. (A) BALB/c mice were fed five doses of OVA-NP containing 50 μg plasmid DNA (days 0 to 9). On day 12, mice were immunized by subcutaneous injection of 100 μg OVA in 100 μl of a 1:1 mixture of PBS and CFA. On day 19, mice were challenged by subcutaneous injection of 50 μg OVA in 30 μl PBS into the right ear pinna, while 30 μl PBS was injected into the left ear pinna as a control. (B) Twenty-four hours later, ear swelling was measured and the DTH response was calculated. (C) On day 21, blood samples were collected and the OVA-specific IgG concentrations were analyzed by endpoint dilution ELISA. Mice of the control groups were fed five times with PBS, OVA-plasmid (50 μg), pure chitosan solution, or EGFP-NP. The OVA protein-fed group received two doses of 25 mg OVA protein on days 0 and 2. Data shown were obtained for 4 to 8 mice per group (*, P < 0.05; n.s., P ≥ 0.05 for OVA-NP compared to controls).
To exclude an unspecific immunosuppressive effect of the OVA-NP or chitosan, control mice were treated either with chitosan-DNA NP encoding enhanced green fluorescence protein (EGFP-NP) as control antigen or with pure chitosan solution. The control group receiving pure chitosan solution showed a strong DTH response (Fig. 2B). In addition, EGFP-NP had no effect on the cell-mediated immune response, indicating an antigen-specific suppression.
To analyze whether nanoparticle formation using chitosan was required or whether plasmid alone would be sufficient, mice were treated with naked OVA-encoding plasmid as encapsulation control. Mice fed with plasmid showed no reduction in the DTH response, indicating that nanoparticle formation using chitosan was indeed necessary (Fig. 2B).
OVA-NP reduce OVA-specific antibody formation.
Next, we analyzed whether oral application of OVA-NP was able to prevent the production of antibodies against OVA. After measuring the DTH response, sera of the treated mice were collected (Fig. 2A) and the OVA-specific IgG antibody concentration was analyzed. Sera of mice treated with OVA-NP showed significantly decreased levels of OVA-specific antibodies compared to control groups receiving PBS, EGFP-NP, chitosan solution, or OVA plasmid (Fig. 2C). The data show that oral application of OVA-NP was also able to suppress the humoral immune response toward the tolerized antigen with effectiveness equal to that of OVA protein.
OVA-NP suppress antigen-specific proliferation.
To further characterize the OVA-specific proliferative response, spleen mononuclear cells (MNC) from tolerized and subsequently immunized mice were cultured with increasing OVA concentrations in vitro. Mice fed with PBS and OVA protein served as controls. Baseline cell proliferation without OVA showed no difference among the three groups (Fig. 3). Culturing spleen MNC in the presence of OVA resulted in a dose-dependent proliferation in the control group, whereas in the two groups receiving either OVA-NP or OVA protein an equally strong reduction of cell proliferation was observed.
Fig 3.
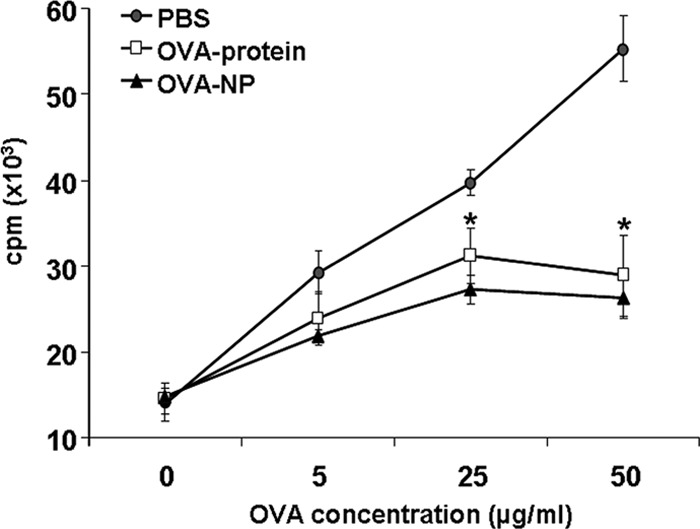
OVA-NP suppress antigen-specific proliferation. OVA-NP-, OVA protein-, or PBS-fed mice were immunized against OVA on day 12, as described for Fig. 1. On day 26, mice received a second immunization. Thirteen days later, spleen MNCs were isolated and cultured with different OVA concentrations in R10+ medium at 37°C with 5% CO2 and humidified atmosphere. After 72 h of incubation, [H3]thymidine (1 μCi/well) was added. Eighteen hours later, cells were harvested and thymidine incorporation was measured. Data shown represent duplicates and were obtained for 3 or 4 mice per group (*, P < 0.05 for OVA-NP or OVA protein compared to PBS).
OVA-NP cause a shift from Th1 to Th2/Th3 response.
To determine the effect of OVA-NP treatment on cytokine production patterns, spleen MNC of OVA-NP-, OVA protein-, or PBS-treated and immunized mice were cultured in the presence of OVA, and the cytokine production was measured in the supernatants. Levels of the Th1 cytokines tumor necrosis factor alpha (TNF-α) and gamma interferon (IFN-γ) were reduced in the groups fed with OVA-NP and OVA protein (Fig. 4). On the other hand, the Th2 and Th3 cytokines interleukin-4 (IL-4), IL-10, and transforming growth factor beta (TGF-β) showed an increased concentration in the OVA-NP-treated mice compared to the control groups. This indicates a shift from a Th1 toward a Th2/Th3 response after OVA-NP application.
Fig 4.
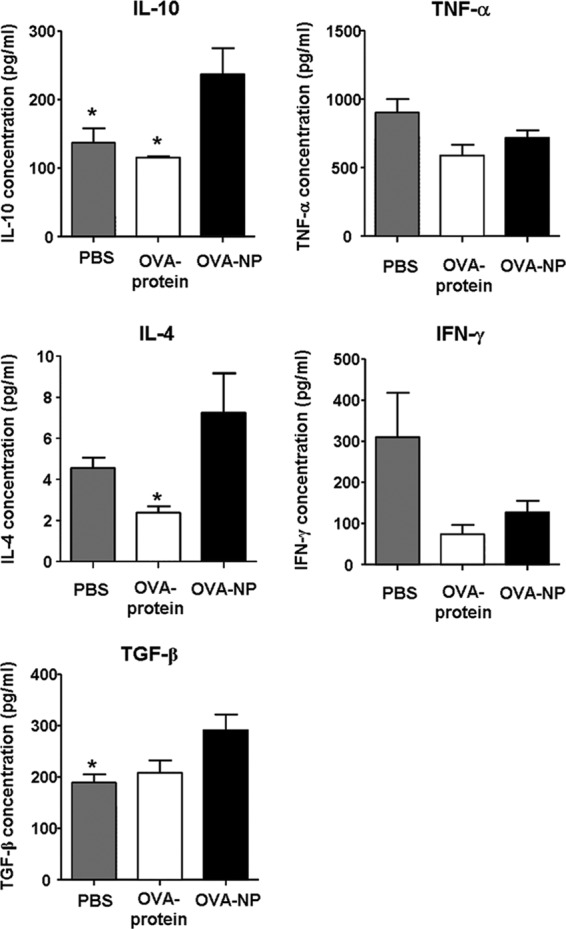
Cytokine production patterns after OVA-NP application. OVA-NP-, OVA protein-, and PBS-fed mice were immunized against OVA on day 12. Eight days later, splenic MNCs were isolated and cultured in the presence of 20 μg/ml OVA. Forty-eight hours later, supernatants were analyzed for cytokine production by cytometric bead assay (CBA) or ELISA. Data shown were obtained for 4 or 5 mice per group (*, P < 0.05 for OVA-NP compared to PBS or OVA protein).
The tolerance induced by OVA-NP is transferable and mediated by CD4+CD25+ T cells.
To investigate whether the nanoparticle-induced tolerance involves the generation of regulatory cells, we adoptively transferred spleen MNC from tolerant into naïve mice. For this purpose, 2 × 107 spleen cells from mice treated with OVA-NP were injected into naïve animals before immunization with OVA (Fig. 5A). Subsequently, the OVA-specific DTH response was measured. As shown in Fig. 5B, the nanoparticle-induced tolerance was transferable as demonstrated by the suppressed OVA-specific ear swelling.
Fig 5.

The tolerance induced by OVA-NP is transferable. (A) Mice were fed five doses of OVA-NP containing 50 μg plasmid DNA (days 0 to 9). The control group received PBS instead of nanoparticles. On day 11, mice were sacrificed and their spleen cells were isolated. (B) Naïve recipient mice were injected with 2 × 107 splenocytes in the tail vein and immunized the next day. Seven days after immunization, the OVA-specific DTH response was analyzed. To identify the phenotype of regulatory cells mediating tolerance, isolated CD4+ T cells or the remaining CD4+-depleted fraction (C) and isolated CD4+CD25+ or CD4+CD25− T cells (D) from OVA-NP- or PBS-treated mice were used for the adoptive transfer. Data shown were obtained for 4 to 6 mice per group (*, P < 0.05; n.s., P ≥ 0.05).
To further analyze the phenotype of the regulatory cells involved in the tolerance induction via chitosan-DNA NP, we transferred isolated CD4+ T cells or CD4+ T cell-depleted splenocytes of OVA-NP-treated mice into naïve recipients. Transfer of CD4+ T cells resulted in a significant suppression of the OVA-specific DTH response, whereas transfer of CD4+ T cell-depleted spleen cells had no effect (Fig. 5C). To characterize the involved T cell subpopulation, we adoptively transferred either CD4+CD25+ or CD4+CD25− T cells. Only mice receiving the CD4+CD25+ subset were able to suppress the OVA-specific DTH response (Fig. 5D), indicating that CD4+CD25+ T cells are required to induce tolerance after oral OVA-NP application. On the other hand, transfer of CD4+CD25+ T cells from mice treated with EGFP-NP or PBS was not able to reduce the OVA-specific DTH response (data not shown), indicating an antigen-specific mechanism.
DISCUSSION
Oral application of antigen is able to induce antigen-specific peripheral tolerance (42) and is a promising approach for the treatment of autoimmune diseases or allograft rejection (3, 16, 26, 34, 44, 46, 48).
Chitosan-DNA NP are of increasing interest as a nonviral gene carrier system (5, 6, 27). Using chitosan-DNA NP for oral application, exploiting oral tolerance mechanisms, would allow the development of DNA-based tolerogenic vaccines. Using DNA-encoded antigen instead of protein or peptide antigens offers potential advantages such as lower costs for synthesis and purification. In addition, application of peptides requires the prior determination of the immunogenic epitopes, and their effect will depend on the presence of specific major histocompatibility complex (MHC) antigens, potentially restricting their use. In contrast, gene delivery systems such as chitosan-DNA NP, which are stable during gastrointestinal passage, allow the direct expression of the encoded antigen in the gut. This allows presentation of the immunogenic peptide within the context of the patient's own MHC antigens, circumventing the need for tailoring the vaccine according to the MHC genotype. Another important aspect of DNA-based vaccines is that the expressed gene of interest will undergo the posttranslational modifications within the host cell. With respect to human antigens being developed for therapeutic use, such as gene substitution or tolerance induction toward autoantigens, this delivers a desired effect, since the DNA vaccine-derived antigen will carry the same posttranslational modifications as the antigen expressed by the patient's cells. The situation for tolerance induction toward foreign antigens, such as allergy treatment, is somewhat different. Here, the posttranslational modifications of the native antigens will most likely differ from the DNA vaccine-derived antigens expressed by the patient's cells. However, despite these differences a therapeutic effect of chitosan-DNA nanoparticles in oral tolerance induction was shown in a mouse model of peanut allergy (35).
In this study, we analyzed chitosan-DNA NP for their ability to induce oral tolerance to ovalbumin. The effect of the chitosan-DNA NP was compared with that obtained with an established regimen of oral OVA-protein application, which has been shown to reduce the levels of the cellular and humoral anti-OVA immune response (1, 25, 41). Application of OVA-NP suppressed the OVA-specific DTH response, IgG formation, and spleen cell proliferation as effectively as the treatment with OVA protein. The immune modulation following OVA-NP was antigen specific, as had been demonstrated previously for protein-induced oral tolerance (29).
Oral tolerance toward protein antigens is mediated by two main mechanisms depending on the doses of administered antigens. High doses of antigen induce clonal deletion and/or anergy of T cells associated with a suppressed Th1 and Th2 response, whereas repeated administration of low doses of antigen induce active suppression, characterized by the generation of regulatory T cells and a shift toward a Th2/Th3 response (11, 17, 19). When we looked at the potential mechanisms involved in the tolerance induction via nanoparticles, we observed an increase of the Th2/Th3-type cytokines IL-4, IL-10, and TGF-β and a reduction of the Th1-type cytokines IFN-γ and TNF-α in the supernatant of OVA-stimulated spleen MNC, indicating a shift from a Th1 toward a Th2/Th3 response. This is in accordance with the cytokine profiles detected in oral tolerance studies using low-dose antigen regimes (10, 11, 17), indicating that related mechanisms promote nanoparticle- and protein-induced low-dose tolerance. In contrast, in the OVA protein group used in our study we observed reduced levels of both Th1 and Th2 cytokines and a basically unchanged TGF-β level, reflecting the high-dose OVA feeding regime used.
Beside the dose of the antigen, the disease model can influence the cytokine pattern in oral tolerance. In Th1 disease models such as experimental autoimmune encephalomyelitis or diabetes, treatment via oral tolerance induction is associated with a shift toward a Th2 immune response (12, 20). In contrast, in allergy models, which are characterized by a Th2 cytokine profile, tolerance induction led to a suppression of the Th2 response (21, 30, 36), which was also observed by Roy et al. and Chew et al. using allergen-encoding chitosan-DNA NP (13, 35).
The three main regulatory cell populations involved in oral tolerance are IL-10-producing CD4+CD25−Foxp3− cells, known as Tr1 cells, TGF-β-producing CD4+CD25−Foxp3− cells, known as Th3 cells, and CD4+CD25+Foxp3+ cells (Tregs), mediating their suppression through CTLA-4 or cytokines such as TGF-β and IL-10 (40). In addition to the thymus-derived naturally occurring Tregs, CD4+CD25+ T cells can also be induced in the gut. TGF-β, abundantly expressed in the gut, was shown to induce the expression of Foxp3 in naïve CD4+T cells, converting them into CD4+CD25+ Tregs (9). Although all of these regulatory T cells were implicated in the induction of oral tolerance, the contribution of the respective subpopulation is not yet clear and may vary between different tolerance models. In our study, we were able to demonstrate that the transfer of CD4+CD25+ T cells was required to reduce the OVA-specific DTH response, indicating a crucial role for the CD4+CD25+ T cell subset in the induction of systemic tolerance via oral chitosan-DNA NP. This is in accordance with previous reports demonstrating the generation of CD4+CD25+ T cells after oral antigen administration (22, 38). Similarly, the nanoparticle-induced cytokine profiles resulting in increased levels of IL-10 and TGF-β are consistent with the induction of CD4+CD25+ T cells in oral tolerance, as demonstrated by Zhang et al. They showed an increase of CD4+CD25+ T cells accompanied by a decrease of CD4+CD25− T cells after oral administration of OVA to OVA T cell receptor (TCR) transgenic mice. These CD4+CD25+ T cells produced high levels of IL-10 and TGF-β, and their adoptive transfer was able to suppress antigen-specific DTH responses (47).
In summary, our data demonstrate the potential of DNA-based tolerogenic vaccines using antigen-encoding chitosan-DNA nanoparticles as therapeutic strategy for the induction of immunological tolerance. Nanoparticle treatment led to a significant reduction of antigen-specific DTH response and antibody formation, which was as effective as the oral application of protein antigen. CD4+CD25+ regulatory T cells were shown to be involved in tolerance induction via nanoparticles. Therefore, using tolerogenic vaccines might be a promising option for the treatment of autoimmune diseases, allergy, or allograft rejection.
ACKNOWLEDGMENT
This work was supported by a grant to S.M.E. and B.M.S. from the Interdisciplinary Centre for Clinical Research (IZKF) at the University Hospital of the University Erlangen-Nürnberg (TP-B15).
Footnotes
Published ahead of print 29 August 2012
REFERENCES
- 1. Afuwape AO, Turner MW, Strobel S. 2004. Oral administration of bovine whey proteins to mice elicits opposing immunoregulatory responses and is adjuvant dependent. Clin. Exp. Immunol. 136:40–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Agnihotri SA, Mallikarjuna NN, Aminabhavi TM. 2004. Recent advances on chitosan-based micro- and nanoparticles in drug delivery. J. Control Release 100:5–28 [DOI] [PubMed] [Google Scholar]
- 3. Bellmann K, et al. 1997. Intervention in autoimmune diabetes by targeting the gut immune system. Int. J. Immunopharmacol. 19:573–577 [DOI] [PubMed] [Google Scholar]
- 4. Bivas-Benita M, et al. 2004. Pulmonary delivery of chitosan-DNA nanoparticles enhances the immunogenicity of a DNA vaccine encoding HLA-A*0201-restricted T-cell epitopes of Mycobacterium tuberculosis. Vaccine 22:1609–1615 [DOI] [PubMed] [Google Scholar]
- 5. Bowman K, Leong KW. 2006. Chitosan nanoparticles for oral drug and gene delivery. Int. J. Nanomedicine 1:117–128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bowman K, Sarkar R, Raut S, Leong KW. 2008. Gene transfer to hemophilia A mice via oral delivery of FVIII-chitosan nanoparticles. J. Control Release 132:252–259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Chaillous L, et al. 2000. Oral insulin administration and residual beta-cell function in recent-onset type 1 diabetes: a multicentre randomised controlled trial. Diabete Insuline Orale group. Lancet 356:545–549 [DOI] [PubMed] [Google Scholar]
- 8. Chen J, et al. 2004. Transfection of mEpo gene to intestinal epithelium in vivo mediated by oral delivery of chitosan-DNA nanoparticles. World J. Gastroenterol. 10:112–116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Chen W, et al. 2003. Conversion of peripheral CD4+CD25-naive T cells to CD4+CD25+ regulatory T cells by TGF-beta induction of transcription factor Foxp3. J. Exp. Med. 198:1875–1886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Chen Y, et al. 1996. Oral tolerance in myelin basic protein T-cell receptor transgenic mice: suppression of autoimmune encephalomyelitis and dose-dependent induction of regulatory cells. Proc. Natl. Acad. Sci. U. S. A. 93:388–391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Chen Y, et al. 1995. Peripheral deletion of antigen-reactive T cells in oral tolerance. Nature 376:177–180 [DOI] [PubMed] [Google Scholar]
- 12. Chen Y, Kuchroo VK, Inobe J, Hafler DA, Weiner HL. 1994. Regulatory T cell clones induced by oral tolerance: suppression of autoimmune encephalomyelitis. Science 265:1237–1240 [DOI] [PubMed] [Google Scholar]
- 13. Chew JL, Wolfowicz CB, Mao HQ, Leong KW, Chua KY. 2003. Chitosan nanoparticles containing plasmid DNA encoding house dust mite allergen, Der p 1 for oral vaccination in mice. Vaccine 21:2720–2729 [DOI] [PubMed] [Google Scholar]
- 14. Choy EH, et al. 2001. Control of rheumatoid arthritis by oral tolerance. Arthritis Rheum. 44:1993–1997 [DOI] [PubMed] [Google Scholar]
- 15. Dhadwar SS, Kiernan J, Wen J, Hortelano G. 2010. Repeated oral administration of chitosan/DNA nanoparticles delivers functional FVIII with the absence of antibodies in hemophilia A mice. J. Thromb. Haemost. 8:2743–2750 [DOI] [PubMed] [Google Scholar]
- 16. Faria AM, et al. 2003. Oral tolerance induced by continuous feeding: enhanced up-regulation of transforming growth factor-beta/interleukin-10 and suppression of experimental autoimmune encephalomyelitis. J. Autoimmun. 20:135–145 [DOI] [PubMed] [Google Scholar]
- 17. Faria AM, Weiner HL. 2005. Oral tolerance. Immunol. Rev. 206:232–259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Frey A, Di Canzio J, Zurakowski D. 1998. A statistically defined endpoint titer determination method for immunoassays. J. Immunol. Methods 221:35–41 [DOI] [PubMed] [Google Scholar]
- 19. Friedman A, Weiner HL. 1994. Induction of anergy or active suppression following oral tolerance is determined by antigen dosage. Proc. Natl. Acad. Sci. U. S. A. 91:6688–6692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hancock WW, Polanski M, Zhang J, Blogg N, Weiner HL. 1995. Suppression of insulitis in non-obese diabetic (NOD) mice by oral insulin administration is associated with selective expression of interleukin-4 and -10, transforming growth factor-beta, and prostaglandin-E. Am. J. Pathol. 147:1193–1199 [PMC free article] [PubMed] [Google Scholar]
- 21. Haneda K, et al. 1997. TGF-beta induced by oral tolerance ameliorates experimental tracheal eosinophilia. J. Immunol. 159:4484–4490 [PubMed] [Google Scholar]
- 22. Hultkrantz S, Ostman S, Telemo E. 2005. Induction of antigen-specific regulatory T cells in the liver-draining celiac lymph node following oral antigen administration. Immunology 116:362–372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Jean M, Alameh M, Buschmann MD, Merzouki A. 2011. Effective and safe gene-based delivery of GLP-1 using chitosan/plasmid-DNA therapeutic nanocomplexes in an animal model of type 2 diabetes. Gene Ther. 18:807–816 [DOI] [PubMed] [Google Scholar]
- 24. Kaneko H, et al. 2010. Efficacy of the slow dose-up method for specific oral tolerance induction in children with cow's milk allergy: comparison with reported protocols. J. Investig. Allergol. Clin. Immunol. 20:538–539 [PubMed] [Google Scholar]
- 25. Kim JH, et al. 2002. Effects of the aqueous extract of Epimedii Herba on the induction of oral tolerance in mice. Biol. Pharm. Bull. 25:1000–1005 [DOI] [PubMed] [Google Scholar]
- 26. Maiti PK, Feferman T, Im SH, Souroujon MC, Fuchs S. 2004. Immunosuppression of rat myasthenia gravis by oral administration of a syngeneic acetylcholine receptor fragment. J. Neuroimmunol. 152:112–120 [DOI] [PubMed] [Google Scholar]
- 27. Mansouri S, et al. 2004. Chitosan-DNA nanoparticles as non-viral vectors in gene therapy: strategies to improve transfection efficacy. Eur. J. Pharm. Biopharm. 57:1–8 [DOI] [PubMed] [Google Scholar]
- 28. Mao HQ, et al. 2001. Chitosan-DNA nanoparticles as gene carriers: synthesis, characterization and transfection efficiency. J. Control Release 70:399–421 [DOI] [PubMed] [Google Scholar]
- 29. Maron R, Slavin AJ, Hoffmann E, Komagata Y, Weiner HL. 2002. Oral tolerance to copolymer 1 in myelin basic protein (MBP) TCR transgenic mice: cross-reactivity with MBP-specific TCR and differential induction of anti-inflammatory cytokines. Int. Immunol. 14:131–138 [DOI] [PubMed] [Google Scholar]
- 30. McMenamin C, Pimm C, McKersey M, Holt PG. 1994. Regulation of IgE responses to inhaled antigen in mice by antigen-specific gamma delta T cells. Science 265:1869–1871 [DOI] [PubMed] [Google Scholar]
- 31. Niu L, Xu YC, Dai Z, Tang HQ. 2008. Gene therapy for type 1 diabetes mellitus in rats by gastrointestinal administration of chitosan nanoparticles containing human insulin gene. World J. Gastroenterol. 14:4209–4215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Niu L, Xu YC, Xie HY, Dai Z, Tang HQ. 2008. Expression of human insulin gene wrapped with chitosan nanoparticles in NIH3T3 cells and diabetic rats. Acta Pharmacol. Sin. 29:1342–1349 [DOI] [PubMed] [Google Scholar]
- 33. Ojeda P, Ojeda I, Pineda F, Alfaya T, Ojeda JA. 2010. Induction of oral tolerance to peanut: a successful home-based protocol. J. Investig. Allergol. Clin. Immunol. 20:524–528 [PubMed] [Google Scholar]
- 34. Petersen JS, et al. 2003. Coupling of oral human or porcine insulin to the B subunit of cholera toxin (CTB) overcomes critical antigenic differences for prevention of type I diabetes. Clin. Exp. Immunol. 134:38–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Roy K, Mao HQ, Huang SK, Leong KW. 1999. Oral gene delivery with chitosan-DNA nanoparticles generates immunologic protection in a murine model of peanut allergy. Nat. Med. 5:387–391 [DOI] [PubMed] [Google Scholar]
- 36. Russo M, et al. 1998. Prevention of lung eosinophilic inflammation by oral tolerance. Immunol. Lett. 61:15–23 [DOI] [PubMed] [Google Scholar]
- 37. Spies B, et al. 2003. Vaccination with plasmid DNA activates dendritic cells via Toll-like receptor 9 (TLR9) but functions in TLR9-deficient mice. J. Immunol. 171:5908–5912 [DOI] [PubMed] [Google Scholar]
- 38. Thorstenson KM, Khoruts A. 2001. Generation of anergic and potentially immunoregulatory CD25+CD4 T cells in vivo after induction of peripheral tolerance with intravenous or oral antigen. J. Immunol. 167:188–195 [DOI] [PubMed] [Google Scholar]
- 39. Trentham DE, et al. 1993. Effects of oral administration of type II collagen on rheumatoid arthritis. Science 261:1727–1730 [DOI] [PubMed] [Google Scholar]
- 40. Tsuji NM, Kosaka A. 2008. Oral tolerance: intestinal homeostasis and antigen-specific regulatory T cells. Trends Immunol. 29:532–540 [DOI] [PubMed] [Google Scholar]
- 41. Wang Y, Mei Y, Bao S, Xu L. 2007. Vasoactive intestinal polypeptide enhances oral tolerance by regulating both cellular and humoral immune responses. Clin. Exp. Immunol. 148:178–187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Weiner HL. 1994. Oral tolerance. Proc. Natl. Acad. Sci. U. S. A. 91:10762–10765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Weiner HL, et al. 1993. Double-blind pilot trial of oral tolerization with myelin antigens in multiple sclerosis. Science 259:1321–1324 [DOI] [PubMed] [Google Scholar]
- 44. Xi C, et al. 2009. A novel recombinant peptide containing only two T-cell tolerance epitopes of chicken type II collagen that suppresses collagen-induced arthritis. Mol. Immunol. 46:729–737 [DOI] [PubMed] [Google Scholar]
- 45. Xu J, et al. 2011. Intranasal vaccination with chitosan-DNA nanoparticles expressing pneumococcal surface antigen a protects mice against nasopharyngeal colonization by Streptococcus pneumoniae. Clin. Vaccine Immunol. 18:75–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Zavazava N, et al. 2000. Oral feeding of an immunodominant MHC donor-derived synthetic class I peptide prolongs graft survival of heterotopic cardiac allografts in a high-responder rat strain combination. J. Leukoc. Biol. 67:793–800 [DOI] [PubMed] [Google Scholar]
- 47. Zhang X, Izikson L, Liu L, Weiner HL. 2001. Activation of CD25(+)CD4(+) regulatory T cells by oral antigen administration. J. Immunol. 167:4245–4253 [DOI] [PubMed] [Google Scholar]
- 48. Zhu P, et al. 2007. Oral administration of type-II collagen peptide 250-270 suppresses specific cellular and humoral immune response in collagen-induced arthritis. Clin. Immunol. 122:75–84 [DOI] [PubMed] [Google Scholar]