

Abstract
Avian influenza virus causes outbreaks in domestic and wild birds around the world, and sporadic human infections have been reported. A DNA vaccine encoding hemagglutinin (HA) protein from the A/Indonesia/5/05 (H5N1) strain was initially tested in two randomized phase I clinical studies. Vaccine Research Center study 304 (VRC 304) was a double-blinded study with 45 subjects randomized to placebo, 1 mg of vaccine, or 4 mg of vaccine treatment groups (n = 15/group) by intramuscular (i.m.) Biojector injection. VRC 305 was an open-label study to evaluate route, with 44 subjects randomized to intradermal (i.d.) injections of 0.5 mg by needle/syringe or by Biojector or 1 mg delivered as two 0.5-mg Biojector injections in the same deltoid or as 0.5 mg in each deltoid (n = 11/group). Injections were administered at weeks 0, 4, and 8 in both studies. Antibody responses to H5 were assessed by hemagglutination inhibition (HAI) assay, enzyme-linked immunosorbent assay (ELISA), and neutralization assay, and the H5 T cell responses were assessed by enzyme-linked immunospot and intracellular cytokine staining assays. There were no vaccine-related serious adverse events, and the vaccine was well tolerated in all groups. At 1 mg, i.d. vaccination compared to i.m. vaccination induced a greater frequency and magnitude of response by ELISA, but there were no significant differences in the frequency or magnitude of response between the i.d. and i.m. routes in the HAI or neutralization assays. T cell responses were more common in subjects who received the 1- or 4-mg dose i.m. These studies demonstrated that the DNA vaccine encoding H5 is safe and immunogenic and served to define the proper dose and route for further studies. The i.d. injection route did not offer a significant advantage over the i.m. route, and no difference was detected by delivery to one site versus splitting the dose between two sites for i.d. vaccine administration. The 4-mg dose (i.m) was further investigated in prime-boost regimens.
INTRODUCTION
Highly pathogenic avian influenza A viruses cause widespread disease in domestic bird populations and have the capacity to cause disease in humans, which poses a threat to public health (1). The World Health Organization reported, as of August 2011, 563 confirmed human H5N1 cases and 330 deaths. The severe illness, high mortality rate (17), and the possibility for human-to-human spread serves as an incentive to develop human vaccines against avian influenza viruses.
Protection against influenza is antibody-mediated and responses to influenza vaccines are typically measured by hemagglutination inhibition (HAI) assays; HAI titers of ≥40 are associated with at least a 50% reduction in influenza illness (16). Enzyme-linked immunosorbent assays (ELISAs) detect binding antibody and neutralizing antibody assays detect antibodies with the capacity to inhibit viral entry into cells in in vitro assays.
Investigation H5N1 influenza vaccines have been studied in clinical trials, and generally the immunogenicity, as assessed primarily by HAI, is less than that seen with traditional seasonal influenza vaccine antigens (4, 7, 19). Attempts to improve the immune response to H5N1 vaccines have included increasing dose, adding additional antigens (NA, NP, or M2), homologous boosting, combining gene-based vaccines with electroporation, and the addition of adjuvants (2, 3, 8, 11, 15, 22), which have resulted in only modest increases in HAI titers in preclinical and clinical studies.
The Vaccine Research Center (VRC) strategy to improving influenza vaccine immunogenicity includes utilizing gene-based vectors in a prime-boost regimen. In the early studies described here, we assessed the safety and immunogenicity of this H5 DNA product. The vaccine was administered as a three-dose regimen without a heterologous vaccine boost, similar to the regimens used to initially evaluate gene-based vaccines against severe acute respiratory syndrome, West Nile virus, and Ebola virus (9, 12–14). In addition to intramuscular (i.m.) administration, we evaluated the potential impact of intradermal (i.d.) administration on immune response. Overall, the vaccine was well tolerated by both routes at all doses. The vaccine is immunogenic, but just as in previous H5N1 vaccine clinical trials, the overall responses were modest when H5 DNA was given without an inactivated vaccine boost. In further evaluations, this vaccine has shown promise as a prime for inactivated vaccine boosting (10).
MATERIALS AND METHODS
Study design.
Clinical trial VRC 304 was a phase I, double-blinded, randomized, placebo-controlled clinical trial conducted from December 2006 through March 2008, and clinical trial VRC 305 was a phase I, open-label, randomized clinical trial conducted from July 2007 through June 2008. The two studies—listed as clinical trials NCT00408109 and NCT00489931 for VRC 304 and VRC 305, respectively—were conducted at a single site at the National Institutes of Health (NIH) Clinical Center by the National Institute of Allergy and Infectious Diseases (NIAID), VRC, National Institutes of Health (Bethesda, MD).
These studies represented the first clinical studies with this product in humans and examined the safety and immunogenicity of this H5 investigational DNA vaccine encoding the HA from Influenza A/Indonesia/5/05 (H5N1), recommended by the World Health Organization as a clade 2.1 H5N1 candidate vaccine virus (http://www.who.int/csr/disease/avian_influenza/guidelines/summaryH520070403.pdf). Doses were administered intramuscularly (i.m.) and intradermally (i.d.) in the VRC 304 and VRC 305 studies, respectively, in healthy adults aged 18 to 60 years. The studies were reviewed and approved by the NIAID Institutional Review Board. U.S. Department of Health and Human Services human experimental guidelines for conducting clinical research were followed.
In the VRC 304 trial, doses of 1 and 4 mg were evaluated, along with a placebo, and 45 volunteers were concurrently randomized to the three study groups, with 15 volunteers per group. For all groups, the study injections were administered i.m. using a Biojector needle-free injection system on days 0, 28, and 56.
In the VRC 305 study, 44 volunteers were concurrently randomized into four study groups, with 11 subjects per group, to receive vaccine on days 0, 28, and 56. Group 1 received 0.5 mg (in 0.125 ml) of H5 DNA vaccine per injection, administered i.d. with the Biojector. Group 2 received 0.5 mg (in 0.125 ml) of H5 DNA vaccine per injection, administered i.d. using a needle and syringe. Group 3 received 1 mg of H5 DNA vaccine administered i.d. by Biojector as two injections of 0.5 mg (in 0.125 ml) in the same deltoid muscle as side-by-side injections. Group 4 received 1 mg of H5 DNA vaccine administered i.d. by Biojector as two injections of 0.5 mg (in 0.125 ml), with one injection in each deltoid.
In both studies, the safety monitoring included laboratory and clinical evaluations at scheduled study visits. Local and systemic reactogenicity was solicited for 5 days after each vaccination. All adverse events were coded using the Medical Dictionary for Regulatory Activities with a graded severity scale ranging from 0 to 5. Subjects were monitored for safety and immunogenicity for a period of 32 weeks.
Vaccines.
The H5 DNA vaccine (VRC-AVIDNA036-00-VP) was manufactured at the VRC/NIAID/Vaccine Pilot Plant operated by SAIC (Frederick, MD) and consists of a single closed-circular plasmid DNA macromolecule (VRC-9123), expressing Influenza A/Indonesia/5/05 HA sequence, derived from a human isolate (influenza virus sequence database no. 125873; Los Alamos National Laboratory Database). The plasmid contained a CMV/R promoter as previously described (14). The plasmid DNA was prepared under Good Manufacturing Practices at 4 mg/ml in phosphate-buffered saline (PBS). The placebo was PBS.
Statistical methods.
For each antibody and T cell response, the positive response rate and the exact 95% confidence interval were reported. The response magnitude from the positive responders was reported with the geometric mean and the 95% confidence intervals. Antibody responses to HA were assessed by ELISA, HAI assay, and neutralization assay and compared between study groups by using the Fisher exact test for response rate and the Wilcoxon test for response magnitude.
Sample processing and cell preparation.
Peripheral blood mononuclear cells (PBMC) were prepared by standard Ficoll-Hypaque density gradient centrifugation (Pharmacia, Uppsala, Sweden). PBMC, frozen in heat-inactivated fetal calf serum containing 10% dimethyl sulfoxide in a Forma CryoMed cell freezer (Marietta, OH), were stored at or below −140°C. All assays were performed on thawed specimens; the average viability was >95%.
Measurement of antibody responses by ELISA.
Endpoint titers of antibodies directed against H5 antigen (Immune Technologies Corp., New York, NY) were determined using 96-well Immulon2 (Dynex Technologies) plates coated with a preparation of purified recombinant proteins according to methods adapted from those previously described (14). The endpoint titer was calculated as the most dilute serum concentration that gave an optical density of >0.2 above background.
Measurement of neutralizing antibody.
H5 neutralizing antibodies were evaluated by the capacity of sera to prevent the infection of 293A cells by replication-incompetent HA-pseudotyped virus (20). The pseudotyped virus expressed the H5-Indonesia antigen and the luciferase reporter gene. Neutralization activity was quantified by relative decrease in the luciferase activity compared to infection of 293A cells in the absence of sera based on previously described methods (21). The 80% inhibition serum titer (ID80) was calculated relative to the signal in the absence of sera using five-parameter curve fitting.
Measurement of antibody responses by HAI assay.
HAI assays were performed in V-bottom 96-well plates using four hemagglutinating units of virus and 1% horse erythrocytes as previously described (18). The virus strain used for the HAI assay is a low-pathogenic, H5N1-PR8 reassortant, obtained from Ruben Donis at the CDC Influenza Branch (Atlanta, GA): Clade 2.1, A/Indo/5/2005(H5N1)/PR8-IBCDC-RG2 (18).
Measurement of T cell responses.
CD4 and CD8 T cell responses to H5 were assessed by intracellular cytokine staining (ICS) for interleukin-2 (IL-2) and gamma interferon (IFN-γ) and by IFN-γ enzyme-linked immunospot assay (ELISPOT) as previously described (5, 6). For both ICS and ELISPOT analyses, stimulations were via vaccine insert-matched peptides (15mers overlapping by 11). A positive response for ICS occurs if a Fisher exact test for the 2×2 table consisting of positive and negative cells by peptide and negative control has a one-sided P value of <0.01 and the percent positive cells for a peptide minus the percent positive cells for the negative control (background-subtracted percentage) exceeds 0.045. A positive ELISPOT response occurs if the if the background-subtracted number of spots per 106 cells exceeds 59 and the non-background-corrected mean is at least 4-fold greater than the mean negative stimulation for the sample.
RESULTS
Study population demographics and vaccine safety.
Baseline demographics are shown in Table 1. In VRC 304, 56% of subjects were women, 80% were white, and 13% were African-American, and the mean age at the time of enrollment was 39.6 years. In VRC 305, 46% were women, 73% were white, and 11% were African-American, and the mean age at the time of enrollment was 36.9 years. In both studies, 93% of subjects reported their highest education level as either college or an advanced degree.
Table 1.
Baseline characteristics of subjects
| Characteristic | No. (%) subjectsa |
|
|---|---|---|
| VRC 304 overall (n = 45) | VRC 305 overall (n = 44) | |
| Gender | ||
| Male | 20 (44.4) | 24 (54.5) |
| Female | 25 (55.6) | 20 (45.5) |
| Age (yr) | ||
| Mean (SD) | 39.6 (10) | 36.9 (11) |
| Range | 19–57 | 22–60 |
| Race | ||
| White | 36 (80.0) | 32 (72.7) |
| Black or African-American | 6 (13.3) | 5 (11.4) |
| Asian | 2 (4.5) | 5 (11.4) |
| All other races | 1 (2.2) | 2 (4.5) |
| Ethnicity | ||
| Non-Hispanic/Latino | 45 (100) | 40 (90.9) |
| Hispanic/Latino | 0 | 4 (9.1) |
| Body-mass index | ||
| Mean (SD) | 28.0 (4.6) | 26.5 (5.3) |
| Range | 20.6–37.5 | 17.5–40.0 |
| Education | ||
| Non-high school graduate | 0 | 0 |
| High school graduate/GED | 3 (6.7) | 3 (6.8) |
| College/university | 23 (51.1) | 20 (45.5) |
| Advanced degree | 19 (42.2) | 21 (47.7) |
Except as noted otherwise in column 1.
In VRC 304, 15 subjects in group 1 received three injections of a 1-mg dose, and one subject was lost to follow-up after receiving all study injections and moving out of the area. Two subjects in group 2 had to discontinue vaccinations after two immunizations because of the need for a systemic corticosteroid (unrelated to vaccination), and two subjects were lost to follow-up in this group. In the placebo group, one participant withdrew after one study injection. One subject in the placebo group completed all visits except week 32 due to moving from the area. The other 40 subjects completed all study visits through week 32 safety monitoring. In VRC 305, nine subjects were withdrawn from the vaccination schedule: two in group 1, three in group 2, one in group 3, and three in group 4. The reasons related to intercurrent illness or a need for a contraindicated medication in five subjects and noncompliance with protocol schedule or lost to follow-up in three subjects. In VRC 305, 80% of the subjects completed all vaccinations, and 91% completed the protocol through week 32 safety monitoring (Fig. 1).
Fig 1.

VRC 304 and VRC 305 study diagram. Study enrollment, allocation, follow-up, and analysis are shown for all subjects screened and enrolled/randomized. These details are shown for VRC 304 on the left and for VRC 305 on the right.
In both studies, the vaccine was well tolerated, and there were no serious adverse events related to vaccine in either study. Adverse events assessed as related to i.d. vaccination in VRC 305 included mild, superficial skin lesion that healed without sequelae in 13/43 (30.2%) subjects and mild “itchiness” (pruritis) reported by 9/43 (20.9%) subjects. Solicited reactogenicity categorized by group is presented in Tables 2 and 3. Across all three groups in VRC 304, the worst severity of local reactogenicity was reported as none by 15.6%, mild by 82.2%, and moderate by 2.2% of subjects with the moderate symptom (pain at the injection site) being reported by a placebo recipient. The worst severity of systemic reactogenicity was reported as none by 40%, mild by 53.3%, and moderate by 4.4% of subjects, with the moderate symptoms being reported in the 1-mg and placebo groups. One placebo subject did not return any diary cards and therefore has missing reactogenicity data. Across all four groups in VRC 305, the worst severity of local reactogenicity was reported as none by 23.3% and mild by 76.7% of subjects. Overall, across both studies, the worst severity of systemic reactogenicity was reported as none by 34.9%, mild by 58.1%, and moderate by 7% of subjects. In addition, there was no apparent trend toward increasing reactogenicity with sequential dosing, even at the highest dose in either study (see Tables S1 and S2 in the supplemental material). There was no severe reactogenicity in either study.
Table 2.
Local reactogenicity by group
| Symptom intensity | No. (%) of subjectsa |
||||||
|---|---|---|---|---|---|---|---|
| 1 mg i.m., Biojector (n = 15) | 4 mg i.m., Biojector (n = 15) | PBS i.m., Biojector (n = 15) | 0.5 mg i.d., needle (n = 10) | 0.5 mg i.d., Biojector (n = 11) | 1 mg i.d. (same arm) (n = 11)* | 1 mg i.d. (different arms) (n = 11)* | |
| Pain/tenderness | |||||||
| None | 0 | 5 (33) | 2 (13) | 5 (50) | 7 (64) | 3 (27) | 4 (36) |
| Mild | 15 (100) | 10 (67) | 12 (80) | 5 (50) | 4 (36) | 8 (73) | 7 (64) |
| Moderate | 0 | 0 | 1 (7) | 0 | 0 | 0 | 0 |
| Severe | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Swelling | |||||||
| None | 9 (60) | 12 (80) | 10 (67) | 7 (70) | 5 (45.5) | 8 (73) | 4 (36) |
| Mild | 6 (40) | 3 (20) | 5 (33) | 3 (30) | 6 (54.5) | 3 (27) | 7 (64) |
| Moderate | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Severe | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Redness | |||||||
| None | 10 (67) | 7 (47) | 11 (73) | 5 (50) | 6 (54.5) | 4 (36) | 4 (36) |
| Mild | 5 (33) | 8 (53) | 4 (27) | 5 (50) | 5 (45.5) | 7 (64) | 7 (64) |
| Moderate | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Severe | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Any local symptom | |||||||
| None | 0 | 5 (33) | 2 (13) | 3 (30) | 4 (36) | 2 (18) | 1 (9) |
| Mild | 15 (100) | 10 (67) | 12 (80) | 7 (70) | 7 (64) | 9 (82) | 10 (91) |
| Moderate | 0 | 0 | 1 (7) | 0 | 0 | 0 | 0 |
| Severe | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
*, 1 mg was administered i.d. by Biojector as two 0.5-mg injections either into the same arm or into different arms, as indicated.
Table 3.
Systemic reactogenicity by group
| Symptom intensity | No. (%) of subjectsa |
||||||
|---|---|---|---|---|---|---|---|
| 1 mg i.m., Biojector (n = 15) | 4 mg i.m., Biojector (n = 15) | PBS i.m., Biojector (n = 15)b | 0.5 mg i.d., needle (n = 10) | 0.5 mg i.d., Biojector (n = 11) | 1 mg i.d. (same arm) (n = 11)* | 1 mg i.d. (different arms) (n = 11)* | |
| Malaise | |||||||
| None | 6 (40) | 12 (80) | 7 (47) | 6 (60) | 7 (64) | 10 (91) | 6 (54.5) |
| Mild | 9 (60) | 3 (20) | 6 (40) | 4 (40) | 2 (18) | 1 (9) | 5 (45.5) |
| Moderate | 0 | 0 | 1 (7) | 0 | 2 (18) | 0 | 0 |
| Severe | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Myalgia | |||||||
| None | 13 (87) | 13 (87) | 12 (80) | 6 (60) | 9 (82) | 8 (73) | 5 (45.5) |
| Mild | 2 (13) | 2 (13) | 2 (13) | 3 (30) | 1 (9) | 3 (27) | 6 (54.5) |
| Moderate | 0 | 0 | 0 | 1 (10) | 1 (9) | 0 | 0 |
| Severe | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Headache | |||||||
| None | 11 (73) | 12 (80) | 10 (67) | 8 (80) | 9 (82) | 4 (36) | 6 (54.5) |
| Mild | 3 (20) | 3 (20) | 3 (20) | 2 (20) | 2 (18) | 7 (64) | 5 (45.5) |
| Moderate | 1 (7) | 0 | 1 (7) | 0 | 0 | 0 | 0 |
| Severe | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Chills | |||||||
| None | 15 (100) | 15 (100) | 12 (80) | 10 (100) | 11 (100) | 11 (100) | 10 (91) |
| Mild | 0 | 0 | 2 (13) | 0 | 0 | 0 | 1 (9) |
| Moderate | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Severe | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Nausea | |||||||
| None | 13 (87) | 15 (100) | 13 (87) | 9 (90) | 9 (82) | 10 (91) | 9 (82) |
| Mild | 2 (13) | 0 | 1 (7) | 1 (10) | 1 (9) | 1 (9) | 2 (18) |
| Moderate | 0 | 0 | 0 | 0 | 1 (9) | 0 | 0 |
| Severe | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Temperature | |||||||
| None | 15 (100) | 15 (100) | 14 (93) | 10 (100) | 10 (91) | 11 (100) | 10 (91) |
| Mild | 0 | 0 | 0 | 0 | 1 (9) | 0 | 1 (9) |
| Moderate | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Severe | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Any systemic symptom | |||||||
| None | 3 (20) | 10 (67) | 5 (33) | 4 (40) | 5 (46) | 3 (27) | 3 (27) |
| Mild | 11 (73) | 5 (33) | 8 (53) | 5 (50) | 4 (36) | 8 (73) | 8 (73) |
| Moderate | 1 (7) | 0 | 1 (7) | 1 (10) | 2 (18) | 0 | 0 |
| Severe | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
*, 1 mg was administered i.d. by Biojector as two 0.5-mg injections either into the same arm or into different arms, as indicated.
One PBS subject (7% of this group) who received only one injection did not provide any systemic solicited reactogenicity data.
Influenza virus-specific antibody responses.
The primary time point for immune assessment in both studies was 4 weeks after the third injection, at week 12 of the study. All subjects were negative for H5 antibodies by HAI assay, ELISA, and neutralizing antibody (NT) assay at baseline. At week 12, the frequency of positive HAI and neutralization responses were slightly higher in subjects who received 4 mg i.m. than in other groups (Fig. 2A and C). When we compared the routes of administration, 1 mg i.d. in VRC 305 (delivered as a divided dose in the same deltoid or divided between each deltoid) induced a higher frequency of response and higher ELISA titers (Fig. 2B) than 1 mg i.m. in VRC 304. This route effect on the 1-mg dose was not seen for HAI or neutralization. The 0.5-mg dose induced negligible HAI and neutralization responses, and none of these responses reached positivity criteria (Fig. 2A and C). There were no significant differences seen by any assay when comparing 1 mg delivered as a divided dose with both injections given in the same deltoid compared to a divided dose with a single 0.5-mg injection given in each deltoid.
Fig 2.

Magnitude of H5 specific antibody responses. The responses determined by HAI assay (A), ELISA (B), and neutralization assay (i.e., the ID80) (C) are shown for all subjects (nonresponders and responders) by group in VRC 304 and VRC 305. Statistical differences are indicated. The frequency of responses meeting positivity criteria are printed on each graph for each group and assay.
Influenza virus specific T-cell responses.
Antigen specific T-cell responses were assessed by ELISPOT assay and ICS at week 12 (Fig. 3). The rate of ELISPOT-positive response was the greatest, at 50%, in groups receiving 1 mg i.m., 4 mg i.m., and 1 mg using Bioject i.d. (two arms). The rate of CD8 (ICS) response was generally low, with the highest frequency of response seen in the 4-mg i.m. group at 17%. H5-specific CD4 T-cell responses were detected by ICS more frequently than H5-specific CD8 T cell responses. The rates of CD4 (ICS) response were highest at 67 and 79% in subjects who received 4 and 1 mg i.m., respectively, and were 50 and 63% in subjects who received 1 mg i.d. by Biojector (one arm and two arms, respectively). The pattern of differences suggests a dose effect and overall T cell responses were more consistent among the groups in the VRC 304 study (i.m. route) than in the VRC 305 study (i.d. route).
Fig 3.
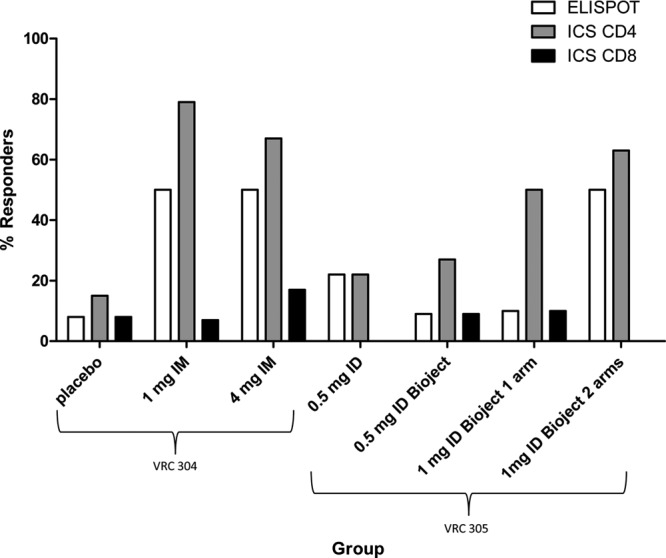
Frequency of H5 T cell responses. The percent response rates are shown for both the VRC 304 and VRC 305 studies as determined by T cell assay (ELISPOT and ICS for CD4 and ICS for CD8) for each dose group.
DISCUSSION
A safe and highly efficacious platform for H5N1 vaccination remains elusive despite attempts to optimize the antigen, vehicle of delivery, regimen, and adjuvant formulation. The H5 antigen is generally less immunogenic than most seasonal influenza antigens and poses a difficult scientific challenge to vaccine researchers. This study demonstrates that this DNA vaccine encoding H5 is safe and immunogenic, but when given as a three-dose regimen without a heterologous boost it induces only modest immunogenicity. Not surprisingly, a dose effect was observed, in that 4 mg i.m. induced higher antibody and T cell responses compared to 1 mg i.m. or 0.5 or 1 mg i.d. In the VRC 305 study, one question focused on the distribution of dosing related to the question of split dosing in different limbs. Two groups received 1 mg i.d. divided into two doses. In one of those groups, both injections were administered in the same deltoid and in the other group, one injection was administered into each deltoid. In the present study, no significant differences were related to safety, antibody, or T cell responses between these two groups. These studies were important to establish the safety and immunogenicity of this vaccine prior to its evaluation in a prime-boost regimen (at a dose of 4 mg i.m.), where more impressive immune responses were seen following H5N1-inactivated vaccine boosting (10).
Some DNA vaccines administered without boosting have been shown to elicit robust immune responses (9, 12, 13), but the most impressive immunogenicity related to DNA vaccines continues to be seen when the DNA vaccine is used as a prime and is boosted by a heterologous vaccine vector or protein (21), including for this H5 DNA vaccine (10). Based on the impressive safety record and improvements in manufacturing and immunogenicity of DNA vaccines, as well as recent findings describing the significant impact of DNA priming on H5N1 monovalent inactivated vaccine boosting (10), further evaluation of DNA vaccines in prime-boost regimens toward improved influenza vaccination is warranted.
Supplementary Material
ACKNOWLEDGMENTS
We thank the vaccine trial volunteers for their contribution and commitment to vaccine research. We also acknowledge the contributions of our NIH Clinical Center and NIAID coworkers, the EMMES corporation, and coworkers at the NIAID Vaccine Research Center. We appreciate assistance from Rick Stout at Bioject (Tualatin, OR).
This clinical trial was funded by the NIAID Intramural program.
The findings and conclusions in this report are those of the authors and do not necessarily reflect the views of the funding agency or collaborators.
The VRC 304 and VRC 305 Study Teams include LaSonji Holman, Laura Novik, Pamela Costner, Brenda Larkin, Cynthia Starr Hendel, Diane Johnson, Sandra Sitar, Olga Vasilenko, Joseph Casazza, Trishna Goswami, Raymond Cruz, Charla Andrews, Phillip Gomez, Rebecca Sheets, Hope Decederfelt, Judith Starling, LaChonne Stanford, and Rhonda Washington-Lewis.
Footnotes
Published ahead of print 5 September 2012
Supplemental material for this article may be found at http://cvi.asm.org/.
REFERENCES
- 1. Belshe RB. 2005. The origins of pandemic influenza: lessons from the 1918 virus. N. Engl. J. Med. 353:2209–2211 [DOI] [PubMed] [Google Scholar]
- 2. Beran J, Abdel-Messih IA, Raupachova J, Hobzova L, Fragapane E. 2010. A phase III, randomized, open-label study to assess the tolerability and immunogenicity of an H5N1 influenza vaccine administered to healthy adults with a 1-, 2-, 3-, or 6-week interval between first and second doses. Clin. Ther. 32:2186–2197 [DOI] [PubMed] [Google Scholar]
- 3. Brady RC, et al. 2009. Safety and immunogenicity of a subvirion inactivated influenza A/H5N1 vaccine with or without aluminum hydroxide among healthy elderly adults. Vaccine 27:5091–5095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bresson JL, et al. 2006. Safety and immunogenicity of an inactivated split-virion influenza A/Vietnam/1194/2004 (H5N1) vaccine: phase I randomised trial. Lancet 367:1657–1664 [DOI] [PubMed] [Google Scholar]
- 5. Catanzaro AT, et al. 2006. Phase 1 safety and immunogenicity evaluation of a multiclade HIV-1 candidate vaccine delivered by a replication-defective recombinant adenovirus vector. J. Infect. Dis. 194:1638–1649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Catanzaro AT, et al. 2007. Phase I clinical evaluation of a six-plasmid multiclade HIV-1 DNA candidate vaccine. Vaccine 25:4085–4092 [DOI] [PubMed] [Google Scholar]
- 7. Goji NA, et al. 2008. Immune responses of healthy subjects to a single dose of intramuscular inactivated influenza A/Vietnam/1203/2004 (H5N1) vaccine after priming with an antigenic variant. J. Infect. Dis. 198:635–641 [DOI] [PubMed] [Google Scholar]
- 8. Laddy DJ, et al. 2009. Electroporation of synthetic DNA antigens offers protection in nonhuman primates challenged with highly pathogenic avian influenza virus. J. Virol. 83:4624–4630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ledgerwood JE, et al. 2011. A West Nile virus DNA vaccine utilizing a modified promoter induces neutralizing antibody in younger and older healthy adults in a phase I clinical trial. J. Infect. Dis. 203:1396–1404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ledgerwood JE, et al. 2011. DNA priming and influenza vaccine immunogenicity: two phase 1 open label randomised clinical trials. Lancet Infect. Dis. 11:916–924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lopez P, et al. 2011. Combined, concurrent, and sequential administration of seasonal influenza and MF59-adjuvanted A/H5N1 vaccines: a phase II randomized, controlled trial of immunogenicity and safety in healthy adults. J. Infect. Dis. 203:1719–1728 [DOI] [PubMed] [Google Scholar]
- 12. Martin JE, et al. 2008. A SARS DNA vaccine induces neutralizing antibody and cellular immune responses in healthy adults in a phase I clinical trial. Vaccine 26:6338–6343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Martin JE, et al. 2007. A West Nile virus DNA vaccine induces neutralizing antibody in healthy adults during a phase 1 clinical trial. J. Infect. Dis. 196:1732–1740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Martin JE, et al. 2006. A DNA vaccine for Ebola virus is safe and immunogenic in a phase I clinical trial. Clin. Vaccine Immunol. 13:1267–1277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Patel A, et al. 2009. Evaluation of conserved and variable influenza antigens for immunization against different isolates of H5N1 viruses. Vaccine 27:3083–3089 [DOI] [PubMed] [Google Scholar]
- 16. Potter CW, Oxford JS. 1979. Determinants of immunity to influenza infection in man. Br. Med. Bull. 35:69–75 [DOI] [PubMed] [Google Scholar]
- 17. Sambhara S, Poland GA. 2006. Avian influenza vaccines: what's all the flap? Lancet 367:1636–1638 [DOI] [PubMed] [Google Scholar]
- 18. Stephenson I, Wood JM, Nicholson KG, Charlett A, Zambon MC. 2004. Detection of anti-H5 responses in human sera by HI using horse erythrocytes following MF59-adjuvanted influenza A/Duck/Singapore/97 vaccine. Virus Res. 103:91–95 [DOI] [PubMed] [Google Scholar]
- 19. Treanor JJ, Campbell JD, Zangwill KM, Rowe T, Wolff M. 2006. Safety and immunogenicity of an inactivated subvirion influenza A (H5N1) vaccine. N. Engl. J. Med. 354:1343–1351 [DOI] [PubMed] [Google Scholar]
- 20. Wei C-J, et al. 2010. Cross-neutralization of 1918 and 2009 influenza viruses: role of glycans in viral evolution and vaccine design. Sci. Translational Med. 2:24ra21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wei CJ, et al. 2010. Induction of broadly neutralizing H1N1 influenza antibodies by vaccination. Science 329:1060–1064 [DOI] [PubMed] [Google Scholar]
- 22. Zangwill KM, Treanor JJ, Campbell JD, Noah DL, Ryea J. 2008. Evaluation of the safety and immunogenicity of a booster (third) dose of inactivated subvirion H5N1 influenza vaccine in humans. J. Infect. Dis. 197:580–583 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.