

Abstract
Intrinsically disordered proteins (IDPs) are increasingly recognized for their important roles in a range of biological contexts, both in normal physiological function and in a variety of devastating human diseases. However, their structural characterization by traditional biophysical methods, for the purposes of understanding their function and dysfunction, has proved challenging. Here, we investigate the model IDPs α-Synuclein (αS) and tau, that are involved in major neurodegenerative conditions including Parkinson’s and Alzheimer’s diseases, using excluded volume Monte Carlo simulations constrained by pairwise distance distributions from single-molecule fluorescence measurements. Using this, to our knowledge, novel approach we find that a relatively small number of intermolecular distance constraints are sufficient to accurately determine the dimensions and polymer conformational statistics of αS and tau in solution. Moreover, this method can detect local changes in αS and tau conformations that correlate with enhanced aggregation. Constrained Monte Carlo simulations produce ensembles that are in excellent agreement both with experimental measurements on αS and tau and with all-atom, explicit solvent molecular dynamics simulations of αS, with much lower configurational sampling requirements and computational expense.
Abbreviations used: AAMD, all-atom molecular dynamics; ECMC, experimentally constrained Monte Carlo; ETeff, energy transfer efficiency; (sm)FRET, (single molecule) Förster resonance energy transfer; IDP, intrinsically disordered protein; LJ, Lennard-Jones; MC, Monte Carlo; MD, molecular dynamics; PRE, paramagnetic relaxation enhancement; SAX(N)S, small-angle x-ray (neutron) scattering; UMC, unconstrained Monte Carlo
Introduction
A growing number of mammalian proteins are classified as natively unstructured or intrinsically disordered, because they lack, in whole or part, stable native secondary structure and tertiary interactions. Many in this class of proteins are involved in signaling functions, where they may act as hubs for information flow within the cell (1). A number of IDPs are also directly implicated in a variety of human diseases, ranging from cancer to Type II diabetes as well as a large spectrum of neurodegenerative disorders (2).
In solution, IDPs populate an ensemble of rapidly interconverting structures, with the relative populations of the various structures determined by the balance between residue-residue interaction energies and chain entropy. IDPs thus span a continuum of dynamic behavior between well-folded proteins that assume a single native state representing a global minimum in the conformational energy landscape (3), and ideal random coils that lack spatial or temporal correlations between residues (4). Characterizing the partially ordered and disordered structures sampled by IDPs remains a challenging problem.
A variety of biophysical techniques have been used to provide indirect structural insight into IDPs (5). Specifically, circular dichroism and infrared spectroscopic techniques can probe the fractions of IDP residues involved in transient α-helical or β-strand secondary structures (6). NMR and electron paramagnetic resonance can provide information on the local chemical environment around particular residues, as well as on mean inter-residue distances and relative orientations (5,7). Fluorescence spectroscopy can probe similar interresidue distances, as well as the dynamics of conformational change and reconfiguration within the solution-state ensemble (8). SAXS or SANS (7) and diffusion-based measurements (9,10) can provide measures of global dimension, such as the radius of gyration (Rg) or hydrodynamic radius (Rh).
Computational approaches have also been frequently employed to study IDP conformational ensembles. Most attempts to simulate IDPs involve one of two approaches: 1), MD simulations and enhanced-sampling variants (11–13), in which a physical representation of a protein chain evolves over time according to Newtonian equations of motion; and 2), stochastic conformational searches in which MC-type algorithms are used to efficiently sample conformations of the protein chain (14). Both types of approaches can be applied to all-atom representations of proteins and solvent, or to models coarse-grained at varying levels of spatial and temporal resolution. The solvent can be represented either as a set of implicit corrections to protein-protein interactions, or as explicit molecules (15,16). Simulated ensembles are also sensitive to the choice of the protein force field. An important caveat is that these force fields are typically parameterized to reproduce the behavior of folded proteins rather than IDPs, and thus they may fail to capture important aspects of IDP ensembles (17–20).
α-Synuclein (αS; Fig. 1 a) is a 140-residue protein whose aggregation is linked directly to the development and pathogenesis of Parkinson’s disease. Tau (Fig. 1 b) is a microtubule-associated protein up to 441 residues long (subject to alternative splicing) that forms characteristic neuronal aggregates in several human disorders collectively called tauopathies, including Alzheimer’s disease, Pick’s disease, and chronic traumatic encephalopathy. Both αS and tau are intrinsically disordered in their unbound monomeric states, but can self-assemble into a variety of partially or highly ordered oligomeric and fibrillar aggregates. Both proteins are subjects of significant interest, not only because of their clinical relevance but also as models for other IDPs that self-assemble into amyloid aggregates. The solution-state ensembles of αS and tau have been studied by NMR (21–25), fluorescence (26–32), SAXS (33–39), and a variety of computational methods (13,23,40–43). Despite this extensive effort, there is significant disagreement about even the most fundamental structural properties of these proteins in solution (e.g., reported Rg values for αS vary nearly twofold, from 2.6 nm (34) to 5.0 nm (36)), possibly due to aggregation or oligomerization in solution at the concentrations (30–800 μM) necessary for SAXS and NMR experiments. Fluorescence techniques such as smFRET circumvent these complications by using much lower protein concentrations (∼50 pM), but provide qualitatively lower resolution data. Given that IDPs are increasingly recognized as viable pharmacological targets (44–46), smFRET may also be particularly well suited to drug candidate screening: the low protein concentrations used should enable the accurate identification of compounds that alter the target conformation by binding with very high affinity (KDs as low as ∼500 pM). On the computational side, MD studies of αS in solution have typically required nonphysiological temperatures (40,41) to generate Rg and Rh values that agree with experiments. Although fragments of tau have been studied by all-atom MD (47,48), the full-length protein is too long (441 residues) for meaningful simulations to be run at reasonable computational expense with current resources. More consistent results have been obtained for both αS and tau by post facto filtering of an unconstrained ensemble to fit experimental data (23,43) and using coarse-grained MC approaches (49).
Figure 1.
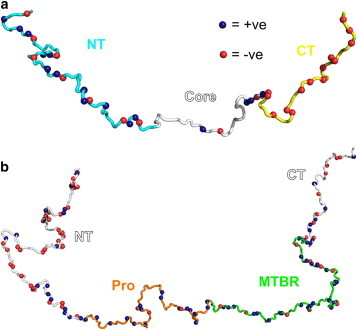
(a) Schematic of 140-residue αS in solution. Residues 60–95 form the core of amyloid fibrils, whereas residues 1–95 form an amphipathic helix in the presence of anionic lipid membranes. The N-terminal region (NT) bears a net positive charge, whereas the C-terminus (CT) is highly negatively charged at pH 7.4 but neutral at pH 3.0. (b) Schematic of the 441-residue isoform of tau. The microtubule-binding region (MTBR; residues 244–369) forms the core of amyloid aggregates and is also the putative binding site for the molecular aggregation inducer heparin, whereas residues 151–243 (Pro) are rich in prolines.
In this work, we describe computational studies based on our extensive experimental smFRET measurements that provide a quantitative description of αS and tau solution-state conformations. We first develop an ECMC method to incorporate long-range pairwise distance constraints from smFRET into MC simulations, and find that remarkably few constraints are sufficient to reproduce the global dimensions of αS and tau found experimentally under various conditions. For αS, we find that AAMD simulations in explicit solvent accurately capture the average pairwise interresidue distances of αS in solution, albeit at significantly higher computational cost than ECMC. The larger size of tau makes the computational cost for analogous AAMD simulations impractically high. Finally, unconstrained UMC simulations that include dihedral angle potentials, hydrogen bonding, effective residue-residue, and residue-environment interactions tuned to match the global dimensions of αS also matched the AAMD ensemble and smFRET observations in terms of polymer scaling behavior and mean interresidue distances. Both UMC and AAMD show large fluctuations about these mean distances that may be important in determining the populations of aggregation-prone states. The ECMC technique can be generalized to other unstructured proteins and will enable efficient characterization of the native state ensembles of IDPs, and of the conformational changes produced by environmental factors, covalent modifications, and small-molecule modulators of function and dysfunction.
Materials and Methods
Details on smFRET data collection, constraint generation, and resampling-based error estimation procedures are provided in the Supporting Material.
MC simulations
All MC calculations were implemented using the PyRosetta (50) and Scientific Python libraries for Python on Yale University High Performance Computing clusters. αS and tau were modeled with all backbone atoms represented explicitly, and each side chain represented by a single bead located at the centroid of the appropriate atoms. For the ECMC simulations, the effective potential consisted of purely repulsive Lennard Jones (LJ) interactions and harmonic restraints with mean distances and variances derived from smFRET measurements (27,51) as detailed in the Supporting Material. For the UMC simulations, the potential included the pair (statistical residue-residue interaction), env (statistical residue-environment interaction), rama (backbone dihedral propensities), hbond_sr_bb and hbond_lr_bb (short- and long-range backbone hydrogen bonding, respectively) terms of the Rosetta force field, each with equal weights. The conformational sampling involved ∼106 backbone moves for each model, with the effective temperature decreased from 10 kbT to the final target temperature (0.1 kbT for ECMC, varied for UMC) over the course of the refinement. Ensembles of at least 400 different structures were generated for each set of parameters.
AAMD simulations
We performed AAMD simulations of αS in explicit water using the fixed NPT ensemble (also known as the isobaric-isothermal ensemble, i.e., with the number of particles, pressure, and temperature held constant) at temperature T = 293 K and pressure P = 1 bar, conditions which closely match the smFRET experiments, to measure the solution-state ensemble of αS. These simulations were implemented in Gromacs 4.5.1 (52) using the Amber99SB (53,54) force field with the TIP4P-Ew water model (55). This parameter set produced markedly better agreement with ECMC Rg values in preliminary simulations than other combinations of force field and solvent model (not shown), and has also been reported to provide superior accuracy in protein simulations (17–19,56,57), especially in describing flexible protein regions (56). Pressure was controlled by the weak coupling method (58) with a 1.0 ps coupling time and temperature by a stochastic velocity distribution thermostat of Bussi et al. (59) that yields the canonical distribution with a 0.1 ps coupling time. The temperature of the protein was controlled independently of the solute and ions. Consistent with the original Amber99SB force field parameterization (53), nonbonded interactions were cutoff at 0.8 nm and the particle mesh Ewald method was employed to calculate the long-ranged electrostatic interactions. A time step of 2 fs and a cubic simulation box with volume (11 nm)3 were used for all simulations. The simulation volume is very large, but it is possible even with this box size for a completely extended protein configuration to interact with itself under periodic boundary conditions. However, such events are rare and were not included in the analysis. Ten different initial configurations randomly selected from the end states of ECMC simulations were run for a total of 574 ns. This ensured independent simulation trajectories for improved configurational sampling. The initial 10 ns of each trajectory were omitted from the analysis to allow for structural relaxation, leaving a total of 474 ns of simulation time used to generate the AAMD ensemble.
Results
ECMC simulations
SmFRET uses the efficiency of energy transfer between two labeled residues in a protein chain as a sensitive probe of protein conformation on the nm scale. We previously reported ETeff values between 12 pairs of residues in αS (27,51), and have also recently measured analogous ETeff between 12 residue pairs in tau (60). These data sets each represents the most extensive set of interresidue distances measured on αS and tau, respectively, at the single-molecule level. In the case of dynamic random coils like αS and tau, the use of polymer models, which reflect the ensemble of conformations sampled by the protein on the timescale of smFRET measurements (∼1 ms), has been shown to lead to more accurate calculations of distances from ETeff than the simple application of the Förster equation (61–63). We used results from excluded volume MC simulations to convert the smFRET measurements of ETeff into distance constraints, as detailed in the Supporting Material.
For all MC simulations, the proteins were modeled using the centroid representation of Rosetta (64), which consists of an all-atom peptide backbone and a single-bead pseudoatom for each residue side chain (accounting for side-chain size, and the location of the average center of mass of favorable side-chain conformations). We performed a MC conformational search implemented in the PyRosetta software package (50,64), with only smFRET-derived constraints and purely repulsive LJ interactions included in the potential, using up to 5 × 106 backbone moves. From this ensemble of structures produced by the ECMC simulations, we generated Rg distributions, interresidue distance maps, and the scaling behavior of the interresidue distance with sequence separation. Totals of 106 and 5 × 106 backbone moves were sufficient to ensure convergence of these three structural characteristics for αS and tau, respectively. These protocols were used for all subsequent simulations.
To study the effects of harmonic constraints on αS ensembles, we performed ECMC with various subsets of the constraints derived from smFRET measurements at pH 7.4. The mean Rg decreased from 5.1 ± 0.9 nm without constraints (the excluded volume random-coil limit for the αS chain), to 3.3 ± 0.3 nm for the fully constrained ensemble (Fig. 2 a). This compaction relative to a random coil involves enhanced local contacts between the N- and C-terminal regions of αS (Fig. 2 b). Polymer conformational statistics are frequently described in terms of the relation between mean interresidue distance r and sequence separation N:
| (1) |
where ν is the Flory scaling exponent, and the prefactor C depends roughly on the size of the monomer unit. The αS ensemble obtained by ECMC possesses a mean scaling exponent of 0.39 (Fig. 2 c), intermediate between the exponents expected for an excluded volume random coil (0.59) and a compact globule (0.33), which suggests the presence of attractive interactions within the disordered αS chain. An important caveat is that the scaling laws in polymer theories apply in the long chain limit; whereas αS and tau have chain lengths of 140 residues and 441 residues, respectively. We are using the scaling form of the interresidue separation in Eq. 1 as an empirical characterization of the size of αS and tau.
Figure 2.
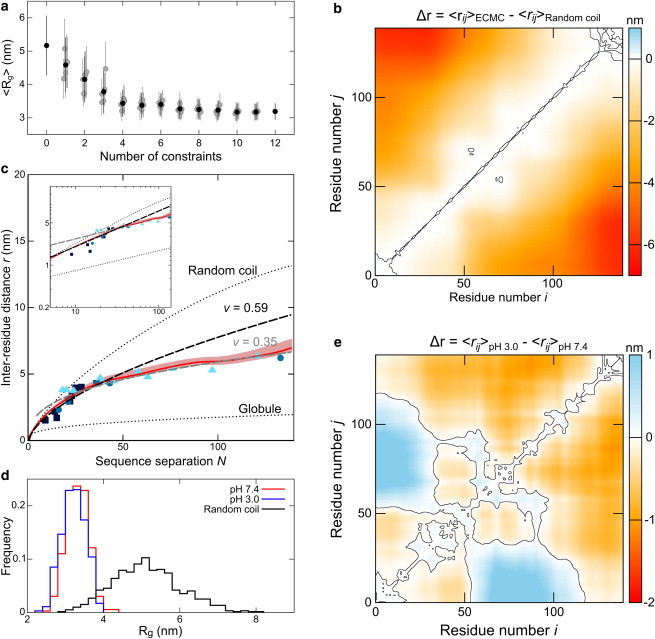
(a) Mean Rg values obtained by ECMC using between 0 and 12 randomly selected smFRET-derived constraints of αS. Gray circles and associated vertical lines indicate means and standard deviations of distributions obtained from a single randomly selected subset of the full constraint set, with the number of constraints indicated on the horizontal axis. Black circles denote means over all subsets for a given number of constraints. (b) An interresidue distance difference map shows the change in mean interresidue distances between the unconstrained random coil ensemble and ECMC-derived αS ensemble at pH 7.4. The black contour separates regions that display compaction (Δr < 0) relative to the random coil from those that display expansion (Δr > 0); compaction is particularly evident between residues 1–30 and 100–140. (c) The ECMC-derived αS ensemble (solid line) shows a polymer scaling intermediate between the excluded volume random-coil limit (obtained from unconstrained MC simulations of αS with purely repulsive LJ interactions and no smFRET constraints) and a compact globule (C = 0.38 nm, ν = 0.33 for Eq. 1 in the text), both shown as dotted lines. The light shaded area indicates the standard deviation in ECMC-derived r values, obtained by resampling error estimation as described in the Supporting Material. Points are discrete values obtained by smFRET (triangles), tryptophan triplet-state lifetime (65) (circles), and time-resolved FRET (29) (squares) measurements. Black and gray dashed lines are fits of Eq. 1 to the ECMC data for N < 40 and N > 40, respectively. The inset shows data replotted on logarithmic axes. (d) Rg histograms obtained by ECMC of αS at pH 7.4 (Rg = 3.3 ± 0.3 nm) and pH 3.0 (3.2 ± 0.3 nm), compared with the excluded volume random-coil model (5.1 ± 0.9 nm). Low pH, which enhances αS aggregation, has a minor effect on Rg. (e) Distance difference map (pH 3.0 – pH 7.4) showing the detailed effect of acidic environments on αS conformation, most notably compaction (Δr < 0) of the C-terminus and core regions.
Major sources of uncertainty in smFRET are experimental error in the determination of ETeff (estimated to be ∼1–2% for our instrumentation based on repeated smFRET measurements of a standard sample), and variation in the Förster radius due to anistropic dye tumbling and/or changes in quantum yield (estimated to be 7–8% based on fluorescence lifetime and anisotropy measurements). We implemented a resampling error estimation protocol described in the Supporting Material to quantify the resulting uncertainties in ECMC results, which are represented by the shaded area in Fig. 2 c. Due to the redundant nature of smFRET-derived constraints, the standard error of the mean in ensemble properties such as Rg or ν is ∼1.5%.
ECMC using smFRET measurements of αS at pH 3.0, a condition that is known to accelerate aggregation (27,67), indicates a slight compaction in terms of the mean Rg (3.2 ± 0.3 nm) relative to the neutral-pH ensemble (Fig. 2 d). However, the low-pH ensemble shows significant changes in the pattern of mean interresidue distances, with decreased contact between the N-terminus and the central region, and increased contact between the C-terminus and much of the rest of the protein (Fig. 2 e).
Similarly, we compared smFRET distance measurements between 12 residue pairs (60) for tau, in the presence and absence of the polyanion heparin, which is known to dramatically enhance the aggregation rate of tau in solution. Like αS, tau displays a progressive compaction relative to an excluded volume random coil of equal length (Rg = 10.9 ± 1.9 nm) as the number of constraints is increased (Fig. S2). The mean Rg of tau increases from 5.1 ± 0.5 nm to 6.0 ± 0.6 nm upon the addition of heparin (see Fig. 3 b), whereas the corresponding Rh values calculated by Hydropro (68) increase from 5.3 ± 0.3 nm to 5.9 ± 0.5 nm. In the absence of heparin, tau demonstrates a polymer scaling intermediate between the random coil and globule limits at small and medium sequence separations, but is markedly more compact at larger separations, so that the overall mean scaling exponent is 0.27. Heparin eliminates this long-range compaction, increasing the mean value of ν to 0.44 (Fig. 3 a). This expansion, particularly evident between residues 1–50 and the span from residues 180–441, is partially offset by compaction between residues 140–220 and residues 340–420 (Fig. 3 c).
Figure 3.
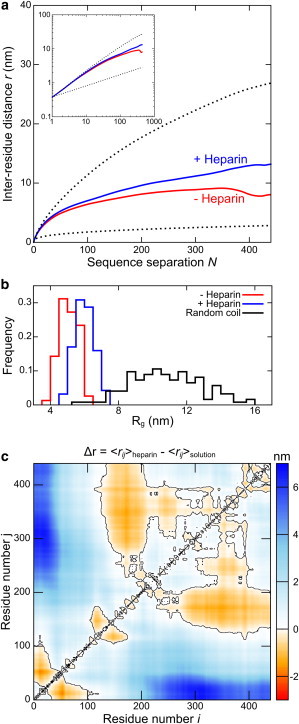
(a) Tau in buffer (- Heparin) displays similar polymer scaling behavior to αS at short and medium sequence separations, between the random coil and globule limits (dotted lines), but displays marked compaction at long sequence separations. The addition of heparin eliminates this long-range compaction, resulting in an increase in the mean scaling exponent from 0.28 to 0.43. (b) The global expansion induced by heparin is reflected in an increase in Rg from 5.1 ± 0.5 nm to 6.0 ± 0.6 nm, compared to 10.9 ± 1.9 nm for an excluded volume random-coil. (c) A distance difference map (heparin - buffer only) reveals that this global expansion is partially compensated for by a compaction of the proline-rich and microtubule-binding regions of tau in the presence of heparin. The black contour separates regions that display compaction (Δr < 0) from those that display expansion (Δr > 0).
Unconstrained MC and AAMD simulations
To further test the relation between Rg and interresidue distances, we investigated how accurately unconstrained simulations that reproduce the global dimensions of αS could predict the mean smFRET-derived distances. UMC simulations using a more complete Rosetta potential were performed over a range of effective temperatures to identify conditions that best reproduced the mean Rg obtained by ECMC. The potential used for UMC simulations included statistically obtained residue-environment and residue-residue interaction terms, backbone dihedral angle propensities, backbone hydrogen-bonding, and repulsive LJ interaction terms, reflecting charge interactions, hydrophobicity and polarity, and the sterics of the protein backbone. The UMC ensemble at 1.2 kbT most accurately captured the mean Rg of αS at neutral pH, and the mean interresidue distances extracted from this ensemble were in excellent agreement with the corresponding distances measured by smFRET (Fig. 4 a). We further performed all-atom, explicit-solvent MD (AAMD) on αS using the Amber99SB (53,54) force field with the TIP4P-Ew water model (55) at 293 K. This combination of parameters produces an ensemble with a Rg of 3.1 ± 0.9 nm, in close agreement with mean experimental values, and reproduces the mean interresidue distances measured by smFRET with high accuracy (Fig. 4 a). The ensemble generated by AAMD resembles that produced by UMC simulations (Fig. 4, b and c), but the computational cost is significantly higher. Properties of the ensembles generated by AAMD, UMC, and ECMC are compared in greater detail in the Supporting Material.
Figure 4.
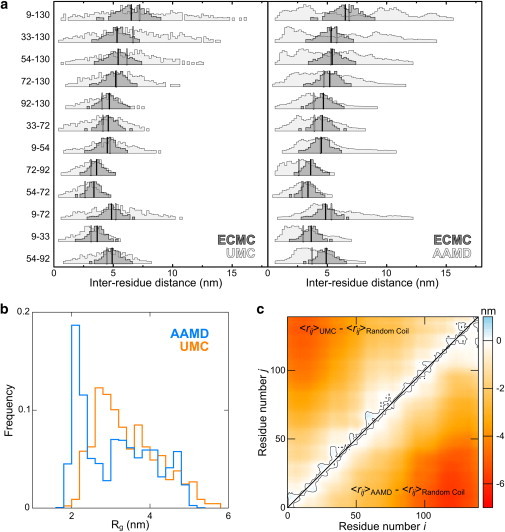
(a) Experimentally derived interresidue distance distributions from ECMC of αS (dark gray histograms with means indicated by black lines) and corresponding distributions from unconstrained simulations (left panel: UMC; right panel: AAMD; light gray histograms with means indicated by gray lines). Both simulation techniques accurately capture the mean pairwise distances but predict broader distance distributions, given either no experimental information (in the case of AAMD), or only the Rg (in the case of UMC). Note that AAMD distance distributions appear to be multimodal, especially for residue pairs spanning the N- and C-terminal regions of αS. (b) Rg distributions from UMC and AAMD simulations match the mean (3.3 nm) from ECMC, but have greater width (cf. Fig. 2d). (c) Distance difference maps comparing a random coil simulation with UMC (top) and AAMD (bottom) ensembles closely resemble each other, and the analogous map for ECMC simulations (cf. Fig. 2b).
Discussion
ECMC simulations
The ECMC protocol we have developed here is an efficient, powerful probe of αS and tau global dimensions, polymer scaling behavior, and local contacts. ECMC accurately reproduces the global dimensions of αS given pairwise distances from smFRET experiments: the mean Rg decreases smoothly and asymptotes to the experimental value as the number of constraints increases, which indicates that the smFRET data are internally self-consistent. Remarkably, as few as two or three constraints are enough to capture much of the compaction of αS, even though the only other term in the effective potential for these simulations is the repulsive LJ interaction. The measurement of Rg by ECMC (3.3 nm) lies within the broad range (2.6 to 5.0 nm) suggested by previous SAXS experiments (33–36), but is substantially more precise than these ensemble measurements (see details of error estimation in the Supporting Material). We recently measured the Rh value of αS to be 2.74 ± 0.04 nm using pulsed field gradient NMR (69). The Rg/Rh ratio of αS is therefore ∼1.2, in excellent agreement with the analogous ratio measured for unfolded protein L using smFRET and fluorescence correlation spectroscopy (62).
The polymer scaling behavior of αS as determined by ECMC also recapitulates very closely the results of tryptophan triplet-state lifetime (65) and time-resolved FRET (29) experiments, even at small sequence separations where the interresidue distances are too short for direct measurement by smFRET. We find that the scaling behavior depends on sequence separation, with the Flory exponent ν changing from ∼0.59 for N < 40 to ∼0.34 for N > 40. The intriguing observation that the scaling exponent for αS shifts from nearly the self-avoiding random-coil value to the globule value as sequence separation increases may reflect the fact that the intrinsic stiffness and excluded volume properties of the polypeptide backbone predominate at small N, whereas intramolecular attractive interactions predominate at large N. The long-range contacts that cause this compaction in the ECMC ensembles resemble those observed in PRE NMR experiments (21,23) and numerical simulations (43), particularly in terms of the enhanced contacts between residues 1–30 and 100–140 (23,43). Approximately 11% of the structures in the ECMC ensemble display end-to-end distances below 5 nm. The interresidue distance distributions obtained from ECMC simulations appear largely unimodal (red histograms in Fig. 4 a).
The polymer scaling of tau in the absence of heparin is qualitatively similar to that of αS, with a progressive compaction with increasing N: the exponent ν changes from ∼0.60 for N < 40, to ∼0.39 for 40 < N < 140, and then to ∼0.09 for N > 140. This marked deviation from excluded volume power-law scaling behavior at high sequence separations to scaling that is below the theoretical globule limit may reflect specific interactions between the N- and C-termini of tau as suggested by Mandelkow and co-workers (31,32) and also observed by PRE NMR (25). Heparin has a dramatic effect on this behavior: the exponent ν changes from ∼0.62 for N < 40, to ∼0.43 for N > 40, whereas the hypercompaction at high N is eliminated. This expansion is also reflected in the Rg and calculated Rh values of tau, which increase by 19% and 11%, respectively, in the presence of heparin. The latter change is in excellent agreement with fluorescence correlation spectroscopy experiments, which show a 14% increase in the diffusion time upon binding of heparin (60).
ECMC is also sensitive to local changes in structure that do not have a major effect on global dimensions. Although decreasing the pH from 7.4 to 3.0 results in only small changes in αS Rg (3.3 nm vs. 3.2 nm), the two ECMC ensembles are significantly different in local structure. The decreased charge on the C-terminus at low pH leads to a marked collapse of this region, and enhanced interactions with residues 25–100. This span includes the aggregation prone region (residues 60–95) of αS, and so this altered interaction may relate to the increased aggregation propensity of αS at low pH. There is a compensatory expansion of the N-terminus that may be due to the increased net positive charge of this region in acidic environments. This observation agrees very well with PRE NMR experiments on αS at low and neutral pH (23,41), which also show a relative collapse of the C-terminus and slight expansion of the N-terminus. Similarly, although the addition of heparin caused a moderate increase in the global dimensions of tau, this masks a compaction of the central microtubule-binding region that forms the core of tau-derived amyloid aggregates. Heparin also eliminates the long-range contacts between the termini of tau that have been previously observed by ensemble FRET (31,32). These conformational changes may be driven by neutralization of the net positive charge on the proline-rich and microtubule-binding regions of tau by the polyanion heparin. Our results illustrate the ability of ECMC to effectively identify local changes in structure, even in the absence of major global changes, which could provide crucial insight into the environmental factors and binding partners that affect the tendency of αS and tau to pathologically self-assemble.
UMC and AAMD simulations
Having demonstrated that ECMC accurately reproduces global dimensions from pairwise constraints, we addressed the inverse problem of whether it is possible to derive pairwise interresidue separations from global dimensions of the chain. UMC simulations with a single free parameter (effective temperature) tuned to match the mean Rg accurately reproduced mean interresidue distances that span nearly the complete sequence of αS. The polymer scaling behavior and interresidue distance map of the UMC ensemble also resemble those of the ECMC ensemble at neutral pH. This remarkable result indicates that it is possible to infer computationally much of the detailed conformational properties of αS given only the mean Rg of the ensemble and the robust knowledge-based potential implemented in Rosetta.
To investigate whether this result is specific to Rosetta, or also applies to other modern simulation techniques, we performed extensive AAMD simulations on αS, which faithfully reflected αS solution-state behavior at physiological temperature, without any bias or post facto filtering. The combination of Amber99SB and TIP4P-Ew solvent appears to simulate the αS conformational ensemble with high accuracy, in good agreement with the extensive results from other groups (17–20,56,57) successfully applying this parameter set to disordered proteins and peptides. The high accuracy in reproducing the mean interresidue separations, as measured by smFRET, is especially noteworthy given that previous MD studies (with other force fields and solvent treatments) had to resort to temperatures 150–300 K above physiological values to achieve similar agreement with experiment (40,41). Recent corrections to the Amber99SB force field (70) used with TIP3P solvent have shown improved accuracy in predicting the folding behavior of several model peptides and proteins (71,72), and in future work we will test whether this parameter set also accurately reproduces αS solution-state behavior.
Conclusions
In this work, we have developed and validated a new, to our knowledge, approach to effectively investigate the conformational behavior of disordered proteins, using MC simulations constrained by smFRET experiments. In particular, we have shown that the global dimensions and polymer conformational statistics of the model protein αS and tau can be accurately determined from a relatively small number of interresidue distance measurements, and produce ensembles in excellent agreement with previous experimental studies, as well as with the AAMD simulations in the case of αS. Conversely, we have shown that recently developed protein energy functions make it possible to predict detailed conformational properties and mean interresidue distances of αS from its global dimensions, and that transient charge-mediated contacts between the residues distant in sequence may underlie the dynamic conformational fluctuations of αS and tau suggested by our findings and others (30,65,73,74).
The ECMC approach is much less computationally intensive than simulation methods that predict IDP conformational ensembles with comparable accuracy, which become rapidly more demanding as the size of the protein of interest increases. For example, comparable AAMD studies on tau would require simulation of a system at least an order of magnitude larger in volume (to accommodate the greater dimensions of tau), for a substantially longer period of time (to sample the increased number of degrees of freedom of the tau chain with a similar level of detail to αS). Given typically available resources, the consequent computational expense makes routine AAMD simulations of large IDPs infeasible. ECMC could therefore serve as an efficient alternative strategy to characterize the detailed conformations of diverse IDPs. ECMC based on smFRET measurements is also well suited to the identification of high-affinity binding partners of IDPs; this is potentially of broad pharmacological relevance, given that IDPs may be effective drug targets for a range of diseases (46). ECMC could therefore prove to be a versatile probe of the effects of environmental factors, drug-like small molecules, and biological ligands on IDP conformations in health and disease.
Acknowledgments
This research was supported by the National Science Foundation under grant Nos. DMS-0835742 (to C.S.O.) and MCB-0919853 (to E.R.), The Ellison Medical Foundation (to E.R.), an American Heart Association Postdoctoral Fellowship (to A.N.), the Academy of Finland (to M.S.), an institutional training grant from the National Institute of Health GM007223 (to D.C.D., A.J.T., and S.E.-G.), and a seed grant from the Raymond and Beverly Sackler Institute for Biological, Physical, and Engineering Sciences (to C.S.O. and E.R.). The research leading to these results has received funding from the European Union Seventh Framework Programme FP7/2007-2013 under grant agreement #293861 (to M.S.). This work also benefited from the facilities and staff of the Yale University Faculty of Arts and Sciences High Performance Computing Center, CSC IT Center for Science in Finland (M.S.), and National Science Foundation (NSF) grant No. CNS-0821132 that partially funded acquisition of the computational facilities.
Contributor Information
Abhinav Nath, Email: abhinav.nath@yale.edu.
Corey S. O’Hern, Email: corey.ohern@yale.edu.
Elizabeth Rhoades, Email: elizabeth.rhoades@yale.edu.
Supporting Material
References
- 1.Dunker A.K., Cortese M.S., Uversky V.N. Flexible nets. The roles of intrinsic disorder in protein interaction networks. FEBS J. 2005;272:5129–5148. doi: 10.1111/j.1742-4658.2005.04948.x. [DOI] [PubMed] [Google Scholar]
- 2.Midic U., Oldfield C.J., Uversky V.N. Unfoldomics of human genetic diseases: illustrative examples of ordered and intrinsically disordered members of the human diseasome. Protein Pept. Lett. 2009;16:1533–1547. doi: 10.2174/092986609789839377. [DOI] [PubMed] [Google Scholar]
- 3.Anfinsen C.B. Principles that govern the folding of protein chains. Science. 1973;181:223–230. doi: 10.1126/science.181.4096.223. [DOI] [PubMed] [Google Scholar]
- 4.Flory P.J. Thermodynamics of high polymer solutions. J. Chem. Phys. 1942;10:51–61. [Google Scholar]
- 5.Eliezer D. Biophysical characterization of intrinsically disordered proteins. Curr. Opin. Struct. Biol. 2009;19:23–30. doi: 10.1016/j.sbi.2008.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Receveur-Bréchot V., Bourhis J.-M., Longhi S. Assessing protein disorder and induced folding. Proteins. 2006;62:24–45. doi: 10.1002/prot.20750. [DOI] [PubMed] [Google Scholar]
- 7.Mittag T., Forman-Kay J.D. Atomic-level characterization of disordered protein ensembles. Curr. Opin. Struct. Biol. 2007;17:3–14. doi: 10.1016/j.sbi.2007.01.009. [DOI] [PubMed] [Google Scholar]
- 8.Chen H., Rhoades E. Fluorescence characterization of denatured proteins. Curr. Opin. Struct. Biol. 2008;18:516–524. doi: 10.1016/j.sbi.2008.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Price W.S. Pulsed-field gradient nuclear magnetic resonance as a tool for studying translational diffusion: Part 1. Basic theory. Concepts Magn. Reson. 1997;9:299–336. [Google Scholar]
- 10.Haustein E., Schwille P. Fluorescence correlation spectroscopy: novel variations of an established technique. Annu. Rev. Biophys. Biomol. Struct. 2007;36:151–169. doi: 10.1146/annurev.biophys.36.040306.132612. [DOI] [PubMed] [Google Scholar]
- 11.Leone V., Marinelli F., Parrinello M. Targeting biomolecular flexibility with metadynamics. Curr. Opin. Struct. Biol. 2010;20:148–154. doi: 10.1016/j.sbi.2010.01.011. [DOI] [PubMed] [Google Scholar]
- 12.Scheraga H.A., Khalili M., Liwo A. Protein-folding dynamics: overview of molecular simulation techniques. Annu. Rev. Phys. Chem. 2007;58:57–83. doi: 10.1146/annurev.physchem.58.032806.104614. [DOI] [PubMed] [Google Scholar]
- 13.Rauscher S., Pomès R. Molecular simulations of protein disorder. Biochem. Cell Biol. 2010;88:269–290. doi: 10.1139/o09-169. [DOI] [PubMed] [Google Scholar]
- 14.Vitalis A., Pappu R.V. Methods for Monte Carlo simulations of biomacromolecules. Annu. Rep. Comput. Chem. 2009;5:49–76. doi: 10.1016/S1574-1400(09)00503-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dill K.A., Truskett T.M., Hribar-Lee B. Modeling water, the hydrophobic effect, and ion solvation. Annu. Rev. Biophys. Biomol. Struct. 2005;34:173–199. doi: 10.1146/annurev.biophys.34.040204.144517. [DOI] [PubMed] [Google Scholar]
- 16.Chen J., Brooks C.L., 3rd, Khandogin J. Recent advances in implicit solvent-based methods for biomolecular simulations. Curr. Opin. Struct. Biol. 2008;18:140–148. doi: 10.1016/j.sbi.2008.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fawzi N.L., Phillips A.H., Head-Gordon T. Structure and dynamics of the Abeta(21–30) peptide from the interplay of NMR experiments and molecular simulations. J. Am. Chem. Soc. 2008;130:6145–6158. doi: 10.1021/ja710366c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Aliev A.E., Courtier-Murias D. Experimental verification of force fields for molecular dynamics simulations using Gly-Pro-Gly-Gly. J. Phys. Chem. B. 2010;114:12358–12375. doi: 10.1021/jp101581h. [DOI] [PubMed] [Google Scholar]
- 19.Lange O.F., van der Spoel D., de Groot B.L. Scrutinizing molecular mechanics force fields on the submicrosecond timescale with NMR data. Biophys. J. 2010;99:647–655. doi: 10.1016/j.bpj.2010.04.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Piana S., Lindorff-Larsen K., Shaw D.E. How robust are protein folding simulations with respect to force field parameterization? Biophys. J. 2011;100:L47–L49. doi: 10.1016/j.bpj.2011.03.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dedmon M.M., Lindorff-Larsen K., Dobson C.M. Mapping long-range interactions in alpha-synuclein using spin-label NMR and ensemble molecular dynamics simulations. J. Am. Chem. Soc. 2005;127:476–477. doi: 10.1021/ja044834j. [DOI] [PubMed] [Google Scholar]
- 22.Sung Y.-H., Eliezer D. Residual structure, backbone dynamics, and interactions within the synuclein family. J. Mol. Biol. 2007;372:689–707. doi: 10.1016/j.jmb.2007.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Salmon L., Nodet G., Blackledge M. NMR characterization of long-range order in intrinsically disordered proteins. J. Am. Chem. Soc. 2010;132:8407–8418. doi: 10.1021/ja101645g. [DOI] [PubMed] [Google Scholar]
- 24.Mukrasch M.D., Markwick P., Blackledge M. Highly populated turn conformations in natively unfolded tau protein identified from residual dipolar couplings and molecular simulation. J. Am. Chem. Soc. 2007;129:5235–5243. doi: 10.1021/ja0690159. [DOI] [PubMed] [Google Scholar]
- 25.Mukrasch M.D., Bibow S., Zweckstetter M. Structural polymorphism of 441-residue tau at single residue resolution. PLoS Biol. 2009;7:e34. doi: 10.1371/journal.pbio.1000034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ferreon A.C.M., Moran C.R., Deniz A.A. Alteration of the alpha-synuclein folding landscape by a mutation related to Parkinson’s disease. Angew. Chem. Int. Ed. Engl. 2010;49:3469–3472. doi: 10.1002/anie.201000378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Trexler A.J., Rhoades E. Single molecule characterization of α-synuclein in aggregation-prone states. Biophys. J. 2010;99:3048–3055. doi: 10.1016/j.bpj.2010.08.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rhoades E., Ramlall T.F., Eliezer D. Quantification of alpha-synuclein binding to lipid vesicles using fluorescence correlation spectroscopy. Biophys. J. 2006;90:4692–4700. doi: 10.1529/biophysj.105.079251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Grupi A., Haas E. Segmental conformational disorder and dynamics in the intrinsically disordered protein α-synuclein and its chain length dependence. J. Mol. Biol. 2011;405:1267–1283. doi: 10.1016/j.jmb.2010.11.011. [DOI] [PubMed] [Google Scholar]
- 30.Grupi A., Haas E. Time-resolved FRET detection of subtle temperature-induced conformational biases in ensembles of α-synuclein molecules. J. Mol. Biol. 2011;411:234–247. doi: 10.1016/j.jmb.2011.04.056. [DOI] [PubMed] [Google Scholar]
- 31.Jeganathan S., von Bergen M., Mandelkow E. Global hairpin folding of tau in solution. Biochemistry. 2006;45:2283–2293. doi: 10.1021/bi0521543. [DOI] [PubMed] [Google Scholar]
- 32.Jeganathan S., Hascher A., Mandelkow E. Proline-directed pseudo-phosphorylation at AT8 and PHF1 epitopes induces a compaction of the paperclip folding of Tau and generates a pathological (MC-1) conformation. J. Biol. Chem. 2008;283:32066–32076. doi: 10.1074/jbc.M805300200. [DOI] [PubMed] [Google Scholar]
- 33.Li J., Uversky V.N., Fink A.L. Conformational behavior of human α-synuclein is modulated by familial Parkinson’s disease point mutations A30P and A53T. Neurotoxicology. 2002;23:553–567. doi: 10.1016/s0161-813x(02)00066-9. [DOI] [PubMed] [Google Scholar]
- 34.Tashiro M., Kojima M., Shimotakahara S. Characterization of fibrillation process of alpha-synuclein at the initial stage. Biochem. Biophys. Res. Commun. 2008;369:910–914. doi: 10.1016/j.bbrc.2008.02.127. [DOI] [PubMed] [Google Scholar]
- 35.Rekas A., Knott R.B., Pham C.L. The structure of dopamine induced alpha-synuclein oligomers. Eur. Biophys. J. 2010;39:1407–1419. doi: 10.1007/s00249-010-0595-x. [DOI] [PubMed] [Google Scholar]
- 36.Giehm L., Svergun D.I., Vestergaard B. Low-resolution structure of a vesicle disrupting α-synuclein oligomer that accumulates during fibrillation. Proc. Natl. Acad. Sci. USA. 2011;108:3246–3251. doi: 10.1073/pnas.1013225108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schweers O., Schönbrunn-Hanebeck E., Mandelkow E. Structural studies of tau protein and Alzheimer paired helical filaments show no evidence for beta-structure. J. Biol. Chem. 1994;269:24290–24297. [PubMed] [Google Scholar]
- 38.Mylonas E., Hascher A., Svergun D.I. Domain conformation of tau protein studied by solution small-angle x-ray scattering. Biochemistry. 2008;47:10345–10353. doi: 10.1021/bi800900d. [DOI] [PubMed] [Google Scholar]
- 39.Shkumatov A.V., Chinnathambi S., Svergun D.I. Structural memory of natively unfolded tau protein detected by small-angle x-ray scattering. Proteins. 2011;79:2122–2131. doi: 10.1002/prot.23033. [DOI] [PubMed] [Google Scholar]
- 40.Allison J.R., Varnai P., Vendruscolo M. Determination of the free energy landscape of alpha-synuclein using spin label nuclear magnetic resonance measurements. J. Am. Chem. Soc. 2009;131:18314–18326. doi: 10.1021/ja904716h. [DOI] [PubMed] [Google Scholar]
- 41.Wu K.-P., Weinstock D.S., Baum J. Structural reorganization of alpha-synuclein at low pH observed by NMR and REMD simulations. J. Mol. Biol. 2009;391:784–796. doi: 10.1016/j.jmb.2009.06.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tsigelny I.F., Sharikov Y., Masliah E. Simulation and modeling of synuclein-based ‘protofibril’ structures: as a means of understanding the molecular basis of Parkinson’s disease. J. Phys. Conf. Ser. 2008;125:012056. [Google Scholar]
- 43.Ullman O., Fisher C.K., Stultz C.M. Explaining the structural plasticity of α-synuclein. J. Am. Chem. Soc. 2011;133:19536–19546. doi: 10.1021/ja208657z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cheng Y., LeGall T., Dunker A.K. Rational drug design via intrinsically disordered protein. Trends Biotechnol. 2006;24:435–442. doi: 10.1016/j.tibtech.2006.07.005. [DOI] [PubMed] [Google Scholar]
- 45.Masuda M., Suzuki N., Hasegawa M. Small molecule inhibitors of alpha-synuclein filament assembly. Biochemistry. 2006;45:6085–6094. doi: 10.1021/bi0600749. [DOI] [PubMed] [Google Scholar]
- 46.Metallo S.J. Intrinsically disordered proteins are potential drug targets. Curr. Opin. Chem. Biol. 2010;14:481–488. doi: 10.1016/j.cbpa.2010.06.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhao J.-H., Liu H.-L., Chuang C.-K., Liu K.-T., Tsai W.-B. Molecular dynamics simulations to investigate the stability and aggregation behaviour of the amyloid-forming peptide VQIVYK from tau protein. Mol. Simul. 2010;36:1013–1024. [Google Scholar]
- 48.Miller Y., Ma B., Nussinov R. Synergistic interactions between repeats in tau protein and Aβ amyloids may be responsible for accelerated aggregation via polymorphic states. Biochemistry. 2011;50:5172–5181. doi: 10.1021/bi200400u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jho Y.S., Zhulina E.B., Pincus P.A. Monte carlo simulations of tau proteins: effect of phosphorylation. Biophys. J. 2010;99:2387–2397. doi: 10.1016/j.bpj.2010.06.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chaudhury S., Lyskov S., Gray J.J. PyRosetta: a script-based interface for implementing molecular modeling algorithms using Rosetta. Bioinformatics. 2010;26:689–691. doi: 10.1093/bioinformatics/btq007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Trexler A.J., Rhoades E. Alpha-synuclein binds large unilamellar vesicles as an extended helix. Biochemistry. 2009;48:2304–2306. doi: 10.1021/bi900114z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hess B., Kutzner C., Lindahl E. GROMACS 4: algorithms for highly efficient, load-balanced, and scalable molecular simulation. J. Chem. Theory Comput. 2008;4:435–447. doi: 10.1021/ct700301q. [DOI] [PubMed] [Google Scholar]
- 53.Hornak V., Abel R., Simmerling C. Comparison of multiple Amber force fields and development of improved protein backbone parameters. Proteins. 2006;65:712–725. doi: 10.1002/prot.21123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sorin E.J., Pande V.S. Exploring the helix-coil transition via all-atom equilibrium ensemble simulations. Biophys. J. 2005;88:2472–2493. doi: 10.1529/biophysj.104.051938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Horn H.W., Swope W.C., Head-Gordon T. Development of an improved four-site water model for biomolecular simulations: TIP4P-Ew. J. Chem. Phys. 2004;120:9665–9678. doi: 10.1063/1.1683075. [DOI] [PubMed] [Google Scholar]
- 56.Showalter S.A., Johnson E., Brüschweiler R. Toward quantitative interpretation of methyl side-chain dynamics from NMR by molecular dynamics simulations. J. Am. Chem. Soc. 2007;129:14146–14147. doi: 10.1021/ja075976r. [DOI] [PubMed] [Google Scholar]
- 57.Wong V., Case D.A. Evaluating rotational diffusion from protein MD simulations. J. Phys. Chem. B. 2008;112:6013–6024. doi: 10.1021/jp0761564. [DOI] [PubMed] [Google Scholar]
- 58.Berendsen H.J.C., Postma J.P.M., Haak J.R. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 1984;81:3684–3690. [Google Scholar]
- 59.Bussi G., Donadio D., Parrinello M. Canonical sampling through velocity rescaling. J. Chem. Phys. 2007;126:014101. doi: 10.1063/1.2408420. [DOI] [PubMed] [Google Scholar]
- 60.Elbaum-Garfinkle S., Rhoades E. Identification of an aggregation-prone structure of tau. J. Am. Chem. Soc. 2012;134:16607–16613. doi: 10.1021/ja305206m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.O’Brien E.P., Morrison G., Thirumalai D. How accurate are polymer models in the analysis of Förster resonance energy transfer experiments on proteins? J. Chem. Phys. 2009;130:124903. doi: 10.1063/1.3082151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sherman E., Haran G. Coil-globule transition in the denatured state of a small protein. Proc. Natl. Acad. Sci. USA. 2006;103:11539–11543. doi: 10.1073/pnas.0601395103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.McCarney E.R., Werner J.H., Plaxco K.W. Site-specific dimensions across a highly denatured protein; a single molecule study. J. Mol. Biol. 2005;352:672–682. doi: 10.1016/j.jmb.2005.07.015. [DOI] [PubMed] [Google Scholar]
- 64.Rohl C.A., Strauss C.E.M., Baker D. Protein structure prediction using Rosetta. Methods Enzymol. 2004;383:66–93. doi: 10.1016/S0076-6879(04)83004-0. [DOI] [PubMed] [Google Scholar]
- 65.Lee J.C., Gray H.B., Winkler J.R. Tertiary contact formation in alpha-synuclein probed by electron transfer. J. Am. Chem. Soc. 2005;127:16388–16389. doi: 10.1021/ja0561901. [DOI] [PubMed] [Google Scholar]
- 66.Reference deleted in proof.
- 67.Uversky V.N., Li J., Fink A.L. Evidence for a partially folded intermediate in alpha-synuclein fibril formation. J. Biol. Chem. 2001;276:10737–10744. doi: 10.1074/jbc.M010907200. [DOI] [PubMed] [Google Scholar]
- 68.García De La Torre J., Huertas M.L., Carrasco B. Calculation of hydrodynamic properties of globular proteins from their atomic-level structure. Biophys. J. 2000;78:719–730. doi: 10.1016/S0006-3495(00)76630-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Sevcsik E., Trexler A.J., Rhoades E. Allostery in a disordered protein: oxidative modifications to α-synuclein act distally to regulate membrane binding. J. Am. Chem. Soc. 2011;133:7152–7158. doi: 10.1021/ja2009554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Best R.B., Hummer G. Optimized molecular dynamics force fields applied to the helix-coil transition of polypeptides. J. Phys. Chem. B. 2009;113:9004–9015. doi: 10.1021/jp901540t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Mittal J., Best R.B. Tackling force-field bias in protein folding simulations: folding of Villin HP35 and Pin WW domains in explicit water. Biophys. J. 2010;99:L26–L28. doi: 10.1016/j.bpj.2010.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Best R.B., Mittal J. Balance between alpha and beta structures in ab initio protein folding. J. Phys. Chem. B. 2010;114:8790–8798. doi: 10.1021/jp102575b. [DOI] [PubMed] [Google Scholar]
- 73.Bernstein S.L., Liu D., Winkler J.R. Alpha-synuclein: stable compact and extended monomeric structures and pH dependence of dimer formation. J. Am. Soc. Mass Spectrom. 2004;15:1435–1443. doi: 10.1016/j.jasms.2004.08.003. [DOI] [PubMed] [Google Scholar]
- 74.Natalello A., Benetti F., Grandori R. Compact conformations of α-synuclein induced by alcohols and copper. Proteins. 2011;79:611–621. doi: 10.1002/prot.22909. [DOI] [PubMed] [Google Scholar]
- 75.Lee J.C., Lai B.T., Winkler J.R. Alpha-synuclein tertiary contact dynamics. J. Phys. Chem. B. 2007;111:2107–2112. doi: 10.1021/jp068604y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Wittkop M., Kreitmeier S., Göritz D. The distribution function of internal distances of a single polymer chain with excluded volume in two and three dimensions: A Monte Carlo study. J. Chem. Phys. 1996;104:351–358. [Google Scholar]
- 77.Caracciolo S., Causo M.S., Pelissetto A. End-to-end distribution function for dilute polymers. J. Chem. Phys. 2000;112:7693–7710. [Google Scholar]
- 78.Dale R., Eisinger J., Blumberg W. The orientational freedom of molecular probes: The orientation factor in intermolecular energy transfer. Biophys. J. 1979;26:161–193. doi: 10.1016/S0006-3495(79)85243-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.