

Abstract
We previously showed that kidney dysfunction/interstitial fibrosis by folate predisposes mice to sepsis mortality (normal/sepsis: 15%; folate/sepsis: 90%); agents that increased survival in normal septic mice were ineffective in the two-stage model. We used a recently characterized 5/6 nephrectomy (Nx) mouse model of progressive chronic kidney disease (CKD) to study how CKD impacts sepsis and acute kidney injury (AKI) induced by cecal ligation-puncture (CLP). CKD intensified sepsis severity (by kidney and liver injury, cytokines, and spleen apoptosis). Accumulation of HMGB1, VEGF, TNF-α, IL-6, or IL-10 was increased in CKD or sepsis alone and to a greater extent in CKD-sepsis, and only part of this effect could be explained by decreased renal clearance. Surprisingly, we found splenic apoptosis in CKD, even in the absence of sepsis. Although sFLT-1 effectively treated sepsis, it was ineffective against CKD-sepsis. Conversely, a single dose of HMGB1-neutralizing antiserum, administered 6h after sepsis alone was ineffective; however, CKD/sepsis was attenuated by anti-HMGB1. Splenectomy transiently decreased circulating HMGB1 levels, which reversed the effectiveness of anti-HMGB1 treatment on CKD/sepsis. We conclude that progressive CKD increases sepsis severity, in part, by reducing renal clearance of cytokines; CKD-induced splenic apoptosis and HMGB1 could be important common mediators for both CKD and sepsis.
Introduction
The rate of mortality from sepsis in critically ill patients is increasing despite improvements in supportive care [1]. The translation of sepsis treatments from animal models into humans has largely failed, in part, because the less complex animal models do not mimic human sepsis [2–6]. Most patients with sepsis have at least one underlying pre-existing (‘co-morbid’) chronic illness [1, 7, 8]. Those with chronic kidney disease (CKD) have a higher prevalence, severity, and mortality of sepsis [9, 10]. This might be caused by uremia-induced leukocyte dysfunction (lymphocyte, monocyte, neutrophil and dendritic cell) [11–16], inflammatory cytokine accumulation from less renal clearance [17–20], or other co-existing illness, etc. [9]. CKD is an important prognostic risk factor in patients with sepsis [21, 22]. Recently, we showed that folate-induced tubulointerstitial kidney fibrosis increases the severity of all sepsis outcomes in a mouse cecal ligation and puncture (CLP) model [23]. However, the folate renal fibrosis model lacks the renal progression that is an essential feature of human CKD [24]. We recently developed a modified surgical approach to inducing chronic kidney disease in mice [25], where 5/6 nephrectomy (5/6 Nx) was performed in two stages: resecting the upper and lower left kidney poles and using Avitene hemostasis, followed by right nephrectomy 1 week later, which mimics many aspects of the progressive natural history of CKD patients. We found that two mouse strains, CD-1 and129S3, developed CKD, but another strain, C57BL/6, did not develop CKD. In contrast, the severity of fibrotic injury in the folate model was strain-independent [25].
In the current study we re-examine the effect of pre-existing chronic kidney disease on the severity of sepsis, using our progressive, 5/6 nephrectomy model. We previously reported initial studies that preexisting 5/6 Nx in CD-1 worsened subsequent sublethal CLP sepsis [23]. We hypothesize that acute-on-chronic kidney disease is a distinct entity that is more than the sum of chronic kidney disease and sepsis-AKI. Therefore, we compare CKD (5/6 Nx), sepsis-AKI (CLP), and acute-on-chronic kidney disease (5/6 Nx + CLP) to determine which sepsis-induced outcomes, including AKI, are exacerbated, and determine whether ongoing inflammation and/or decreased clearance of pro-inflammatory cytokines can account for amplified/accelerated disease progression. We focus on two cytokines that are implicated in both CKD and sepsis: HMGB1, a late proinflammatory cytokine released from apoptotic cells [26–29] and VEGF, an angiogenesis factor that promotes vascular leakage [23, 30–32].
Results
Increased severity of sepsis after 5/6 Nx
We performed CLP at a time of advanced CKD: four weeks after 5/6 Nx in CD-1 mice (Supplemental Fig 1) [25]. CKD mice had more severe sepsis at 18 h after CLP compared with mice subjected to CLP alone, as measured by kidney injury (BUN and renal tubular vacuolization), liver injury (ALT, AST), serum inflammatory cytokine levels (TNF-α, IL-6, IL-10), and spleen apoptosis (Fig 1–3). While there was a similar trend for serum creatinine (Scr) (Fig 1A), it was not statistically significant; however, interpretation of serum creatinine levels are not straightforward because of sepsis-induced reduction in creatinine production [8]. Although the set of organ-specific and systemic manifestations of sepsis that we measured was quite diverse, 5/6 Nx increased the entire set, almost uniformly.
Figure 1. Widespread exacerbation of sepsis outcomes by CKD.

CKD was induced by 5/6Nx in CD-1 mice, and CLP surgery was performed 4 weeks later. Organ injury was measured at 18 h after sham (white bar) or CLP (black bar) surgery. Renal function was determined by serum creatinine (A) and BUN (B), liver function was determined by ALT (C), AST (D), inflammation was determined by serum cytokine levels (TNF-α, IL-6, IL-10) (E–G), renal injury was determined by semi-quantitative measurement of renal vacuolized tubules (H), and splenic apoptosis (I) was measured by activated caspase 3 (n=6–7). *, P<0.05 vs. control sham; #, P<0.05 vs. control CLP; +, P<0.05 vs. 5/6 Nx sham.
Figure 3. CKD enhances sepsis-induced splenic apoptosis.

CD-1 mice were untreated (normal, A, B) or subjected to 5/6 Nx (C, D) 4 weeks prior to sham (A, C) or CLP surgery (B, D). Representative images of spleen stained for activated caspase3 are shown (A–D); black bar corresponds to 200 μm. Number of activated caspase3 positive cells per high powered field in spleen of composite control at 16 weeks (n=9); C57BL/6 5/6 Nx at 16 weeks (n=4); 129S3 5/6 Nx at 4, 8, 12 weeks (n= 4/group); CD-1 5/6 Nx at 2, 4 weeks (n=4/group)
Increases in spleen apoptosis, serum HMGB1 and VEGF after 5/6 Nx
Spleen apoptosis is a well-documented characteristic of sepsis, associated with immune depression, that is associated with increased mortality in mouse models, and also in humans [33]. Compared to CLP alone, spleen apoptosis in 5/6 Nx + CLP was substantially greater, however there was a significant increase in spleen apoptosis after 5/6 Nx alone (Fig 1, 3). Because there were no reports in the literature of CKD-associated spleen apoptosis, we measured spleen apoptosis (by activated caspase 3 staining) at different times during the course of CKD. To confirm this finding, we took advantage of the strain-dependent susceptibility toward developing CKD after 5/6 Nx, where C57BL/6 mice were not susceptible, but 129S3 and CD-1 developed maximal CKD at 12 and 4 weeks, respectively [25]. The amount of spleen apoptosis (without sepsis) increased in CD-1 mice and progressively increased in 129S3 mice (Fig 3E). Spleen apoptosis was greater in 129S3 and CD-1 mice, compared to C57BL/6 mice, and appeared to rise more rapidly in CD-1 mice than in 129S3 mice (Fig 3E), corresponding to the strain-dependent onset/severity of CKD [26]. Spleen apoptosis in the composite 2/6 Nx control group was not higher than in normal mice (not shown), indicating that the healing process from the resected kidney wounds was not enough to trigger spleen apoptosis. Because VEGF and HMGB1 increase in human CKD [26, 34, 35], and splenic apoptosis is associated with systemic accumulation of HMGB1 [27, 36], we measured both VEGF and HMGB1. Serum HMGB1 and VEGF increased after 5/6 Nx (Fig 4A,B), that corresponded to progressive albuminuria seen previously [25] (and Supplemental Fig 1). VEGF appeared to increase more rapidly than HMGB1 (as early as 1 week) after 5/6 Nx (Fig 4A,B), consistent with a faster response of VEGF to injury. Other inflammatory cytokines (TNF-α, IL-6, IL-10) were not elevated until 4 weeks (Fig 4C, D, E, data not shown).
Figure 4. Accumulation of HMGB1, and VEGF, but not TNF-α, IL-6 and IL-10 during CKD (5/6 Nx) progression.

Time course of serum HMGB1 (A); and VEGF (B) during the course of 5/6 Nx-induced CKD (n= 4–6/group). Repeated measures ANOVA with post-hoc (Tukey) comparisons were performed (## P<0.0001 vs. 2/6 Nx, #P<0.05 vs. 2/6 Nx). Serum levels of TNF-α (C), IL-6 (D), IL-10 (E) were measured in a composite of normal and 2/6 Nx controls vs. 5/6 Nx at 4 weeks (+ P<0.05 vs. control by one-way ANOVA).
Influence of kidney injury or removal on cytokine production after sepsis
Inflammatory cytokines, including VEGF and HMGB1, have been implicated as markers and/or pathogenic mediators that contribute to the severity of sepsis [27, 32, 36–38]. We determined the dynamic changes of these cytokines during post-CKD sepsis. Additionally, bilateral nephrectomy plus CLP was compared to 5/6 Nx plus CLP to determine the renal-specific contribution of these cytokine level changes in the CKD-sepsis model. Serum HMGB1 increased late after CLP alone or after bilateral Nx plus CLP (Fig 5A). In contrast, in 5/6 Nx mice, HMGB1 was elevated at baseline, and increased early after CLP (Fig 5A). Other cytokines (VEGF, TNF-α, IL-6, IL-10) were less affected by 5/6 Nx alone (Fig 4C–E), or 5/6 Nx plus CLP compared with CLP alone (ranging from 1.4- to 2.4-fold), but 5/6 Nx plus CLP had larger increases relative to 5/6 Nx alone (3- to 28-fold), comparable to increases observed with bilateral Nx plus CLP (Fig 5B–E). The cytokines were often higher in bilateral Nx because of increased cytokine production and/or less renal elimination, as described by others [39, 40]. To determine if kidney function was important for cytokine elimination, bilateral Nx or 5/6 Nx was performed, then exogenous cytokines were intravenously injected and measured frequently to calculate cytokine clearance and half-life values. The half-life of HMGB1 increased by 60% and the half-life of TNF-α, IL-6, IL-10 increased by 2–3 fold in 5/6 Nx compared to normal. The half-life for HMGB1, TNF-α, IL-6, and IL-10 further increased 31–67% once all renal function was removed with bilateral Nx (Fig 6). The pharmacokinetics of VEGF in bilateral Nx could not be calculated because endogenous VEGF increased as early as 30 min after bilateral Nx (Supplemental Fig 2). A rapid accumulation of VEGF after bilateral Nx is consistent the kidney being a dominant organ of VEGF elimination. Cytokine accumulation in sepsis with pre-existing impaired kidney function, was a result, in part, of impaired renal cytokine elimination which can contribute to the higher sepsis severity after 5/6 Nx.
Figure 5. Time course of serum cytokines (HMGB1, VEGF, TNF-α, IL-6 and IL-10) following CLP with or without pre-existing kidney impairment.

Serum HMGB1 (A), VEGF (B), TNF-α (C), IL-6 (D) and IL-10 (E) at indicated time points in CD-1 mice after normal-CLP, 5/6 Nx-CLP or bilateral Nx-CLP. (n = 4–5/time point). By repeated measures ANOVA all except IL-6 had significant time x nephrectomy interactions; post-hoc (Tukey) comparisons: * P<0.05 vs. normal-CLP, # P<0.05 vs. 5/6 Nx-CLP.
Figure 6. Comparison of cytokine clearance/half-life in normal, 5/6 Nx and bilateral Nx mice.

After injection of exogenous recombinant HMGB1 (A), VEGF (B), TNF-α (C), IL-6 (D) or IL-10 (E) serum cytokine concentrations were measured at different times for area under the curve (AUC), clearance, and half-life calculations. (n = 4–5/group) * P<0.05 vs. normal, ** P<0.03 vs. normal, # P <0.05 vs. 5/6 Nx.
Neutralizing HMGB1, but not VEGF, attenuated sepsis severity in mice with pre-existing CKD (5/6 Nx)
To compare the early VEGF or late HMGB1 contributions to the severity of sepsis after 5/6 Nx, we used neutralizing therapies in normal and 5/6 Nx CD-1 mice. Soluble FLT-1 (sFLT-1) is an endogenous, circulating VEGF receptor splice variant that can serve as a VEGF antagonist [41]. We attempted to counteract the decreased clearance of VEGF by neutralizing it with sFLT-1, which was administered immediately after CLP, then at 3, 6, and 9 h, which attenuated sepsis severity in normal mice as described previously [23, 32, 37] but was not effective after 5/6 Nx-CLP (Fig 7), similar to our previous report using a folate kidney fibrosis model combined with CLP [23]. In contrast, a single dose of anti-HMGB1 neutralizing antiserum at 6h after CLP attenuated sepsis severity in 5/6 Nx mice but not in normal mice, as measured by kidney injury (Scr, BUN), liver injury (ALT, AST), and inflammatory cytokines (TNF-α, IL-6, IL-10), but not splenic apoptosis (Fig 8). Furthermore, a single dose of anti-HMGB1 also improved systemic hemodynamics and delayed sepsis mortality in pre-existing 5/6 Nx with CLP (Fig 9).
Figure 7. sFLT-1 attenuated sepsis severity in normal-CLP but not in 5/6 Nx-CLP mice.

CD-1 mice 4 weeks after 5/6 Nx or normal controls were subjected to sham surgery (white bars) or CLP, then injected at 0, 3, 6, and 9 h after CLP with saline (NSS, black bars) or soluble FLT-1 (sFLT-1, 33.3 mg/kg i.v., gray bars). The following were measured 18 h post-CLP: renal injury (Scr, BUN) (A, B), liver injury (ALT, AST) (C, D), inflammatory cytokines (TNF-α, IL-6, IL-10) (E–G), and splenic apoptosis (H) (n = 4–6/group). By ANOVA, there was no significant interaction between CKD-sepsis and treatment; post-hoc comparisons (Holm-Sidak) comparisons: * P<0.05 vs. normal-CLP + normal saline.
Figure 8. Anti-HMGB1 attenuated sepsis in 5/6 Nx but not normal mice.

CD-1 mice 4 weeks after 5/6 Nx or normal controls were subjected to sham surgery (white bars) or CLP, then injected at 6 h after CLP with control rabbit IgG (IgG, black bars) or anti-HMGB1 (3.6 mg/kg, i.p., gray bars). The following were measured 18 h post-CLP: renal injury (Scr, BUN) (A, B), liver injury (ALT, AST) (C, D), inflammatory cytokines (TNF-α, IL-6, IL-10) (E–G), and splenic apoptosis (H) (n = 4–6/group). By ANOVA, there was significant interaction between CKD-sepsis and treatment for BUN, AST, TNFα, IL-6, and splenic apoptosis; post-hoc comparisons (Holm-Sidak) comparisons: * P<0.05 vs. normal-CLP + rabbit IgG, # P<0.05 vs. 5/6 Nx-CLP + rabbit IgG.
Figure 9. Anti-HMGB1 improved sepsis-induced hypotension, bradycardia and survival after 5/6 Nx and sepsis.

Telemetric recording of conscious mean arterial pressure (MAP) (A) and heart rate (HR) (B) of normal (red, pink) or 5/6 Nx mice (blue, black) subjected to CLP, and after 6 hours injected with rabbit IgG control (red, blue) or anti-HMGB1 (pink, black) (n=4/group). By repeated measures ANOVA p<0.0001 for either MAP or HR; post-hoc (Tukey) comparisons 5/6 Nx-CLP + IgG control vs. 5/6 Nx-CLP + anti-HMGB1 was significant 20–38 h post-CLP (MAP) and 15–38 h, except 30,35, and 36 h (HR). Survival curve (C) of 5/6 Nx mice subjected to CLP, then treatment 6 h later with rabbit IgG control (gray) or anti-HMGB1 (black) (n = 7/group). # P<0.05 anti-HMGB1 vs. rabbit IgG control (Kaplan-Meier analysis)
Acute splenectomy reduces circulating HMGB1 and the effectiveness of HMGB1 neutralizing antiserum
Spleen apoptosis can be an important source of serum HMGB1 both in vitro and in vivo following sepsis [27, 36]. Because 5/6 Nx accentuates CLP-induced increases in HMGB1, and anti-HMGB1 therapy was more effective in 5/6 Nx-CLP than CLP alone, we used splenectomy to determine the contribution of the spleen to the heightened intensity of disease in the CKD-sepsis state. Four weeks after 5/6 Nx splenectomy decreased the levels of HMGB1 after CKD by 78% at 1 day, but the HMGB1 levels returned to control levels within 5 days (Fig 10A). When we performed 5/6 Nx, then acute splenectomy after 4 weeks, and then CLP 3 days later, the CKD-sepsis-induced increase in HMGB1 was almost completely diminished (Fig 10B). Further, all other CKD-sepsis-induced parameters, except AST, were significantly reduced by acute splenectomy (Supplemental Fig 3). After acute splenectomy, anti-HMGB1 therapy was ineffective at reducing CKD-sepsis-induced Scr, ALT, TNFα (Fig 10C–E), as well as BUN, AST, IL-6, and IL-10 (Supplemental Fig 4).
Figure 10. Splenectomy transiently decreases HMGB1 and renders anti-HMGB1 ineffective in treating CKD-sepsis.

CD-1 mice were subjected to 5/6 Nx, and after 4 weeks, splenectomy was performed at day 0. Serum HMGB1 (A) was measured in 5/6 Nx controls (black squares) or 5/6 Nx-splenectomy (white squares). Loss of CKD-sepsis-stimulated serum HMGB1 (B): four weeks after 5/6 Nx mice were subjected to splenectomy (gray bar), and after three days sham surgery (white bar) or CLP (black bar, gray bar) was performed, and serum HMGB1 was measured after 18 h. Outcomes of CKD-sepsis (see also Supplemental Fig 4): four weeks after 5/6 Nx mice were subjected to splenectomy (day 0), and after three days sham surgery (white bars) or CLP (black bars, gray bars) was performed, followed by administration of control IgG (black bars) or anti-HMGB1 (gray bars) 6 h later, then measurement of Scr (C), ALT (D), or TNFα (E) at 18 h.
Discussion
We recently developed a two-stage model of sepsis with pre-existing folate-induced kidney injury and showed that this comorbidity affected the severity of sepsis [23]. Unfortunately, the transient course of kidney injury after folate does not match the progressive worsening of human CKD. In the present study, we used a remnant kidney model that more closely mimics CKD with respect to progressive hypertension, glomerulosclerosis, and albuminuria [25]. Not only were we able to confirm our previous findings in a bona fide CKD model, we also explored mechanistic pathways that accelerate the sepsis-AKI phase.
As expected, we found that CKD increased the severity of all sepsis outcomes that we tested, including splenic apoptosis. To our surprise, we found that 5/6 Nx CKD itself increased splenic apoptosis, in contrast to the folate model, which did not [23]; correspondingly, HMGB1 levels did not increase in the folate model (data not shown). We demonstrated that elevated serum HMGB1 was associated with more severe sepsis. By neutralizing HMGB1 but not VEGF, sepsis severity could be reduced in mice with CKD, but not in normal mice. Further, by removing the spleen we could transiently decrease HMGB1 levels, reduce the severity of CKD-sepsis, and eliminate the benefit of anti-HMGB1 therapy.
Pre-existing chronic kidney disease predisposes mice to more severe sepsis
We found that the severity of sepsis: kidney, liver, and spleen injury, or inflammatory cytokines (data not shown) corresponded to the strain-dependent susceptibility of CKD after 5/6Nx in C57BL/6, 129S3, or CD-1 mice [25]. Thus, underlying chronic kidney injury predisposed mice to more severe sepsis (either with folic acid as previously reported [23], or 5/6 Nx in this study), consistent with previous studies of host defense defects in CKD, where a variety of immune cell functions are impaired, by yet undefined factors that presumably include circulating uremic toxins [9]. Because serum inflammatory cytokines, VEGF [23, 32, 37], and HMGB1 [27, 36] are all increased following sepsis in healthy mice, we first explored the importance of these factors in CKD alone, sepsis alone, and the amplification caused by the sequential combination of pre-existing CKD and sepsis.
Inflammatory cytokines
Human and animal studies have indicated that CKD includes an inflammatory state, with corresponding increases in inflammatory cytokines (TNF-α, IL-6, IL-10) that may enhance protein catabolism, malnutrition and atherosclerosis [18–20, 42]. These cytokines are also elevated in sepsis patients and animal models of sepsis [2, 43, 44]. We found relatively mild accumulation of these cytokines in serum, approximately two-fold above baseline, even at late stages of CKD following 5/6 Nx. This was reflected in a two-fold increase in TNF-α, IL-6 and IL-10 after CKD + sepsis vs. sepsis alone. Increases of these cytokines during CKD-sepsis can be attributed, in part, to reduced renal excretion; 70–80% of the cytokine elimination was via a renal route. However, strain-dependent severity of CKD [25] or CKD + sepsis did not change the levels of these cytokines (data not shown). Thus, a model where impaired renal function amplifies inflammation by passively increasing prototypical pro-(TNF-α, IL-6) and anti-inflammatory (IL-10) cytokine accumulation could only partially account for the enhanced severity of CKD-sepsis in our experimental model. It is likely that mild inflammation during CKD can also interact with the acute inflammation during sepsis to drive cytokine production. The contribution of these cytokines to sepsis and CKD-sepsis may not be straightforward, as treatment with TNF-α can decrease sepsis severity [45]. The higher inflammatory cytokine levels that have been reported in CKD patients may not be central to the pathogenesis of CKD, but might explain the susceptibility to infections in CKD [46, 47].
VEGF
VEGF is increased in CKD both in animals and humans [23, 34]. High levels of VEGF can cause vascular endothelial leakage and have been associated with a higher severity of human sepsis [30, 31]. VEGF increased very early after 5/6 Nx surgery and continued to increase in parallel to CKD progression. Some of this early increase might promote healing of the surgical wound [48, 49]. Most exogenously administered VEGF was cleared via the kidney, suggesting that the VEGF increase after CKD was primarily derived by decreased renal elimination rather than increased production. In contrast to prototypical pro- and anti-inflammatory cytokines, both VEGF and HMGB1 differed among strains during the progression of CKD (not shown), and the circulating levels of both mediators corresponded to the severity of CKD. VEGF increased to very high levels after sepsis, with or without CKD, as previously described [23, 30–32, 34, 37]. We found that exogenously administered sFLT-1, an endogenous neutralizing, soluble VEGF receptor, did not attenuate sepsis severity after CKD by 5/6 Nx nor after folate-induced fibrotic injury [23], despite effectiveness of this treatment in mouse CLP sepsis without pre-existing kidney injury [32, 37]. While VEGF participates in both CKD progression and uncomplicated sepsis-AKI does not synergistically amplify the severity of sepsis after CKD.
HMGB1
The pro-inflammatory cytokine HMGB1 is passively released from dying cells that are either undergoing apoptosis or necrosis [50], and it induces the release of other cytokines from macrophages and other cell types [51–54]. HMGB1 can induce additional release of HMGB1 in macrophage-like RAW 264.7 cells [27], thus HMGB1 can amplify inflammation by positive-feedback. In sepsis, HMGB1 has been proposed as a late-appearing pro-inflammatory cytokine, and HMGB1 neutralizing therapy improved mortality in normal mice [27, 28, 51, 54, 55], even when it was started 24 hrs after surgery [29]. We found that serum HMGB1 increased in the late phase of CKD and was especially elevated at the peak of the kidney injury and albuminuria, similar to observations in a few patients with CKD [26]. This time course suggests that HMGB1 alone does not drive CKD progression, as HMGB1 levels have probably not reached a critical mass for positive feedback, but rather accumulates as renal function deteriorates. Indeed, about half of HMGB1 clearance was via a renal route. HMGB1 was elevated following sepsis, but the timing depended on how much renal function remained prior to sepsis. HMGB1 was a late-appearing cytokine (increased at 12 h) as previously demonstrated [28, 29]; whereas, in sepsis following CKD, HMGB1 was an early appearing cytokine (6 h) (Fig 5A). From our data we propose the following framework: because HMGB1 levels are higher during the progression of CKD, and the capacity for renal clearance of HMGB1 is reduced, the small increases in HMGB1 during the early phase of sepsis can trigger an autocrine or positive feedback loop where HMGB1 is high enough to induce more HMGB1 release [28, 29, 55]. This view is supported by a temporal shift in the HMGB1 appearance from late in sepsis alone to early after CKD-sepsis. On the other hand, an earlier increase in HMGB1 could be an indicator of more severe injury as recently mentioned in moribund trauma patients [56]. We found that a single dose of anti-HMGB1 neutralizing antibody started at 6 h after CLP—when HMGB1 increased—attenuated sepsis severity and improved survival in 5/6 Nx-CLP but not in normal-CLP mice. Because neutralizing HMGB1, but not VEGF (by sFLT-1), was effective, the attenuation of pro-inflammatory response might be more beneficial than decreasing vascular leakage in CKD-sepsis. Alternatively, HMGB1 might drive VEGF, high levels of VEGF could induce a beneficial adaptation in endothelial cells, and/or sFLT-1 could have additional toxic effects; whereas high levels of HMGB1 may have only harmful effects, including positive feedback. Since the same dose of anti-HMGB1 showed effectiveness only in CLP with CKD but not CLP alone, HMGB1 might only induce tissue damage once it is above a critical threshold. However, co-induced factors often complicate any simple interpretation. The higher mean arterial pressure (MAP) in CKD from 5/6 Nx did not help maintain blood pressure after sepsis. The MAP started to drop as early as 3 hr in both normal and 5/6 Nx mice. Anti-HMGB1 treatment appeared to maintain cardiovascular function in the ‘late’ sepsis phase (after 18 hr) as previously reported [57–59]. However, some of the mice with improved MAP died after 40–48 hr, and additional doses of anti-HMGB1 might be needed to maximize efficacy [29]. Thus, HMGB1 seems to be more central to the amplification seen in CKD-sepsis. Anti-HMGB1 treatment, which showed therapeutic benefit in previous studies on sepsis from healthy mice [27, 29, 36], might have a greater impact on sepsis with underlying CKD. We summarize how our findings fit into our current understanding in Figure 11. Considering the increasing prevalence of CKD patients with a higher rate of sepsis [9] and longer lengths of hospital stay [10], HMGB1 may be a promising target as part of a combination pre-emption and/or treatment strategy.
Figure 11. Proposed framework for Acute-on-Chronic kidney disease.

Chronic kidney disease progresses slowly (A), and after a septic insult the trajectory accelerates (C) relative to uncomplicated sepsis-AKI (B). This acceleration may be attributable, in part, to 1) to decreased GFR, which increases levels of cytokines, VEGF, and HMGB1, and 2) existing spleen apoptosis and/or HMGB1 during CKD may enhance the sepsis-induced increase in HMGB1 (dashed arrow). A mechanistic shift occurs when CKD and sepsis-AKI are combined; Acute-on-Chronic kidney disease is distinct from the sum of its parts. Anti-VEGF treatment, which is effective for sepsis-AKI, is no longer effective in Acute-on-Chronic kidney disease; and anti-HMGB1 treatment, which was ineffective for uncomplicated sepsis-AKI, is effective for Acute-on-Chronic kidney disease.
Role of spleen apoptosis in HMGB1 accumulation in CKD-sepsis
We then turned to cellular immune defects, since both CKD and sepsis are associated with immunomodulatory and immune depression defects [9, 60] including uremia-induced immune cell apoptosis in CKD [11, 61–65]. Because HMGB1 is released from apoptotic cells [29], we explored the role of the spleen, the largest lymphoid organ [66–68] and a major site of apoptosis following sepsis [36]. We surprisingly found spleen apoptosis in CKD (before CLP surgery) that started as early as 4 and 8 weeks after 5/6 Nx in CD-1 and 129S3 mice, respectively. Although CKD and uremia can induce immune cell apoptosis, we could not find any previous animal or human studies of spontaneous cell apoptosis in lymphoid organs that could support our findings. Splenic apoptosis appears to be upstream of HMGB1, as recombinant HMGB1 administration (6 mg/kg) did not induce splenic apoptosis even after bilateral Nx (data not shown), HMGB1 neutralizing antibody treatment of CKD-sepsis did not decrease splenic apoptosis, and HMGB1 levels were decreased transiently by splenectomy. Because splenectomy eliminated the effectiveness of anti-HMGB1 treatment, our data is consistent with spleen apoptosis being an important source of HMGB1 during CKD-sepsis; however, because splenectomy does not distinguish between apoptotic and non-apoptotic cells, we cannot rule out the impact of other functions of the spleen on CKD-sepsis.
The role of spleen apoptosis in the pathogenesis and progression of CKD remains an open question because splenectomy only transiently reduced HMGB1 levels. We speculate that the CKD- and/or sepsis-induced trigger(s) of lymphocyte apoptosis remain after splenectomy, and induce apoptosis in other lymphoid cells in lymph nodes, thymus, liver, or the circulation, with subsequent increases in HMGB1.
Conclusion
Successfully translating treatments from pre-clinical animal models to human patients has been exceedingly difficult in sepsis [2–6]. Some of this difficulty may arise because the animal models do not faithfully reproduce enough features of human sepsis; alternatively, candidate drugs are typically tested in young healthy animals, whereas a majority of sepsis occurs in the setting of pre-existing medical conditions [9]. We modified the standard CLP model to incorporate a common pre-existing condition – progressive chronic kidney disease, thereby creating a more clinically relevant model that more closely resembles the complexity of human sepsis. We found that pre-existing, progressive CKD amplified sepsis, and that a major amplification factor was HMGB1, which was not cleared by the injured kidney. In addition, we found that 5/6 Nx CKD increased spleen apoptosis, which appeared to be an important source of elevated serum HMGB1 level in CKD. A high baseline level of HMGB1 in CKD might trigger additional HMGB1 release after sepsis, resulting in a positive feedback loop. Interception with anti-HMGB1 reduced sepsis after CKD but not in normal animals or CKD animals shortly after splenectomy. Thus, the two-stage CKD sepsis model has dramatically increased mortality and divergent mechanisms/therapeutic targets than a sepsis model in young mice. Models that more closely replicate the underlying co-morbidity might be more suitable for testing biomarkers and therapeutic agents for use in human sepsis.
The interplay between CKD and sepsis is inherently complex. By comparing both CKD and sepsis components vs. CKD-sepsis, we can begin to isolate which progression factors for CKD and sepsis act in concert, which factors act independently, and which factors may even counteract. For example, routine examination of baseline values of typical sepsis outcomes post-CKD/pre-sepsis led to our discovery of splenic apoptosis as a potentially important mediator of CKD progression. This unconventional line of thinking was made possible by dissecting the complex clinical problem of CKD-sepsis into two distinct and disparate components, then systematically examining the models for each component alone or in combination.
Methods
Animals and animal models
Animal care and experiments were performed according to the National Institutes of Health (NIH) criteria for the care and use of laboratory animals in research. Male, 6–8 weeks CD-1 (Charles River Laboratories, MA), 129S3 or C57BL/6 (Frederick, MD) mice had free access to water and chow. Morbidly ill mice were euthanized per protocol. All surgical procedures were performed under isoflurane anesthesia.
Nephrectomy
The 5/6 nephrectomy (Nx) was performed in two stages: upper and lower pole kidney removal (2/6 Nx); followed by right nephrectomy (3/6 Nx) 1 week later) as previously described [25]. Mice were used only if adequate kidney mass was removed, (a ratio of removed left kidney fragments to right kidney between 0.55 – 0.72). In partial nephrectomy controls (2/6 Nx), the same 1st stage procedure was followed 1 week later by right flank incision and identification of both poles of the right kidney. Similarly, sham normal mice had two flank incisions, one week apart, where both poles of left and right kidney were identified. Bilateral nephrectomy was performed via bilateral flank approach as previously described [69]. The renal capsule was peeled away before partial or total nephrectomy to avoid adrenal gland injury.
Polymicrobial cecal ligation and puncture (CLP)
CLP was performed as previously described [70]. In short, we ligated the cecum at 12 mm in length and punctured twice with a 21-gauge needle then gently squeezed to express a small amount of fecal material and returned to the central abdominal cavity. In sham-operated animals, the cecum was isolated, but neither ligated nor punctured. Pre-warmed normal saline (30 ml/kg) was immediately given intraperitoneally after surgery and antibiotic was given subcutaneously (imipenem/cilastatin; 14 mg/kg in 1 mL of normal saline) 6h later. Eighteen hours after surgery, blood was collected by cardiac puncture for measurement of serum markers of organ injury and cytokine response. Kidneys, liver and spleen were fixed in 10% neutral buffered formalin for histology
Splenectomy
Splenectomy was performed as previously described [70] via left flank three days before or immediately before CLP in normal or 5/6 Nx mice and immediately after bilateral nephrectomy (via left flank after left kidney removal).
Drug administration
Recombinant human soluble FLT-1 domain D1–3 (sFLT-1: Cell Sciences, Canton, MA, USA) 1 mg per 30 g mouse (or an equal volume of NSS for sham treatment) was injected intravenously every 3 h (four doses), starting immediately after CLP [23]. A single dose of anti-HMGB1 neutralizing antibody, which was generously supplied by Dr. Kevin J. Tracey (Feinstein Institute for Biomedical Research, Long Island, NY) [29], or purified rabbit IgG (Sigma-Aldrich, St. Louis MO), was injected intraperitoneally (3.6 mg/kg) [29] 6 h after CLP in CD-1 mice either untreated (“normal”) or 4 weeks after 5/6 Nx.
Blood chemistries and cytokine measurements
At week 0 (immediately before 2nd stage operation: 1 week after both pole resection), 1, 2, 4, 8, 12 h after CLP surgery, 60 μl of capillary tube blood was collected by retro-orbital approach under isoflurane anesthesia then centrifuged at 1,000 × g for 8 min to remove cells. VEGF, TNF-α, IL-6, IL-10 (R&D Systems, Minneapolis, MN) and HMGB1 (Shino-Test Corporation, Kanagawa, Japan) [71] were measured by ELISA. Blood urea nitrogen (BUN), aspartate transaminase (AST), alanine transaminase (ALT), was measured by an autoanalyzer (Hitachi 917, Boehringer Mannheim, Indianapolis, IN). Serum creatinine was measured by high-performance liquid chromatography (HPLC) [72].
Histology
The 10% formalin-fixed, paraffin-embedded kidney sections were stained with periodic acid-Schiff (PAS) reagent (Sigma-Aldrich). Renal tubular damage caused by CLP-induced sepsis was assessed by counting vacuolized tubules per 200 total tubules per field using 10 randomly selected fields from each animal at 400x magnification. Immunohistochemical staining of 4 μm paraffin sections was performed as previously described with anti-active caspase-3 antibody (Cell Signaling Technology, Beverly, MA), a marker of apoptosis, was examined in 5 randomly chosen 200X fields and expressed as positive cells per high power field [23, 70].
Pharmacokinetics (PK) of cytokines
A single dose of recombinant cytokine was injected intravenously in CD-1 mice immediately after sham surgery in normal, 5/6 Nx at 4 weeks and immediately after bilateral Nx: mouse TNF-α, IL-6, IL-10, VEGF (eBioscience, San Diego, CA) and recombinant human HMGB1 (R&D Systems, Minneapolis, MN) at the dose 0.03 μg; 3 μg; 0.3 μg; 0.03 μg and 6 mg/kg respectively. Each dose of cytokine was validated to have no effect on endogenous cytokine production (data not shown). Sixty μl of capillary blood collected via retro-orbital sinus at least 1 day before injection as a baseline and at 5 min, 0.5, 1, 3, 5, 8, or 24 h after injection were used to measure an individual cytokine. Area under the concentration time curve from 0–24 hours (AUC0-24 hr) was calculated by trapezoidal rule [73]. Elevated levels of HMGB1 and VEGF after 5/6 Nx were assumed to be in steady state, and PK parameters were calculated after subtraction of the baseline level. Previously described equations [74, 75]; were used for pharmacokinetic parameters 1) Ke = maximal concentration/AUC0-24 hr; 2) half-life (t1/2) = 0.693/Ke 3); volume of distribution (Vd) = known injected dose/maximal concentration; 4) Clearance (CL) = 0.693*Vd/t1/2. The cytokine level 5 min after injection was used as maximal concentration.
Measurement of blood pressure
Mean arterial pressure (MAP) was measured by radiotelemetry as previously described [23, 76]. A telemeter transmitter (model TA11PA-C10, Data Sciences International, St Paul, MN) was implanted one week before subsequent data acquisition.
Survival study
Survival after CLP surgery was determined by conventional methods and confirmed by telemetric recordings of mean arterial pressure and heart rate. Imipenem/cilastatin (14 mg/kg) and fluid resuscitation (1 ml of normal saline) were started 6 h after CLP by subcutaneous injection and then repeated with 7 mg/kg imipenem/cilastatin in 1 ml of normal saline) every 12 h for 4 days. Morbidly ill animals were euthanized.
Statistical analysis
Differences between the groups were examined for statistical significance by student t-test or analysis of variance (ANOVA) with appropriate multiple comparison corrections (SigmaStat 3.1, Systat Software, Inc., Point Richmond, CA); longitudinal measurements were analyzed by repeated measures analysis of variance (ANOVA) to test time-dependent interactions (SAS, Cary, NC, USA); When the statistical model was significant, further post-hoc analyses were performed by Tukey’s method for multiple comparisons. A P value < 0.05 was accepted as statistically significant.
Supplementary Material
Figure 2. CKD increases sepsis-induced renal tubular vacuolization.
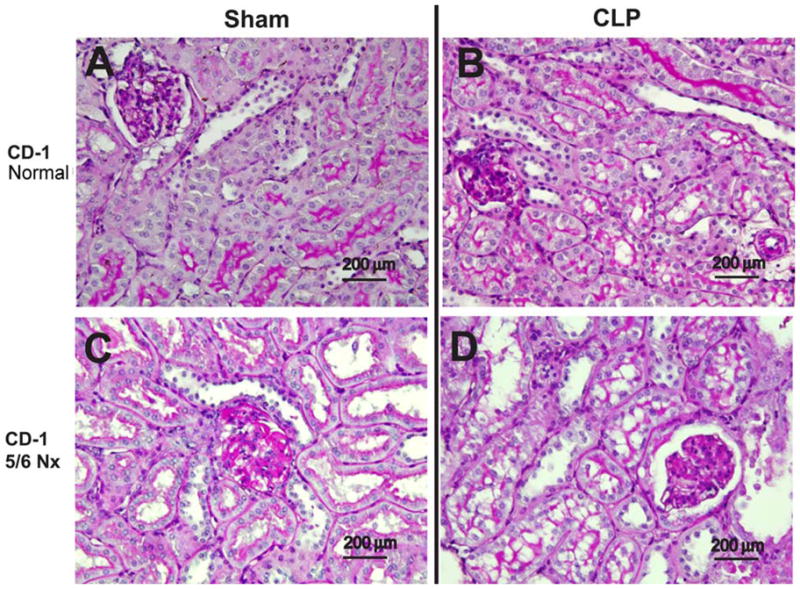
Representative images of periodic acid-Schiff-stained renal cortex in normal CD-1 mice (A, B), or CD-1 mice 4 weeks after 5/6 Nx (G, H) in sham (A, C) and CLP (B, D). Black bar corresponds to 200 μm.
Acknowledgments
This research was supported by the Intramural Research Program of the NIH, NIDDK. We thank Kevin J. Tracey (Feinstein Institute for Biomedical Research, Long Island, NY) for generously supplying HMGB1 neutralizing antibody.
References
- 1.Martin GS, Mannino DM, Eaton S, Moss M. The epidemiology of sepsis in the United States from 1979 through 2000. N Engl J Med. 2003 Apr 17;348(16):1546–54. doi: 10.1056/NEJMoa022139. [DOI] [PubMed] [Google Scholar]
- 2.Doi K, Leelahavanichkul A, Yuen PS, Star RA. Animal models of sepsis and sepsis-induced kidney injury. J Clin Invest. 2009 Oct;119(10):2868–78. doi: 10.1172/JCI39421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dyson A, Singer M. Animal models of sepsis: why does preclinical efficacy fail to translate to the clinical setting? Crit Care Med. 2009 Jan;37(1 Suppl):S30–7. doi: 10.1097/CCM.0b013e3181922bd3. [DOI] [PubMed] [Google Scholar]
- 4.Esmon CT. Why do animal models (sometimes) fail to mimic human sepsis? Crit Care Med. 2004 May;32(5 Suppl):S219–22. doi: 10.1097/01.ccm.0000127036.27343.48. [DOI] [PubMed] [Google Scholar]
- 5.Riedemann NC, Guo RF, Ward PA. The enigma of sepsis. J Clin Invest. 2003 Aug;112(4):460–7. doi: 10.1172/JCI19523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rittirsch D, Hoesel LM, Ward PA. The disconnect between animal models of sepsis and human sepsis. J Leukoc Biol. 2007 Jan;81(1):137–43. doi: 10.1189/jlb.0806542. [DOI] [PubMed] [Google Scholar]
- 7.Angus DC, Linde-Zwirble WT, Lidicker J, Clermont G, Carcillo J, Pinsky MR. Epidemiology of severe sepsis in the United States: analysis of incidence, outcome, and associated costs of care. Crit Care Med. 2001 Jul;29(7):1303–10. doi: 10.1097/00003246-200107000-00002. [DOI] [PubMed] [Google Scholar]
- 8.Dhainaut JF, Claessens YE, Janes J, Nelson DR. Underlying disorders and their impact on the host response to infection. Clin Infect Dis. 2005 Nov 15;41( Suppl 7):S481–9. doi: 10.1086/432001. [DOI] [PubMed] [Google Scholar]
- 9.Dalrymple LS, Go AS. Epidemiology of acute infections among patients with chronic kidney disease. Clin J Am Soc Nephrol. 2008 Sep;3(5):1487–93. doi: 10.2215/CJN.01290308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Naqvi SB, Collins AJ. Infectious complications in chronic kidney disease. Adv Chronic Kidney Dis. 2006 Jul;13(3):199–204. doi: 10.1053/j.ackd.2006.04.004. [DOI] [PubMed] [Google Scholar]
- 11.Cohen G, Haag-Weber M, Horl WH. Immune dysfunction in uremia. Kidney Int Suppl. 1997 Nov;62:S79–82. [PubMed] [Google Scholar]
- 12.Eleftheriadis T, Antoniadi G, Liakopoulos V, Kartsios C, Stefanidis I. Disturbances of acquired immunity in hemodialysis patients. Semin Dial. 2007 Sep-Oct;20(5):440–51. doi: 10.1111/j.1525-139X.2007.00283.x. [DOI] [PubMed] [Google Scholar]
- 13.Lim WH, Kireta S, Leedham E, Russ GR, Coates PT. Uremia impairs monocyte and monocyte-derived dendritic cell function in hemodialysis patients. Kidney Int. 2007 Nov;72(9):1138–48. doi: 10.1038/sj.ki.5002425. [DOI] [PubMed] [Google Scholar]
- 14.Minnaganti VR, Cunha BA. Infections associated with uremia and dialysis. Infect Dis Clin North Am. 2001 Jun;15(2):385–406. viii. doi: 10.1016/s0891-5520(05)70152-5. [DOI] [PubMed] [Google Scholar]
- 15.Pesanti EL. Immunologic defects and vaccination in patients with chronic renal failure. Infect Dis Clin North Am. 2001 Sep;15(3):813–32. doi: 10.1016/s0891-5520(05)70174-4. [DOI] [PubMed] [Google Scholar]
- 16.Vanholder R, Ringoir S. Infectious morbidity and defects of phagocytic function in end-stage renal disease: a review. J Am Soc Nephrol. 1993 Mar;3(9):1541–54. doi: 10.1681/ASN.V391541. [DOI] [PubMed] [Google Scholar]
- 17.Carrero JJ, Yilmaz MI, Lindholm B, Stenvinkel P. Cytokine dysregulation in chronic kidney disease: how can we treat it? Blood Purif. 2008;26(3):291–9. doi: 10.1159/000126926. [DOI] [PubMed] [Google Scholar]
- 18.Stenvinkel P, Heimburger O, Paultre F, Diczfalusy U, Wang T, Berglund L, et al. Strong association between malnutrition, inflammation, and atherosclerosis in chronic renal failure. Kidney Int. 1999 May;55(5):1899–911. doi: 10.1046/j.1523-1755.1999.00422.x. [DOI] [PubMed] [Google Scholar]
- 19.Stenvinkel P, Ketteler M, Johnson RJ, Lindholm B, Pecoits-Filho R, Riella M, et al. IL-10, IL-6, and TNF-alpha: central factors in the altered cytokine network of uremia--the good, the bad, and the ugly. Kidney Int. 2005 Apr;67(4):1216–33. doi: 10.1111/j.1523-1755.2005.00200.x. [DOI] [PubMed] [Google Scholar]
- 20.Zoccali C, Mallamaci F, Tripepi G. Inflammation and atherosclerosis in end-stage renal disease. Blood Purif. 2003;21(1):29–36. doi: 10.1159/000067852. [DOI] [PubMed] [Google Scholar]
- 21.Alberti C, Brun-Buisson C, Goodman SV, Guidici D, Granton J, Moreno R, et al. Influence of systemic inflammatory response syndrome and sepsis on outcome of critically ill infected patients. Am J Respir Crit Care Med. 2003 Jul 1;168(1):77–84. doi: 10.1164/rccm.200208-785OC. [DOI] [PubMed] [Google Scholar]
- 22.Hutchinson TA, Thomas DC, MacGibbon B. Predicting survival in adults with end-stage renal disease: an age equivalence index. Ann Intern Med. 1982 Apr;96(4):417–23. doi: 10.7326/0003-4819-96-4-417. [DOI] [PubMed] [Google Scholar]
- 23.Doi K, Leelahavanichkul A, Hu X, Sidransky KL, Zhou H, Qin Y, et al. Pre-existing renal disease promotes sepsis-induced acute kidney injury and worsens outcome. Kidney Int. 2008 Oct;74(8):1017–25. doi: 10.1038/ki.2008.346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Klingler EL, Jr, Evan AP, Anderson RE. Folic acid-induced renal injury and repair. Correlation of structural and functional abnormalities. Arch Pathol Lab Med. 1980 Feb;104(2):87–93. [PubMed] [Google Scholar]
- 25.Leelahavanichkul A, Yan Q, Hu X, Eisner C, Huang Y, Chen R, et al. Angiotensin II overcomes strain-dependent resistance of rapid CKD progression in a new remnant kidney mouse model. Kidney Int. 2010 Dec;78(11):1136–53. doi: 10.1038/ki.2010.287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bruchfeld A, Qureshi AR, Lindholm B, Barany P, Yang L, Stenvinkel P, et al. High Mobility Group Box Protein-1 correlates with renal function in chronic kidney disease (CKD) Mol Med. 2008 Mar-Apr;14(3–4):109–15. doi: 10.2119/2007-00107.Bruchfeld. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Qin S, Wang H, Yuan R, Li H, Ochani M, Ochani K, et al. Role of HMGB1 in apoptosis-mediated sepsis lethality. J Exp Med. 2006 Jul 10;203(7):1637–42. doi: 10.1084/jem.20052203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang H, Bloom O, Zhang M, Vishnubhakat JM, Ombrellino M, Che J, et al. HMG-1 as a late mediator of endotoxin lethality in mice. Science. 1999 Jul 9;285(5425):248–51. doi: 10.1126/science.285.5425.248. [DOI] [PubMed] [Google Scholar]
- 29.Yang H, Ochani M, Li J, Qiang X, Tanovic M, Harris HE, et al. Reversing established sepsis with antagonists of endogenous high-mobility group box 1. Proc Natl Acad Sci U S A. 2004 Jan 6;101(1):296–301. doi: 10.1073/pnas.2434651100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pickkers P, Sprong T, Eijk L, Hoeven H, Smits P, Deuren M. Vascular endothelial growth factor is increased during the first 48 hours of human septic shock and correlates with vascular permeability. Shock. 2005 Dec;24(6):508–12. doi: 10.1097/01.shk.0000190827.36406.6e. [DOI] [PubMed] [Google Scholar]
- 31.van der Flier M, van Leeuwen HJ, van Kessel KP, Kimpen JL, Hoepelman AI, Geelen SP. Plasma vascular endothelial growth factor in severe sepsis. Shock. 2005 Jan;23(1):35–8. doi: 10.1097/01.shk.0000150728.91155.41. [DOI] [PubMed] [Google Scholar]
- 32.Yano K, Liaw PC, Mullington JM, Shih SC, Okada H, Bodyak N, et al. Vascular endothelial growth factor is an important determinant of sepsis morbidity and mortality. J Exp Med. 2006 Jun 12;203(6):1447–58. doi: 10.1084/jem.20060375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hotchkiss RS, Nicholson DW. Apoptosis and caspases regulate death and inflammation in sepsis. Nat Rev Immunol. 2006 Nov;6(11):813–22. doi: 10.1038/nri1943. [DOI] [PubMed] [Google Scholar]
- 34.Harper SJ, Downs L, Tomson CR, Dwight JS, Bolton C. Elevated plasma vascular endothelial growth factor levels in non-diabetic predialysis uraemia. Nephron. 2002 Mar;90(3):341–3. doi: 10.1159/000049071. [DOI] [PubMed] [Google Scholar]
- 35.Pawlak K, Pawlak D, Mysliwiec M. Possible association between circulating vascular endothelial growth factor and oxidative stress markers in hemodialysis patients. Med Sci Monit. 2006 Apr;12(4):CR181–5. [PubMed] [Google Scholar]
- 36.Huston JM, Wang H, Ochani M, Ochani K, Rosas-Ballina M, Gallowitsch-Puerta M, et al. Splenectomy protects against sepsis lethality and reduces serum HMGB1 levels. J Immunol. 2008 Sep 1;181(5):3535–9. doi: 10.4049/jimmunol.181.5.3535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tsao PN, Chan FT, Wei SC, Hsieh WS, Chou HC, Su YN, et al. Soluble vascular endothelial growth factor receptor-1 protects mice in sepsis. Crit Care Med. 2007 Aug;35(8):1955–60. doi: 10.1097/01.CCM.0000275273.56547.B8. [DOI] [PubMed] [Google Scholar]
- 38.Ulloa L, Tracey KJ. The “cytokine profile”: a code for sepsis. Trends Mol Med. 2005 Feb;11(2):56–63. doi: 10.1016/j.molmed.2004.12.007. [DOI] [PubMed] [Google Scholar]
- 39.Kim do J, Park SH, Sheen MR, Jeon US, Kim SW, Koh ES, et al. Comparison of experimental lung injury from acute renal failure with injury due to sepsis. Respiration. 2006;73(6):815–24. doi: 10.1159/000095588. [DOI] [PubMed] [Google Scholar]
- 40.Zurovsky Y, Eligal Z, Grossman S. Increased sensitivity to bilateral nephrectomy in rat caused by endotoxemia. Exp Toxicol Pathol. 1995 Nov;47(5):353–8. doi: 10.1016/S0940-2993(11)80347-7. [DOI] [PubMed] [Google Scholar]
- 41.Ferrara N, Gerber HP, LeCouter J. The biology of VEGF and its receptors. Nat Med. 2003 Jun;9(6):669–76. doi: 10.1038/nm0603-669. [DOI] [PubMed] [Google Scholar]
- 42.Fleet M, Osman F, Komaragiri R, Fritz A. Protein catabolism in advanced renal disease: role of cytokines. Clin Nephrol. 2008 Aug;70(2):91–100. doi: 10.5414/cnp70091. [DOI] [PubMed] [Google Scholar]
- 43.Casey LC, Balk RA, Bone RC. Plasma cytokine and endotoxin levels correlate with survival in patients with the sepsis syndrome. Ann Intern Med. 1993 Oct 15;119(8):771–8. doi: 10.7326/0003-4819-119-8-199310150-00001. [DOI] [PubMed] [Google Scholar]
- 44.Blackwell TS, Christman JW. Sepsis and cytokines: current status. Br J Anaesth. 1996 Jul;77(1):110–7. doi: 10.1093/bja/77.1.110. [DOI] [PubMed] [Google Scholar]
- 45.Alexander HR, Sheppard BC, Jensen JC, Langstein HN, Buresh CM, Venzon D, et al. Treatment with recombinant human tumor necrosis factor-alpha protects rats against the lethality, hypotension, and hypothermia of gram-negative sepsis. J Clin Invest. 1991 Jul;88(1):34–9. doi: 10.1172/JCI115298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kimmel PL, Phillips TM, Simmens SJ, Peterson RA, Weihs KL, Alleyne S, et al. Immunologic function and survival in hemodialysis patients. Kidney Int. 1998 Jul;54(1):236–44. doi: 10.1046/j.1523-1755.1998.00981.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pertosa G, Grandaliano G, Gesualdo L, Schena FP. Clinical relevance of cytokine production in hemodialysis. Kidney Int Suppl. 2000 Aug;76:S104–11. doi: 10.1046/j.1523-1755.2000.07613.x. [DOI] [PubMed] [Google Scholar]
- 48.Szabo S, Vincze A. Growth factors in ulcer healing: lessons from recent studies. J Physiol Paris. 2000 Mar-Apr;94(2):77–81. doi: 10.1016/s0928-4257(00)00146-7. [DOI] [PubMed] [Google Scholar]
- 49.Barrientos S, Stojadinovic O, Golinko MS, Brem H, Tomic-Canic M. Growth factors and cytokines in wound healing. Wound Repair Regen. 2008 Sep-Oct;16(5):585–601. doi: 10.1111/j.1524-475X.2008.00410.x. [DOI] [PubMed] [Google Scholar]
- 50.Scaffidi P, Misteli T, Bianchi ME. Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature. 2002 Jul 11;418(6894):191–5. doi: 10.1038/nature00858. [DOI] [PubMed] [Google Scholar]
- 51.Andersson U, Wang H, Palmblad K, Aveberger AC, Bloom O, Erlandsson-Harris H, et al. High mobility group 1 protein (HMG-1) stimulates proinflammatory cytokine synthesis in human monocytes. J Exp Med. 2000 Aug 21;192(4):565–70. doi: 10.1084/jem.192.4.565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bonaldi T, Talamo F, Scaffidi P, Ferrera D, Porto A, Bachi A, et al. Monocytic cells hyperacetylate chromatin protein HMGB1 to redirect it towards secretion. EMBO J. 2003 Oct 15;22(20):5551–60. doi: 10.1093/emboj/cdg516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Li J, Kokkola R, Tabibzadeh S, Yang R, Ochani M, Qiang X, et al. Structural basis for the proinflammatory cytokine activity of high mobility group box 1. Mol Med. 2003 Jan-Feb;9(1–2):37–45. [PMC free article] [PubMed] [Google Scholar]
- 54.Yang H, Wang H, Czura CJ, Tracey KJ. The cytokine activity of HMGB1. J Leukoc Biol. 2005 Jul;78(1):1–8. doi: 10.1189/jlb.1104648. [DOI] [PubMed] [Google Scholar]
- 55.Wang H, Yang H, Tracey KJ. Extracellular role of HMGB1 in inflammation and sepsis. J Intern Med. 2004 Mar;255(3):320–31. doi: 10.1111/j.1365-2796.2003.01302.x. [DOI] [PubMed] [Google Scholar]
- 56.Peltz ED, Moore EE, Eckels PC, Damle SS, Tsuruta Y, Johnson JL, et al. HMGB1 is markedly elevated within 6 hours of mechanical trauma in humans. Shock. 2009 Jul;32(1):17–22. doi: 10.1097/shk.0b013e3181997173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Xu H, Su Z, Wu J, Yang M, Penninger JM, Martin CM, et al. The alarmin cytokine, high mobility group box 1, is produced by viable cardiomyocytes and mediates the lipopolysaccharide-induced myocardial dysfunction via a TLR4/phosphatidylinositol 3-kinase gamma pathway. J Immunol. 2010 Feb 1;184(3):1492–8. doi: 10.4049/jimmunol.0902660. [DOI] [PubMed] [Google Scholar]
- 58.Li W, Li J, Ashok M, Wu R, Chen D, Yang L, et al. A cardiovascular drug rescues mice from lethal sepsis by selectively attenuating a late-acting proinflammatory mediator, high mobility group box 1. J Immunol. 2007 Mar 15;178(6):3856–64. doi: 10.4049/jimmunol.178.6.3856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Tzeng HP, Fan J, Vallejo JG, Dong JW, Chen X, Houser SR, et al. Negative inotropic effects of high-mobility group box 1 protein in isolated contracting cardiac myocytes. Am J Physiol Heart Circ Physiol. 2008 Mar;294(3):H1490–6. doi: 10.1152/ajpheart.00910.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hotchkiss RS, Karl IE. The pathophysiology and treatment of sepsis. N Engl J Med. 2003 Jan 9;348(2):138–50. doi: 10.1056/NEJMra021333. [DOI] [PubMed] [Google Scholar]
- 61.Cendoroglo M, Jaber BL, Balakrishnan VS, Perianayagam M, King AJ, Pereira BJ. Neutrophil apoptosis and dysfunction in uremia. J Am Soc Nephrol. 1999 Jan;10(1):93–100. doi: 10.1681/ASN.V10193. [DOI] [PubMed] [Google Scholar]
- 62.Heidenreich S, Schmidt M, Bachmann J, Harrach B. Apoptosis of monocytes cultured from long-term hemodialysis patients. Kidney Int. 1996 Mar;49(3):792–9. doi: 10.1038/ki.1996.110. [DOI] [PubMed] [Google Scholar]
- 63.Jaber BL, Cendoroglo M, Balakrishnan VS, Perianayagam MC, King AJ, Pereira BJ. Apoptosis of leukocytes: basic concepts and implications in uremia. Kidney Int Suppl. 2001 Feb;78:S197–205. doi: 10.1046/j.1523-1755.2001.59780197.x. [DOI] [PubMed] [Google Scholar]
- 64.Matsumoto Y, Shinzato T, Amano I, Takai I, Kimura Y, Morita H, et al. Relationship between susceptibility to apoptosis and Fas expression in peripheral blood T cells from uremic patients: a possible mechanism for lymphopenia in chronic renal failure. Biochem Biophys Res Commun. 1995 Oct 4;215(1):98–105. doi: 10.1006/bbrc.1995.2438. [DOI] [PubMed] [Google Scholar]
- 65.Perianayagam MC, Murray SL, Balakrishnan VS, Guo D, King AJ, Pereira BJ, et al. Serum soluble Fas (CD95) and Fas ligand profiles in chronic kidney failure. J Lab Clin Med. 2000 Oct;136(4):320–7. doi: 10.1067/mlc.2000.109318. [DOI] [PubMed] [Google Scholar]
- 66.Beilhack A, Rockson SG. Immune traffic: a functional overview. Lymphat Res Biol. 2003;1(3):219–34. doi: 10.1089/153968503768330256. [DOI] [PubMed] [Google Scholar]
- 67.Jirillo E, Mastronardi ML, Altamura M, Munno I, Miniello S, Urgesi G, et al. The immunocompromised host: immune alterations in splenectomized patients and clinical implications. Curr Pharm Des. 2003;9(24):1918–23. doi: 10.2174/1381612033454306. [DOI] [PubMed] [Google Scholar]
- 68.Timens W. The human spleen and the immune system: not just another lymphoid organ. Res Immunol. 1991 May;142(4):316–20. doi: 10.1016/0923-2494(91)90081-s. [DOI] [PubMed] [Google Scholar]
- 69.Doi K, Yuen PS, Eisner C, Hu X, Leelahavanichkul A, Schnermann J, et al. Reduced production of creatinine limits its use as marker of kidney injury in sepsis. J Am Soc Nephrol. 2009 Jun;20(6):1217–21. doi: 10.1681/ASN.2008060617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Leelahavanichkul A, Yasuda H, Doi K, Hu X, Zhou H, Yuen PS, et al. Methyl-2-acetamidoacrylate, an ethyl pyruvate analog, decreases sepsis-induced acute kidney injury in mice. Am J Physiol Renal Physiol. 2008 Dec;295(6):F1825–35. doi: 10.1152/ajprenal.90442.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Yamada S, Maruyama I. HMGB1, a novel inflammatory cytokine. Clin Chim Acta. 2007 Jan;375(1–2):36–42. doi: 10.1016/j.cca.2006.07.019. [DOI] [PubMed] [Google Scholar]
- 72.Yuen PS, Dunn SR, Miyaji T, Yasuda H, Sharma K, Star RA. A simplified method for HPLC determination of creatinine in mouse serum. Am J Physiol Renal Physiol. 2004 Jun;286(6):F1116–9. doi: 10.1152/ajprenal.00366.2003. [DOI] [PubMed] [Google Scholar]
- 73.Tse FL, Nedelman JR. Serial versus sparse sampling in toxicokinetic studies. Pharm Res. 1996 Jul;13(7):1105–8. doi: 10.1023/a:1016079228995. [DOI] [PubMed] [Google Scholar]
- 74.Farris FF, Dedrick RL, King FG. Cisplatin pharmacokinetics: applications of a physiological model. Toxicol Lett. 1988 Oct;43(1–3):117–37. doi: 10.1016/0378-4274(88)90024-0. [DOI] [PubMed] [Google Scholar]
- 75.Leelahavanichkul A, Areepium N, Vadcharavivad S, Praditpornsilpa K, Avihingsanon Y, Karnjanabuchmd T, et al. Pharmacokinetics of sirolimus in Thai healthy volunteers. J Med Assoc Thai. 2005 Sep;88( Suppl 4):S157–62. [PubMed] [Google Scholar]
- 76.Doi K, Hu X, Yuen PS, Leelahavanichkul A, Yasuda H, Kim SM, et al. AP214, an analogue of alpha-melanocyte-stimulating hormone, ameliorates sepsis-induced acute kidney injury and mortality. Kidney Int. 2008 Jun;73(11):1266–74. doi: 10.1038/ki.2008.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.