

Abstract
A group of synthetic antimicrobial oligomers, inspired by naturally occurring antimicrobial peptides, were analyzed for the ability to modulate innate immune responses to Toll-like receptor (TLR) ligands. These synthetic mimics of antimicrobial peptides (SMAMPs) specifically reduced cytokine production in response to Staphylococcus aureus and the S. aureus component lipoteichoic acid (LTA), a TLR2 agonist. Anti-inflammatory SMAMPs prevented the induction of tumor necrosis factor (TNF), interleukin 6 (IL-6), and IL-10 in response to S. aureus or LTA, but no other TLR2 ligands. We show that these SMAMPs bind specifically to LTA in vitro and prevent its interaction with TLR2. Importantly, the SMAMP greatly reduced the induction of TNF and IL-6 in vivo in mice acutely infected with S. aureus while simultaneously reducing bacterial loads dramatically (4 log10). Thus, these SMAMPs can eliminate the damage induced by pathogen-associated molecular patterns (PAMPs) while simultaneously eliminating infection in vivo. They are the first known SMAMPs to demonstrate anti-inflammatory and antibacterial activities in vivo.
INTRODUCTION
Infectious diseases have become the most pressing global health care problem in recent years (31). The increasing occurrence of multidrug-resistant infections and the low discovery rate of conventional antibiotics create an urgent need for new strategies to fight bacterial infections (42). Antimicrobial peptides (AMPs) isolated from organisms across the phylogenetic spectrum form part of the innate immune system and serve as the first line of defense against microbial infection in many species. Over the last decade, there has been considerable interest in developing AMPs as potent antibiotics (12, 39, 50). However, despite extensive efforts in the pharmaceutical and biotechnology industries, this goal has proven difficult to achieve. The size, stability, tissue distribution, and toxicity of AMPs have made them difficult therapeutic candidates. Furthermore, the native structures of AMPs remain relatively complex, and efforts are being made to study their biological activities in simpler structures. This has led several research groups to develop synthetic mimics of antimicrobial peptides (SMAMPs), which capture the essential biological properties of AMPs (10, 38, 40, 44). A number of SMAMPs, designed on peptidomimetic principles, have been identified as potent and selective compounds that demonstrate broad-spectrum antimicrobial activities in vitro and low cytotoxicities against mammalian cells (38).
Recent efforts have been dedicated to altering the structural and physicochemical properties of SMAMPs in order to improve their antimicrobial activity and to reduce their toxicity to host cells. One particular class of SMAMPs, based on small aromatic oligomers, has been extensively studied (38), leading to good in vivo activity (6, 38). In fact, PolyMedix Inc. is currently conducting a phase 2 clinical trial for the treatment of Staphylococcus infections with PMX-30063, a SMAMP structurally similar to several of those tested in this study. For example, SMAMP 02, a close structural analogue of PMX-30063, induced a 5 log10 reduction in bacterial-cell counts (CFU) in a neutropenic mouse model of Staphylococcus aureus infection (6).
Apart from the ability of AMPs/SMAMPs to directly kill bacteria, certain AMPs can bind the pathogen-associated molecular pattern (PAMP) molecules originating from Gram-negative and Gram-positive bacteria, which in turn reduces their interactions with Toll-like receptors (TLRs). This interaction of AMPs with selective PAMPs has been implicated in the anti-inflammatory functions of those AMPs (11, 34). For example, human cathelicidin peptide LL-37 binds lipopolysaccharide (LPS) and prevents its interaction with LPS binding protein, thereby reducing its ability to stimulate macrophages and inhibiting proinflammatory cytokine secretion (33, 36). However, these AMPs, including LL-37, have very moderate antimicrobial activities (MIC of LL-37, >32 μM against both Escherichia coli and S. aureus) (8, 33) and likely are not produced in high enough concentrations to be antimicrobial in vivo (3). This and the other limitations of AMPs described above render LL-37 a poor therapeutic candidate. We hypothesized that SMAMPs, with their excellent in vitro and in vivo antibacterial activity, may also exhibit (AMP-like) specific PAMP binding, reducing inflammatory activity. Although the antimicrobial activity of SMAMPs has been extensively studied, their interactions with PAMPs and ensuing anti-inflammatory activity have not been explored.
We selected a group of antibacterial SMAMPs that are nontoxic to host cells and screened them as possible modulators of proinflammatory cytokine production by RAW 264.7 cells in both the absence and presence of the bacterial components LPS and lipoteichoic acid (LTA). We identified SMAMPs with the capacity to modulate cytokine responses of murine macrophages both in vitro and in vivo. Our results describe a group of novel synthetic antimicrobial compounds that also involve the blockade of PAMPs and can act as anti-inflammatory compounds, as well. These novel SMAMPs open new therapeutic opportunities to target prevalent infections and at the same time arrest the inflammatory response caused by the infection and/or the antibacterial treatment to cure the infection.
MATERIALS AND METHODS
Antimicrobial molecules.
All SMAMPs were provided by Polymedix Inc., PA. Their structures, antimicrobial activities, and cytotoxicities are available in Scheme S1 and Tables S1 and S2 in the supplemental material. A 10-mg/ml stock solution was prepared in dimethyl sulfoxide (DMSO) (Sigma-Aldrich Inc., St. Louis, MO) and diluted in Hanks balanced salt solution (HBSS) (Lonza, Walkersville, MD) as necessary.
Reagents, bacterial strains, and cells.
Ultrapurified LPS from Salmonella enterica subsp. enterica serovar Minnesota R595 (S. Minnesota) was from List Biological Laboratories, Inc. (Campbell, CA). All other TLR ligands were purchased from InvivoGen (San Diego, CA). A recombinant murine TLR2-Fc chimera was obtained from R&D systems, Inc. (Minneapolis, MN). Goat anti-human IgG-alkaline phosphatase was from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA).
S. aureus isolates (ATCC 13709 and ATCC 19636) were grown overnight in Bacto Heart Infusion (BHI) broth (BD Biosciences, San Diego, CA) and washed in HBSS. Bacterial concentrations were calculated from optical density at 600 nm (OD600) values (Genesys 2 Spectronic; Thermo Fisher Scientific Inc., Madison, WI). S. aureus isolates were plated in Difco Heart Infusion (DHI) agar (BD Biosciences).
RAW264.7 cells gamma NO− (ATCC, Manassas, VA) were cultured in RPMI 1640 (Lonza) containing 10% fetal bovine serum (FBS). Mouse bone marrow-derived macrophages (BMDMs) were obtained as described previously (51) (see the supplemental material). HEK-hTLR2 cells (InvivoGen) were cultured in Dulbecco's modified Eagle's medium (DMEM) (ATCC) containing 4.5 g/liter glucose and 10% FBS.
Stimulation with TLR ligands and S. aureus.
The following general stimulation program was followed unless otherwise stated. RAW 264.7 cells (1 × 106) or BMDMs (see the supplemental material) were seeded in 12-well plates in 1 ml of complete RPMI medium. After 3 h, the cells were incubated with 1 μg/ml SMAMP for 1 h and then stimulated with the following concentrations of TLR ligands: LTA (10 μg/ml), LPS (100 ng/ml), poly(IC) (1.0 μg/ml), R848 (3.1 μg/ml), CpG oligonucleotide (10 μg/ml), and ODN control (10 μg/ml). The cells were incubated for 12 to 14 h, and the supernatants were assessed for tumor necrosis factor (TNF), interleukin 6 (IL-6), and IL-10 by enzyme-linked immunosorbent assay (ELISA) (BD Biosciences) according to the manufacturer's instructions.
For S. aureus stimulation, RAW 264.7 cells or BMDMs (1 × 106/ml) were seeded in 12-well plates in antibiotic-free RPMI medium. The cells were treated with 1 μg/ml SMAMP for 1 h prior to the addition of S. aureus (multiplicity of infection [MOI] = 10). After 4.5 h or 12 h of incubation, cytokine levels were assessed in the supernatants by ELISA.
Screening of TLR2 ligands by NF-κB reporter assay.
HEK-Blue-hTLR2/CD14 cells were seeded onto 96-well plates (1 × 105 cells/well) in 200 μl complete DMEM. After 2 h, the cells were incubated with 1 μg/ml SMAMP for 30 min, followed by stimulation with different TLR2 ligands: FSL-1 (synthetic diacylated lipoprotein; 10 ng/ml), Pam2CSK4 (synthetic diacylated lipopeptide; 10 ng/ml), Pam3CSK4 (synthetic triacylated lipopeptide; 100 ng/ml), PgLPS (LPS from Porphyromonas gingivalis; 1.0 μg/ml), LM-MS (lipomannan from Mycobacterium smegmatis; 100 ng/ml), and LTA (500 ng/ml). After 5 h of incubation at 37°C (5% CO2), 20 μl supernatant was added to 180 μl Quanti-Blue solution in a 96-well plate and incubated for 1 to 2 h for color development. Secreted embryonic alkaline phosphatase (SEAP) activity was assessed by spectrometry at OD655 in a Multi-Mode Microplate Reader Synergy 2 (BioTek Instrumentns Inc., Winooski, VT).
UV-visible (Vis) spectroscopy.
Serial dilutions (0 to 100 μg/ml) of LTA or Pam3CSK4 in HBSS were mixed with vehicle (1% DMSO) or SMAMP 02 (100 μg/ml) in 96-well plates. The plates were incubated at room temperature for 20 min with constant shaking. The absorption spectra were then recorded at 250 to 500 nm in a Multi-Mode Microplate Reader Synergy 2.
TLR2 binding competition assay.
The abilities of SMAMPs to compete for the binding of LTA and Pam3CSK4 to a recombinant mouse TLR2-Fc chimera were determined by a microtiter assay. Ninety-six-well plates were coated with LTA and Pam3CSK4 (0.5 μg/well) in chloroform-ethanol (1:9). The solvent was removed by evaporation at 37°C. The plates were then washed with phosphate-buffered saline (PBS)-0.1% Tween 20 (PBS-Tween) and blocked for 1 h with PBS-1% bovine serum albumin (BSA) at room temperature. The coated wells were then incubated with SMAMP 02 (1 μg/ml) or DMSO (control), followed by the addition of recombinant mouse TLR2-Fc chimera (0.5 μg/ml). After 3 h of incubation at 4°C, the plates were washed with PBS-Tween and incubated with goat anti-human IgG-AP (0.04 μg per well in PBS-1% BSA) for 1 h at room temperature. The plates were then washed with PBS-Tween, followed by incubation with 100 μl BluePhos Microwell Substrate (Kirkegaard & Perry Laboratories, Inc., Gaithersburg, MD). Color development was allowed for 10 to 20 min, followed by absorbance determination at 620 nm.
qRT-PCR.
RNA isolated from cells was analyzed for gene expression by quantitative real-time reverse transcription (RT)-PCR (qRT-PCR). Fold changes for tnf gene expression were normalized to actin and are relative to the gene expression in vehicle (0.01% DMSO)-stimulated cells using the ΔΔCT method (28). The primers used were as follows: actin, forward, 5′-GAC GAT GCT CCC CGG GCT GTA TTC-3′, and reverse, 5′-TCT CTT GCT CTG GGC CTC GTC ACC-3′; tnf, forward, 5′-AGC CCA CGT CGT AGC AAA CCA C-3′, and reverse, 5′-ATC GGC TGG CAC CAC TAG TTG GT-3′.
Acute infection of mice with S. aureus.
Six- to 8-week-old C57BL/6 mice were infected by intramuscular injection of 1 × 105 S. aureus ATCC 13709 cells (in 0.1 ml HBSS) in the right posterior thigh. The mice were treated (intraperitoneally [i.p.]) twice (at 1 h and 7 h postinfection) with 0.1 ml of SMAMP 02 (1 mg/kg of body weight) or vehicle (HBSS containing 2% DMSO). Twenty-five hours after infection, the thigh muscle at the site of infection was aseptically collected for quantification of the bacterial burden. The preweighed muscle was homogenized in 250 μl HBSS with a Kontes Pellet Pestle, and 20-μl aliquots of serial dilutions were plated on DHI agar plates. The plates were incubated at 37°C for 10 to 12 h. Colony counts were used to calculate the number of CFU per g of tissue. Murine sera were also collected at sacrifice and analyzed for cytokine levels by ELISA. All experiments involving mice were approved by the Institutional Animal Care and Use Committee at the University of Massachusetts, Amherst.
Data analysis.
The results were analyzed using GraphPad Prism version 5.01. Data are reported as the average ± SEM. The means were compared using the Student t test. A P value of ≤0.05 was considered significant.
RESULTS
Identification of SMAMPs that thwart the stimulatory capacity of LTA in RAW264.7 cells.
Twenty-five SMAMP candidates were selected based on their antimicrobial activities and lack of cytotoxicity (see Tables S1 and S2 in the supplemental material for antimicrobial activity in terms of the MICs and cytotoxicity values of the SMAMPs screened). RAW264.7 cells were preincubated with 1.0 μg/ml SMAMP or 0.01% DMSO vehicle (control) for 1 h and stimulated with LTA or LPS for 12 h. TNF production was then evaluated by ELISA, and the percent reductions of TNF induced by LTA or LPS in the presence of these SMAMPs are shown (see Fig. S1 in the supplemental material). A group of SMAMPs (01 to 05) reduced LTA-induced TNF production by >65% (see Fig. S1a and c, solid bars, in the supplemental material); specifically, SMAMPs 01, 02, and 03 inhibited TNF production more than 70% (see Fig. S1c in the supplemental material). In contrast, the inhibitory effects of all SMAMPs tested in response to LPS stimulation were below 40% (see Fig. S1b and c, gray bars, in the supplemental material). None of the SMAMPs showed any stimulatory effect on RAW264.7 cells in the absence of LTA or LPS (Fig. 1A shows SMAMPs 01 to 03).
Fig 1.
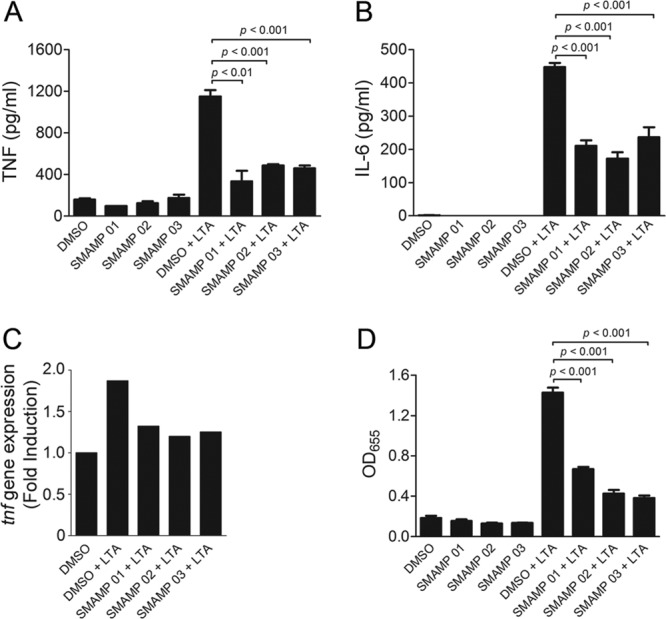
SMAMPs specifically inhibit TLR2-induced responses. (A and B) RAW 264.7 cells (106 per ml) were preincubated with SMAMPs (1.0 μg/ml) or 0.01% DMSO (control) for 1 h and stimulated with LTA (10 μg/ml) for 12 h. The stimulation supernatants were analyzed for TNF and IL-6 production by ELISA. The data are presented as the average and SEM of triplicate samples and are representative of at least 3 independent experiments. (C) RAW 264.7 cells (106 per ml) were preincubated with the indicated SMAMPs (1.0 μg/ml) or DMSO (control) for 1 h and stimulated with LTA (10 μg/ml) for 4 h. The cells were used to extract RNA and analyze tnf gene expression levels by qRT-PCR. Fold changes for tnf gene expression levels were normalized to actin and are relative to tnf expression in 0.01% DMSO-treated cells using the ΔΔCT method. The data are representative of 2 independent determinations. (D) HEK-hTLR2 cells (0.5 × 106/ml) were pretreated with 1.0 μg/ml SMAMPs or DMSO (control) for 30 min and stimulated with 500 ng/ml LTA. After 5 h of incubation, NF-κB-induced SEAP production was determined by alkaline phosphatase activity in the supernatants using Quanti-Blue as the substrate. The reactions were quantified at OD655. The data are presented as the average and SEM of triplicate samples and are representative of at least 3 independent determinations.
SMAMPs 01 to 03 inhibit TLR2-mediated signals and the response of macrophages to S. aureus.
SMAMPs 01, 02, and 03, which showed the greatest TNF reduction capacity in LTA-stimulated RAW264.7 cells, were then chosen to further characterize their immunomodulatory activities. These SMAMPs significantly inhibited the production of TNF in response to LTA (Fig. 1A). The same inhibitory effect was observed for the proinflammatory cytokine IL-6 (Fig. 1B). The inhibition of TNF production occurred at the transcriptional level, as assessed by real-time RT-PCR of tnf transcripts in RAW264.7 cells stimulated with LTA (Fig. 1C). Indeed, the analysis of NF-κB activation in response to LTA in SEAP reporter HEK-Blue-hTLR2 cells showed significant SEAP activity inhibition (Fig. 1D). Overall, these results demonstrated that SMAMPs 01 to 03 inhibit TLR2-induced NF-κB activation and proinflammatory cytokine gene expression.
The specificity of SMAMPs to suppress the agonistic effect of S. aureus LTA prompted us to evaluate their activities against the proinflammatory response to whole bacteria. RAW264.7 cells were preincubated with 1.0 μg/ml of SMAMPs 01 to 03 and challenged with S. aureus at an MOI of 10. After 16 h, TNF and IL-6 present in the supernatants were measured by ELISA. The preincubation with the SMAMPs resulted in an ∼95% inhibition of TNF production (Fig. 2A), while IL-6 production was inhibited ∼65% (Fig. 2B). We next evaluated the inhibitory activities of the SMAMPs on murine BMDMs to ensure that primary cells responded similarly. SMAMP-mediated suppression of TNF and IL-6 production in response to S. aureus challenge was readily observed (Fig. 2C-1 and D). In addition, SMAMP induced 45-fold suppression of tnf expression as assessed by real-time RT-PCR of BMDMs stimulated with S. aureus (Fig. 2C-2). Interestingly, production of the anti-inflammatory cytokine IL-10 in response to S. aureus was also suppressed in the presence of the SMAMPs (Fig. 2E), indicating that the observed inhibitory effect on TNF and IL-6 production induced by these oligomers does not involve the induction of IL-10.
Fig 2.
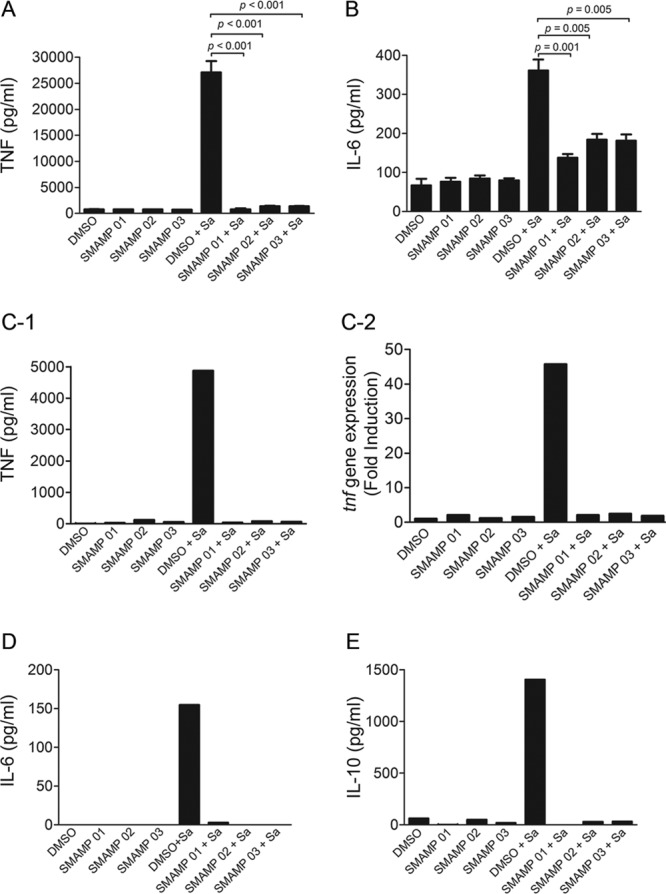
SMAMPs 01 to 03 inhibit macrophage proinflammatory responses to S. aureus. RAW 264.7 cells (A and B) and BMDMs (C-1 and C-2) (106 per ml) were stimulated with S. aureus (Sa; ATCC 19636; 1 × 107 CFU/ml) for 12 h and 4.5 h, respectively, in the presence or absence of SMAMPs (1.0 μg/ml). (A and B) TNF and IL-6 were measured in the stimulation supernatants by ELISA. The data are averages and SEM of three replicates and represent three independent experiments. (C-1) TNF production was assessed in the stimulation supernatants by ELISA. (C-2) RNA was isolated, and qRT-PCR was performed. Fold changes for tnf were normalized to actin and are relative to the tnf expression in 0.01% DMSO-treated cells. (D and E) BMDMs (106 per ml) were stimulated with S. aureus (MOI = 10) in the presence or absence of SMAMP (1.0 μg/ml) for 4.5 h. IL-6 and IL-10 were measured in the stimulation supernatants by ELISA. Under all conditions, the cells were pretreated with the SMAMPs for 1 h before S. aureus stimulation; 0.01% DMSO was used as a control. (C, D, and E) The data represent two independent experiments.
The anti-inflammatory activity of SMAMPs is specific for LTA.
As previously observed (Fig. 3A; see Fig. S1b in the supplemental material), SMAMPs 01 to 03 did not affect the TNF production induced by the TLR4 ligand LPS. Therefore, in order to determine whether the effects of these SMAMPs were specific for TLR2 agonists or more broadly active against other TLRs, we tested the TLR3 ligand poly(I · C), the TLR7/8 ligand R848, and the TLR9 ligand CpG DNA. Preincubation of RAW264.7 cells with the three SMAMPs did not affect the production of TNF in response to these TLR agonists (Fig. 3A), indicating that the SMAMPs are specific for LTA/TLR2-induced responses.
Fig 3.

SMAMP-mediated cytokine inhibition is specific for LTA. (A) RAW 264.7 cells (106 per ml) were stimulated with LPS (100 ng/ml), poly(I · C) (1.0 μg/ml), R848 (3.1 μg/ml), and CpG oligonucleotide and ODN (10.0 μg/ml) in the presence of three different SMAMPs (1.0 μg/ml) for 12 h. TNF production was measured in the stimulation supernatants by ELISA. RAW 264.7 cells (106 per ml) were pretreated with SMAMPs or DMSO (control) for 1 h before stimulation. (B) HEK-hTLR2 cells (0.5 × 106 per ml) were pretreated with 1.0 μg/ml SMAMPs or DMSO (control) for 30 min and stimulated with different TLR2 ligands: FSL-1 (10 ng/ml), Pam2CSK4 (10 ng/ml), Pam3CSK4 (100 ng/ml), PgLPS (1.0 μg/ml), LM-MS (100 ng/ml), and LTA (500 ng/ml). After 5 h of incubation, NF-κB-induced SEAP production was determined by alkaline phosphatase activity in the stimulation supernatants using Quanti-Blue as the substrate. The data shown represent the averages and SEM of triplicate determinations and are representative of three independent experiments.
The abilities of these novel oligomers to inhibit TLR2-mediated stimulation in the presence of several TLR2 ligands were investigated to determine if inhibition was LTA specific or general for TLR2 ligands. We preincubated HEK-Blue-hTLR2/CD14 cells with SMAMPs 01 to 03 for 30 min, followed by 5 h of stimulation with different TLR2 ligands. Both TLR1/TLR2 and TLR2/TLR6 ligands were used (1, 25, 26, 30): FSL-1, Pam3CSK4, Pam2CSK4, LM, and PgLPS. The results (Fig. 3B) show that SMAMPs inhibited the stimulatory effects of only LTA and not the other TLR2 ligands tested.
SMAMPs interact with S. aureus LTA and prevent its interaction with TLR2.
Our results suggested that SMAMPS exert their inhibitory activities via a specific interaction with LTA. To further investigate this possibility, RAW264.7 cells were preincubated with SMAMPs, washed, and resuspended in fresh antibiotic-free medium, allowing the elimination of any SMAMPs not associated with the cells. The cells were then challenged with S. aureus (MOI = 10), and after 14 h, the production of TNF and IL-6 in the stimulation supernatants was analyzed by ELISA (Fig. 4A and B, denoted preincubation [open bars]). As a control, RAW 264.7 cells that had been incubated with SMAMP for 1 h were challenged directly with S. aureus without replacing the medium (Fig. 4A and B, solid bars) (similar to the previously described procedure for Fig. 2A and B). The elimination of the SMAMPs by washing prior to bacterial challenge resulted in the disappearance of their inhibitory effects. These data indicated that the inhibitory effects of the SMAMPs are not due to their interaction with the cells but primarily to that with S. aureus or its components.
Fig 4.
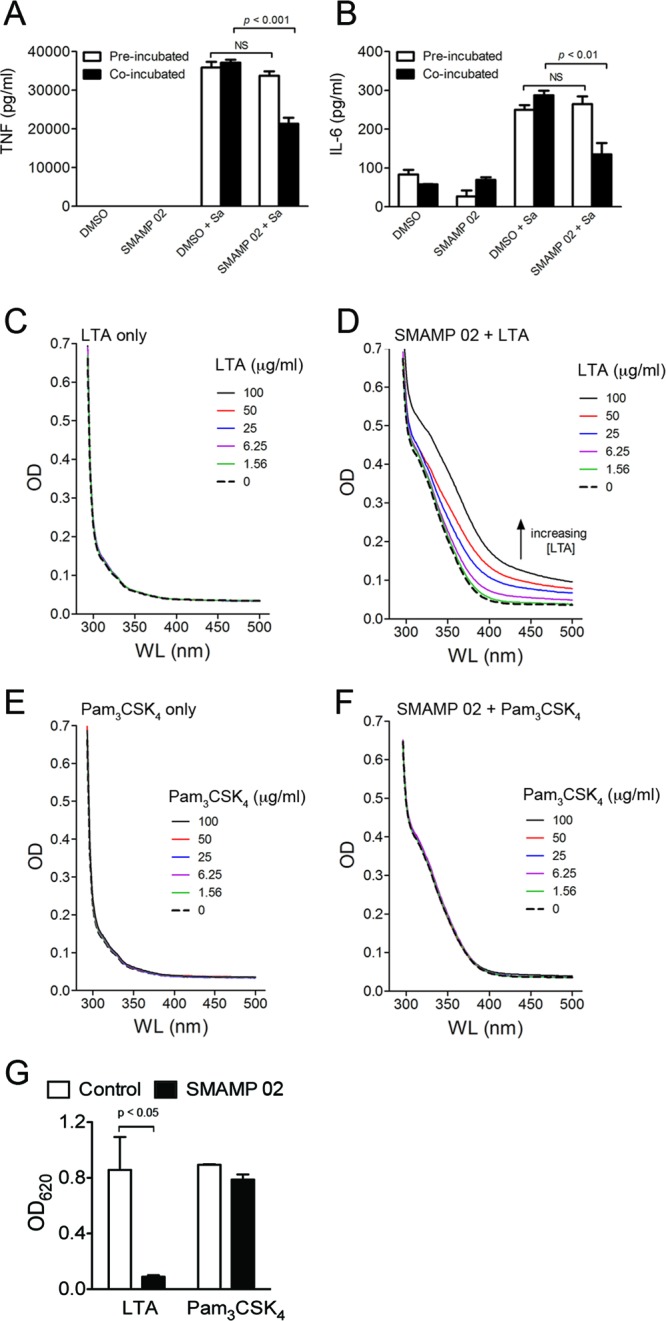
SMAMPs interact with LTA. (A and B) RAW 264.7 cells (106 per ml) (open bars) were pretreated with SMAMPs (1.0 μg/ml) or DMSO (control). The cells were then washed and incubated in antibiotic-free RPMI for 1 h before stimulation with S. aureus (MOI = 10) for 14 h. The cells (solid bars) were also pretreated with SMAMPs and stimulated with S. aureus (MOI = 10) for 14 h without medium change. The supernatants were assessed for TNF and IL-6 production by ELISA. The data shown represent the averages and SEM of triplicate determinations and are representative of three independent experiments. (C to F) UV titration of 1% DMSO (C and E) and 100 μg/ml SMAMP 02 (D and F) with increasing concentrations of LTA (C and D) or Pam3CSK4 (E and F). The results are representative of three independent determinations. (G) TLR2 binding competition assay. Wells were coated with LTA or Pam3CSK4 (0.5 μg/well) in chloroform-ethanol (1:9), followed by incubation with SMAMP (1 μg/ml) and TLR2-Fc (0.5 μg/ml). Alkaline phosphatase activity was determined using BluePhos phosphatase substrate. The data shown represent the averages and SEM of triplicate determinations and are representative of three independent experiments.
Further evidence of SMAMP-LTA interaction was obtained using UV-Vis absorption spectroscopy to measure their association in solution. LTA is devoid of any chromophore in the visible range (350 to 500 nm) and did not show significant absorption as a function of the concentration (Fig. 4C). In contrast, the unique aromatic chromophore of the SMAMP shows a distinct, broad absorption band starting at ∼375 nm (Fig. 4D, dashed line). This allowed us to detect LTA-SMAMP interactions by analyzing the changes in the SMAMP absorption spectra as a function of the LTA concentration. Increasing the LTA concentration while holding the SMAMP concentration constant resulted in a dose-dependent increase in the absorption spectrum of SMAMP 02 (Fig. 4D). As a control, we also analyzed the UV absorption of SMAMP 02 in the presence of the TLR1/2 ligand Pam3CSK4, whose proinflammatory activity was not inhibited by the SMAMP. Similar to LTA, Pam3CSK4 showed no absorption between 350 and 500 nm (Fig. 4E). In contrast to LTA, the presence of increasing concentrations of Pam3CSK4 did not result in measurable changes in the absorption spectra of SMAMP 02 (Fig. 4F). These UV-Vis experiments confirmed that SMAMP 02 interacts specifically with LTA and not other TLR2 ligands, such as Pam3CSK4.
These results suggested that SMAMPs may prevent the interaction of LTA with TLR2. To assess whether this is the case, we performed TLR2 binding competition assays. Microtiter plates were coated with LTA and Pam3CSK4 as a control, treated with SMAMP 02, and then exposed to a soluble form of recombinant TLR2 (TLR2-Fc chimera). Complexes were then detected using anti-human IgG. The preincubation of LTA with SMAMP 02 significantly reduced its ability to bind soluble TLR2, while no effect was observed when Pam3CSK4 was used (Fig. 4G). Overall, these results demonstrate that SMAMP 02 binds specifically to LTA, resulting in its inability to interact with TLR2.
SMAMPs prevent inflammation in vivo, in addition to clearing bacteria.
Our results strongly argue that treatment with SMAMPs 01 to 03 during infection with S. aureus could exert an anti-inflammatory effect, in addition to their proven antibacterial activities (6). We further analyzed the anti-inflammatory and antibacterial capacities of SMAMP 02 in immunocompetent mice in order to analyze innate immune cell function during infection. C57BL/6 mice were infected with 105 CFU S. aureus (ATCC 13709) in the thigh muscle and treated (i.p.) with SMAMP 02 (1 mg/kg, well below the tolerated dose of 20 mg/kg [6]) in two doses, at 1 h and 7 h postinfection. Control infected mice were treated with vehicle (HBSS containing 2% DMSO). The thighs were harvested 25 h after infection, and the bacterial burdens were quantified by CFU enumeration on BHI agar plates. This thigh muscle infection model did not result in sepsis or any other visible disease symptoms in animals within the time frame of our studies. An average bacterial burden of 104 CFU/g of thigh tissue was detected in the vehicle-treated control mice (Fig. 5A). In contrast, SMAMP 02-treated animals showed bacterial burdens below the detection limit of the assay (Fig. 5A), demonstrating that this SMAMP is highly efficient in clearing infections in immunocompetent mice. At the same time, the sera from SMAMP 02-treated infected mice contained significantly lower levels of TNF and IL-6 (Fig. 5B and C) than control infected mice, indicating that the treatment resulted in both the control of infection and the suppression of deleterious proinflammatory systemic responses.
Fig 5.

Antibacterial and anti-inflammatory effects of SMAMPs in a mouse model of acute infection with S. aureus. (A) C57BL/6 mice were infected intramuscularly with 1 × 105 S. aureus cells (ATCC 13709) in the right posterior thigh. The infected mice were injected intraperitoneally with 1 mg/kg SMAMP 02 or vehicle (HBSS containing 2% DMSO; control) at 1 h and 7 h after infection. The thighs were harvested 25 h after infection, and the bacterial burden was quantified by CFU enumeration. (B and C) Sera from the infected mice were assessed for TNF and IL-6 by ELISA. The results represent 4 to 5 mice per group in three independent experiments. The error bars indicate SEM.
DISCUSSION
Infectious diseases, especially those caused by multidrug-resistant bacteria, remain one of the most serious global health care problems. While traditional antibiotics revolutionized medical care in the last century, they have two serious drawbacks. Bacteria have developed resistance to our best antibiotics, and in fact, bacterial strains that are resistant to all but one commonly prescribed antibiotic are known (9, 14, 22, 24, 41, 45). In addition, the use of antibiotics often stimulates the release of bacterial components that can contribute to toxic shock (29). For example, the use of β-lactam antibiotics, such as imipenem (a carbapenem), flucloxacillin (a penicillin), or cefamandole (a cephalosporin), enhances LTA release from Gram-positive cocci (17, 47). Toxic shock is a major problem; more than half a million people suffer toxic shock (sepsis) every year in North America alone (13). Gram-negative sepsis is caused by the outer membrane component LPS, whereas Gram-positive sepsis is associated with exotoxin production, as well as cell wall components, such as LTA (16, 19, 46). An in vivo study reported that cell wall components, including LTA, from S. aureus induced proinflammatory cytokine production and multiple organ dysfunction syndrome (MODS) associated with septic shock (7). Therefore, it would be ideal when developing new weapons to combat 21st century infections if these potential new drugs could make antibiotic resistance development more difficult while simultaneously limiting sepsis and its related pathologies.
Here, we demonstrate for the first time that a nonpeptidic mimic of antimicrobial peptides effectively eliminates the proinflammatory activity associated with S. aureus infection while simultaneously clearing the microorganism in the animal. One of the hallmarks of AMPs and our SMAMPs is a low propensity for the development of bacterial resistance (6, 27, 39, 40, 50). Our results show that these SMAMPs specifically inhibit TLR2-dependent, LTA-induced responses by macrophages, while they do not affect the response of these cells to other TLR agonists. Our results show that the effect of SMAMPs on LTA-induced TLR2-mediated signaling results in the inhibition of NK-κB-mediated transcriptional responses (Fig. 3B). This results in an effect on the production of the proinflammatory cytokines TNF and IL-6, as well as the anti-inflammatory cytokine IL-10. However, due to the effect of SMAMPs on NF-κB-mediated transcriptional activity, we expect that other cytokines regulated by the transcription factor would also be affected by these SMAMPs. Furthermore, the inhibition of LTA-induced TNF production was also observed in human peripheral blood mononuclear cells (PBMCs) in the presence of SMAMP 02 (see Fig. S2 in the supplemental material). Importantly, these SMAMPs showed a high level of anti-inflammatory activity in response to whole S. aureus cells. These results demonstrate that by effectively blocking LTA-induced proinflammatory responses, these novel synthetic compounds prevent the overall response of macrophages to the bacterium. Further ongoing studies based on our screening experiment (see Fig. S1 in the supplemental material) have identified a few other SMAMPs that may have the potential to suppress both LPS- and LTA-induced TNF production.
The murine macrophage cell line RAW264.7 and murine BMDMs responded similarly, showing strong inhibition of TNF, IL-6, and IL-10. This inhibition was controlled at the transcription level, as demonstrated by qRT-PCR. Perhaps most important were the in vivo experiments performed with immunocompetent animals. Previous studies showed SMAMP 02 was able to reduce S. aureus bacterial counts by ∼4 to 5 log10 units in neutropenic mice (6). Here, SMAMP 02 was able to completely clear the S. aureus infection compared to the control, which retained a bacterial burden of 3.8 log10. These results, coupled with those from the neutropenic animals, conclusively show that SMAMP 02 is antibacterial. A structurally similar SMAMP (PMX-30063) completed a phase 2 clinical trial for the treatment of acute bacterial skin and skin structure infections caused by S. aureus; the trial achieved its objectives of meeting efficacy and safety standards at all evaluated doses of PMX-30063. At the same time, in this report, SMAMP 02 effectively eliminated the production of the proinflammatory cytokines TNF and IL-6. The reduction was more than 98%. This is critical, as TNF is known to be associated with deleterious effects of toxic shock (15, 20, 34). To our knowledge, this is the first example of a specific, nonpeptidic synthetic PAMP blocker that can specifically prevent proinflammatory responses while simultaneously clearing infections in vivo. In a previous report, the pharmacokinetics study of SMAMP 02 was performed, and the plasma half-life (t1/2 = 0.83 to 0.94 h), clearance (CL = 108 to 135 ml/h/kg), and volume of distribution (Vz = 146 to 162 ml/kg) values were reported (6). The clearance values correspond to a small percentage of liver blood flow in the mouse, and the volume of distribution range suggests that SMAMP 02 is being distributed primarily in plasma and extracellular fluid (6).
Our results show that immunomodulatory SMAMPs prevent LTA-induced responses mediated by TLR2 through a direct interaction with the pathogen molecular pattern. The interaction between SMAMPs and LTA occurs in solution, as well as with whole bacterial cells in vivo. Importantly, our results demonstrate that SMAMPs thwart the accessibility of LTA to TLR2. The result is the effective blockade of the interaction of LTA with TLR2, both on the surfaces of macrophages and within endosomal compartments, upon bacterial phagocytosis (18). Previous research has shown the capacity of a group of peptide-based antimicrobials to bind LPS and neutralize its ability to stimulate the production of proinflammatory cytokines (11, 35). Synthetic AMPs (e.g., cecropin-melittin-related peptides) also exhibit LTA-binding capacity and inhibit LTA-stimulated cytokine production from RAW 264.7 cells (2, 34, 35). A combination of synthetically altered AMPs, temporin B and temporin A, also exerted in vivo antibacterial and anti-inflammatory activities (5). However, these AMPs exhibited only moderate antimicrobial activity in in vitro assays, with MICs of >10 μg/ml against both Gram-positive and Gram-negative bacteria. Indeed, the first reported synthetic immunomodulatory peptide based on an AMP template, innate defense regulator 1 (IDR-1), was devoid of any direct antimicrobial activity and instead induced protection against bacterial challenge by selectively modulating innate immune responses (37).
Most AMPs, including those discussed above, are comprised of 12 to 50 amino acid residues with molecular masses between 2,500 and 5,000 Da (e.g., LL-37 is a 4,500-Da peptide) and a formal secondary structure. Shorter synthetic peptides with helical conformation, containing 7 to 11 amino acid residues, are also known, and some of these peptides displayed substantial antimicrobial activity, e.g., a 9-residue peptide displayed potent antibacterial activity (with a MIC as low as <1.3 μg/ml against Gram-positive bacteria), although it was severely hemolytic (minimal hemolytic concentration [MHC], 5 μg/ml) (21, 49). No in vivo activity was reported for these short AMPs, which is not surprising, since such short peptides are expected to have short half-lives due to rapid degradation in vivo. However, our design of SMAMPs goes beyond amino acid-derived peptidomimetics to completely abiotic molecular scaffolds (23, 40, 44). This reduced the molecular mass (SMAMPs range between 600 and 1,000 Da) and dramatically expands the structural derivatives that can be envisaged by employing the entire range of modern synthetic chemistry. This strategy has eliminated many of the problems related to the use of AMPs as intravenously administered antibiotics, such as poor tissue distribution, toxicity, and proteolysis.
Natural AMPs are important components of the innate defense systems of all animals, and therefore, their synthetic variants hold great potential as weapons against antibiotic-resistant bacteria (13). Although several new approaches to treating Gram-negative sepsis have been reported in recent years (32, 48), very little progress has been achieved in finding therapies targeting sepsis mediated by Gram-positive bacteria (34). This, coupled with the additional benefit of using artificial compounds to which resistance is unlikely to arise (4, 6, 38, 43, 45), makes these SMAMPs ideal candidates for the treatment of acute infections with S. aureus. Our results present the first proof that a nonpeptidic small molecule can confer protection against infection by a Gram-positive pathogen without toxicity while simultaneously suppressing harmful proinflammatory responses. It is worth noting that the PAMP-binding antibacterial SMAMPs we evaluated here were not originally designed to control PAMP-mediated anti-inflammatory activity. Therefore, we anticipate that a significant research opportunity exists to develop PAMP binding SMAMPs with the capacity to prevent secondary inflammation, in addition to fighting prevalent infectious agents.
Supplementary Material
ACKNOWLEDGMENTS
We gratefully acknowledge Dylan Clements for microbiological susceptibility assays and Yongjiang Xu, Haizhong Tang, Dahui Liu, Xiaodong Fan, Trevor Young, and Carol Mulrooney for SMAMP synthesis.
This work was supported by NIH contract U01 AI082192 (to G.N.T.) and a Grand Challenges Explorations grant from the Gates Foundation (to G.N.T. and J.A.).
We declare competing financial interests. G.N.T. is on the Advisory Board of Polymedix, Inc., and R.W.S. is an employee of PolyMedix, Inc., and a participant in the company's stock option plan.
Footnotes
Published ahead of print 5 September 2012
Supplemental material for this article may be found at http://cvi.asm.org.
REFERENCES
- 1. Aliprantis AO, et al. 1999. Cell activation and apoptosis by bacterial lipoproteins through Toll-like receptor-2. Science 285:736–739 [DOI] [PubMed] [Google Scholar]
- 2. Boman HG, Wade D, Boman IA, Wahlin B, Merrifield RB. 1989. Antibacterial and antimalarial properties of peptides that are cecropin-melittin hybrids. FEBS Lett. 259:103–106 [DOI] [PubMed] [Google Scholar]
- 3. Bowdish DME, Davidson DJ, Hancock REW. 2005. A re-evaluation of the role of host defence peptides in mammalian immunity. Curr. Protein Pept. Sci. 6:35–51 [DOI] [PubMed] [Google Scholar]
- 4. Bragonzi A. 2010. Fighting back: peptidomimetics as a new weapon in the battle against antibiotic resistance. Sci. Transl. Med. 2:21ps9. [DOI] [PubMed] [Google Scholar]
- 5. Capparelli R, et al. 2009. Synergistic antibacterial and anti-inflammatory activity of temporin a and modified temporin b in vivo. Plos ONE 4:e7191 doi:10.1371/journal.pone.0007191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Choi S, et al. 2009. De novo design and in vivo activity of conformationally restrained antimicrobial arylamide foldamers. Proc. Natl. Acad. Sci. U. S. A. 106:6968–6973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. De Kimpe SJ, Kengatharan M, Thiemermann C, Vane JR. 1995. The cell-wall components peptidoglycan and lipoteichoic acid from Staphylococcus aureus act in synergy to cause shock and multiple organ failure. Proc. Natl. Acad. Sci. U. S. A. 92:10359–10363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Durr UHN, Sudheendra US, Ramamoorthy A. 2006. Ll-37, the only human member of the cathelicidin family of antimicrobial peptides. Biochim. Biophys. Acta 1758:1408–1425 [DOI] [PubMed] [Google Scholar]
- 9. Fernandes P. 2006. Antibacterial discovery and development: the failure of success? Nat. Biotechnol. 24:1497–1503 [DOI] [PubMed] [Google Scholar]
- 10. Giuliani A, et al. 2008. Antimicrobial peptides: natural templates for synthetic membrane-active compounds. Cell. Mol. Life Sci. 65:2450–2460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Gough M, Hancock REW, Kelly NM. 1996. Antiendotoxin activity of cationic peptide antimicrobial agents. Infect. Immun. 64:4922–4927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hancock REW, Lehrer R. 1998. Cationic peptides: a new source of antibiotics. Trends Biotechnol. 16:82–88 [DOI] [PubMed] [Google Scholar]
- 13. Hancock REW, Scott MG. 2000. The role of antimicrobial peptides in animal defenses. Proc. Natl. Acad. Sci. U. S. A. 97:8856–8861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hancock REW. 2007. The end of an era? Nat. Rev. Drug Discov. 6:28 [Google Scholar]
- 15. Hennessy EJ, Parker AE, O'Neill LAJ. 2010. Targeting Toll-like receptors: emerging therapeutics? Nat. Rev. Drug Discov. 9:293–307 [DOI] [PubMed] [Google Scholar]
- 16. Heumann D, Barras C, Severin A, Glauser MP, Tomasz A. 1994. Gram-positive cell walls stimulate synthesis of tumor necrosis factor alpha and interleukin-6 by human monocytes. Infect. Immun. 62:2715–2721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Horne D, Tomasz A. 1979. Release of lipoteichoic acid from Streptococcus sanguis: stimulation of release during penicillin treatment. J. Bacteriol. 137:1180–1184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ip WKE, Takahashi K, Moore KJ, Stuart LM, Ezekowitz RAB. 2008. Mannose-binding lectin enhances Toll-like receptors 2 and 6 signaling from the phagosome. J. Exp. Med. 205:169–181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kengatharan KM, De Kimpe S, Robson C, Foster SJ, Thiemermann C. 1998. Mechanism of gram-positive shock: identification of peptidoglycan and lipoteichoic acid moieties essential in the induction of nitric oxide synthase, shock, and multiple organ failure. J. Exp. Med. 188:305–315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kopf M, Bachmann MF, Marsland BJ. 2010. Averting inflammation by targeting the cytokine environment. Nat. Rev. Drug Discov. 9:703–718 [DOI] [PubMed] [Google Scholar]
- 21. Lee SH, et al. 2011. De novo generation of short antimicrobial peptides with simple amino acid composition. Regul. Pept. 166:36–41 [DOI] [PubMed] [Google Scholar]
- 22. Levy SB, Marshall B. 2004. Antibacterial resistance worldwide: causes, challenges and responses. Nat. Med. 10:S122–S129 [DOI] [PubMed] [Google Scholar]
- 23. Liu DH, et al. 2004. Nontoxic membrane-active antimicrobial arylamide oligomers. Angew. Chem., Int. Ed. Engl. 43:1158–1162 [DOI] [PubMed] [Google Scholar]
- 24. Moran GJ, et al. 2006. Methicillin-resistant S. aureus infections among patients in the emergency department. N. Engl. J. Med. 355:666–674 [DOI] [PubMed] [Google Scholar]
- 25. Nemoto E, Darveau RP, Foster BL, Nogueira GR, Somerman MJ. 2006. Regulation of cementoblast function by P. gingivalis lipopolysaccharide via tlr2. J. Dent. Res. 85:733–738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Okusawa T, et al. 2004. Relationship between structures and biological activities of mycoplasmal diacylated lipopeptides and their recognition by Toll-like receptors 2 and 6. Infect. Immun. 72:1657–1665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Perron GG, Zasloff M, Bell G. 2006. Experimental evolution of resistance to an antimicrobial peptide. Proc. Biol. Sci. 273:251–256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Pfaffl MW. 2001. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 29:e45 doi:10.1093/nar/29.9.e45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Prins JM, Kuijper EJ, Mevissen MLCM, Speelman P, Vandeventer SJH. 1995. Release of tumor necrosis factor alpha and interleukin-6 during antibiotic killing of Escherichia coli in whole blood: influence of antibiotic class, antibiotic concentration, and presence of septic serum. Infect. Immun. 63:2236–2242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Quesniaux VJ, et al. 2004. Toll-like receptor 2 (tlr2)-dependent-positive and tlr2-independent-negative regulation of proinflammatory cytokines by mycobacterial lipomannans. J. Immunol. 172:4425–4434 [DOI] [PubMed] [Google Scholar]
- 31. Rappuoli R. 2004. From Pasteur to genomics: progress and challenges in infectious diseases. Nat. Med. 10:1177–1185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Rogy MA, et al. 1994. Antiendotoxin therapy in primate bacteremia with ha-1a and bpi. Ann. Surg. 220:77–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Rosenfeld Y, Papo N, Shai Y. 2006. Endotoxin (lipopolysaccharide) neutralization by innate immunity host-defense peptides: peptide properties and plausible modes of action. J. Biol. Chem. 281:1636–1643 [DOI] [PubMed] [Google Scholar]
- 34. Scott MG, Gold MR, Hancock REW. 1999. Interaction of cationic peptides with lipoteichoic acid and gram-positive bacteria. Infect. Immun. 67:6445–6453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Scott MG, Yan H, Hancock REW. 1999. Biological properties of structurally related alpha-helical cationic antimicrobial peptides. Infect. Immun. 67:2005–2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Scott MG, Vreugdenhil ACE, Buurman WA, Hancock REW, Gold M. 2000. Cationic antimicrobial peptides block the binding of lipopolysaccharide (LPS) to LPS binding protein. J. Immunol. 164:549–553 [DOI] [PubMed] [Google Scholar]
- 37. Scott MG, et al. 2007. An anti-infective peptide that selectively modulates the innate immune response. Nat. Biotechnol. 25:465–472 [DOI] [PubMed] [Google Scholar]
- 38. Scott R, Degrado W, Tew G. 2008. De novo designed synthetic mimics of antimicrobial peptides. Curr. Opin. Biotechnol. 19:620–627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Shai Y. 2002. From innate immunity to de novo designed antimicrobial peptides. Curr. Pharm. Des. 8:715–725 [DOI] [PubMed] [Google Scholar]
- 40. Som A, Vemparala S, Ivanov I, Tew GN. 2008. Synthetic mimics of antimicrobial peptides. Biopolymers 90:83–93 [DOI] [PubMed] [Google Scholar]
- 41. Spellberg B, Powers JH, Brass EP, Miller LG, Edwards JE. 2004. Trends in antimicrobial drug development: implications for the future. Clin. Infect. Dis. 38:1279–1286 [DOI] [PubMed] [Google Scholar]
- 42. Spellberg B, et al. 2008. The epidemic of antibiotic-resistant infections: a call to action for the medical community from the Infectious Diseases Society of America. Clin. Infect. Dis. 46:155–164 [DOI] [PubMed] [Google Scholar]
- 43. Tew GN, Clements D, Tang HZ, Arnt L, Scott RW. 2006. Antimicrobial activity of an abiotic host defense peptide mimic. Biochim. Biophys. Acta 1758:1387–1392 [DOI] [PubMed] [Google Scholar]
- 44. Tew GN, Scott RW, Klein ML, Degrado WF. 2010. De novo design of antimicrobial polymers, foldamers, and small molecules: from discovery to practical applications. Acc. Chem. Res. 43:30–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Theuretzbacher U, Toney JH. 2006. Nature's clarion call of antibacterial resistance: are we listening? Curr. Opin. Investig. Drugs. 7:158–166 [PubMed] [Google Scholar]
- 46. Van Amersfoort ES, Van Berkel TJC, Kuiper J. 2003. Receptors, mediators, and mechanisms involved in bacterial sepsis and septic shock. Clin. Microbiol. Rev. 16:379–414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Van Langevelde P, et al. 1998. Antibiotic-induced release of lipoteichoic acid and peptidoglycan from Staphylococcus aureus: quantitative measurements and biological reactivities. Antimicrob. Agents Chemother. 42:3073–3078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Van Zee KJ, et al. 1996. Protection against lethal Escherichia coli bacteremia in baboons (Papio anubis) by pretreatment with a 55-kda TNF receptor (cd120a)-Ig fusion protein, ro 45–2081. J. Immunol. 156:2221–2230 [PubMed] [Google Scholar]
- 49. Won HS, Jung SJ, Kim HE, Seo MD, Lee BJ. 2004. Systematic peptide engineering and structural characterization to search for the shortest antimicrobial peptide analogue of gaegurin 5. J. Biol. Chem. 279:14784–14791 [DOI] [PubMed] [Google Scholar]
- 50. Zasloff M. 2002. Antimicrobial peptides of multicellular organisms. Nature 415:389–395 [DOI] [PubMed] [Google Scholar]
- 51. Zhang X, Goncalves R, Mosser DM. 2008. The isolation and characterization of murine macrophages. Curr. Protoc. Immunol. Chapter 14:Unit 14.1 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.