

Abstract
Deficiency of the vital muscle protein dystrophin triggers Duchenne/Becker muscular dystrophy, but the structure-function relationship of dystrophin is poorly understood. To date, molecular structures of three dystrophin domains have been determined, of which the N-terminal actin-binding domain (N-ABD or ABD1) is of particular interest. This domain is composed of two calponin-homology (CH) domains, which form an important class of ABDs in muscle proteins. A previously determined x-ray structure indicates that the dystrophin N-ABD is a domain-swapped dimer, with each monomer adopting an extended, open conformation in which the two CH domains do not interact. This structure is controversial because it contradicts functional studies and known structures of similar ABDs from other muscle proteins. Here, we investigated the solution conformation of the dystrophin N-ABD using a very simple and elegant technique of pyrene excimer fluorescence. Using the wild-type protein, which contains two cysteines, and the corresponding single-cysteine mutants, we show that the protein is a monomer in solution and is in a closed conformation in which the two CH domains seem to interact, as observed from the excimer fluorescence of pyrene-labeled wild-type protein. Excimer fluorescence was also observed in its actin-bound form, indicating that the dystrophin N-ABD binds to F-actin in a closed conformation. Comparison of the dystrophin N-ABD conformation with other ABDs indicates that the tandem CH domains in general may be monomeric in solution and predominantly occur in closed conformation, whereas their actin-bound conformations may differ.
Introduction
Muscular dystrophy (MD) is the most common genetic lethal disorder in children (1). Mutations in the gene encoding the vital muscle protein dystrophin trigger the two most common forms of MD: Duchenne and Becker. Dystrophin stabilizes the muscle cell membrane against the mechanical forces associated with muscle contraction and stretch by connecting actin filaments to the sarcolemmal glycoprotein complex (2,3). Despite its well-established role in MD, the relationship between the structure and function of dystrophin is poorly understood.
Dystrophin is a long, rod-shaped molecule containing 3685 amino acids (4). Because of its large size, it is currently not possible to determine the molecular structure of the complete protein. Therefore, research focus has shifted toward determining the structures of individual domains. So far, the molecular structures have been determined for <14% of the full-length protein (3), which stresses the need for more structural studies on dystrophin to understand how its structure controls function. Of the three dystrophin domains whose structures are known (5,6), the N-terminal actin-binding domain (N-ABD or ABD1) is of particular interest because of its role in the most important function of dystrophin, i.e., actin binding. In addition, a major fraction of disease-causing missense mutations occur in this domain (7,8), and probing its structure-function relationship and the effect of mutations might help in understanding the mechanisms by which the disease is triggered. A previously determined x-ray crystal structure of the dystrophin N-ABD indicates that it is an antiparallel domain-swapped dimer (Fig. 1 a) (5), with each monomer existing in an extended, open conformation. This dimeric crystal structure is controversial. Biochemical (9–11) and recent biophysical studies (7,12) indicate that the dystrophin N-ABD might be a monomer in solution. In addition, similar ABDs from other muscle proteins, such as utrophin (13,14), fimbrin (15), plectin (16), and α-actinin (17), are monomers. Therefore, we must first resolve the controversy regarding the solution structure of the dystrophin N-ABD, before determining its structure-function relationship and the effects of disease-causing mutations.
Figure 1.
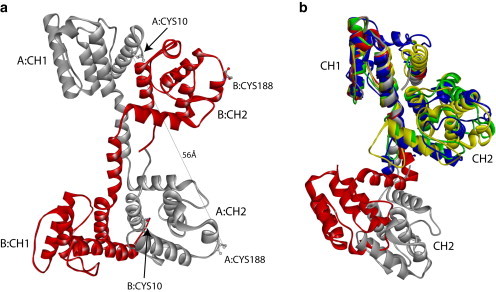
(a) X-ray crystal structure of the N-ABD of human dystrophin (1DXX.pdb) indicates an antiparallel, domain-swapped dimer (5). The two monomers, labeled A and B, are shown in gray and red, respectively. In the dimer, the CH1 domain from one monomer interacts with the CH2 domain from the other monomer. Each monomer contains two cysteines (Cys-10 and Cys-188), one in each CH domain, at an intramolecular distance of 56 Å. (b) Structural alignment of x-ray crystal structures of tandem CH domains of dystrophin (1DXX.pdb; gray), utrophin (1QAG.pdb; red), fimbrin (1AOA.pdb; blue), α-actinin (1TJT.pdb; green), and plectin (1MB8.pdb; yellow) using the MultiProt program (47) (http://bioinfo3d.cs.tau.ac.il/MultiProt/). Dystrophin and utrophin tandem CH domains crystallize in an open conformation where the two CH domains do not interact, whereas tandem CH domains of fimbrin, α-actinin, and plectin crystallize in a closed conformation with significant inter-CH domain interactions. The N-terminal (CH1) and C-terminal (CH2) CH domains are labeled in the figure. Molecular structures were drawn using the program Accelrys Discovery Studio Visualizer (http://accelrys.com/products/discovery-studio/visualization-download.php).
In addition, studying the structure-function relationship of the dystrophin N-ABD might improve the efficacy of mini-dystrophins. These proteins are currently under clinical trials to treat the loss of functional dystrophin in MD patients (18,19), but they tend to have decreased stability, functionality, and in-vivo half-life (20–22) compared to the full-length dystrophin. All these proteins contain the N-ABD. Therefore, one method of stabilizing the mini-dystrophins might be to increase the stability of the component domains, for example, that of N-ABD. Understanding the structural principles that govern N-ABD function might lead to improving actin-binding efficiency of mini-dystrophins.
Determining the solution conformation of the dystrophin N-ABD might provide new insights into the structural plasticity of tandem calponin-homology (CH) domains, which form an important class of ABDs in muscle proteins (10,23). The dystrophin N-ABD contains two CH domains in tandem (5). In its dimeric crystal structure (Fig. 1 a), each monomer is in an open, extended conformation with no significant interactions between the N-terminal (CH1) and C-terminal (CH2) CH domains. However, this open conformation of the dystrophin N-ABD contradicts the structures of other tandem CH domains, for example, those of fimbrin (15), plectin (16), and α-actinin (17), which crystallize in a closed, compact conformation with significant interactions between the individual CH domains (Fig. 1 b). The tandem CH domain from another homologous protein, utrophin, crystallizes as an antiparallel dimer similar to that of dystrophin with each monomer in an open conformation (14) (Fig. 1 b), however, recent solution studies show that it exists in both closed and open conformations (24,25). These recent studies (24,25) also indicate that the solution conformation of tandem CH domains might differ from their crystal structures, and stresses the need to determine their solution structures. Similar to the conformational differences of tandem CH domains, their mode of binding to F-actin and associated conformational changes are also controversial. No high-resolution molecular structures are available for these proteins in their actin-bound form, and the low-resolution cryo-electron microscopy (cryo-EM) data have been used to deduce contradicting conclusions (10,23,26). For example, cryo-EM data on the actin-bound tandem CH domain of utrophin have led to the conclusion that it is in a closed conformation (27), whereas similar data have been used to conclude that it is in an open conformation (28,29). Thus, a controversy has arisen as to whether the tandem CH domains bind to F-actin in a closed or open conformation (26). The cryo-EM data on the two tandem CH domains of fimbrin suggest that they bind to F-actin in closed conformations (30,31). Cryo-EM studies on α-actinin’s tandem CH domain indicate that it binds to F-actin in an open conformation (32). In the case of cryo-EM studies on actin-bound dystrophin N-ABD (27), the extent of labeling was poor; however, the available data suggest that it might bind to F-actin in a closed conformation, although further definitive experiments are needed to determine its actin-bound conformation. In light of these controversies associated with the crystal structures and the actin-bound conformations of tandem CH domains interpreted from cryo-EM studies, we were in search of a very simple method that could help determine whether the tandem CH domains exist in a closed or open conformation in solution. We found our answer in the technique of pyrene excimer fluorescence.
The main principle behind pyrene excimer fluorescence is that when an excited pyrene molecule is close in three-dimensional (3D) space to a neighboring, ground-state pyrene, their aromatic rings stack against each other to form a dimer (excited-state dimer or excimer). This results in a new fluorescence emission band at ∼470 nm (33–35), which is quite distinct from the fluorescence of monomeric pyrene that occur at ∼375 nm. Pyrenes can be covalently linked to cysteine residues in proteins using maleimide chemistry (33). Considering the length of the linkers, if the two labeled positions are within a distance of ≤23 Å in the 3D protein structure (33,34,36), excimer fluorescence can be expected. Unlike the commonly used fluorescence resonance energy transfer techniques, pyrene excimer fluorescence does not provide any information about the exact distances, and in that sense, it provides more of a yes/no answer where the observation of excimer fluorescence indicates that the two labeled positions are close in 3D space. This technique has been extensively used to probe conformational transitions of proteins (33,36–39), and also the dynamics of actin and actin-binding proteins (11,34,40–42). The wild-type (WT) dystrophin N-ABD contains cysteines at positions 10 in the CH1 domain and 188 in the CH2 domain, which are not involved in a disulfide bond (Fig. 1 a). We therefore designed two single-cysteine mutants, namely C10S and C188S, by mutating one of the cysteines to serine. We labeled the WT and the mutants using pyrene maleimide. Using the excimer fluorescence of these proteins, we addressed three important issues: 1), whether the dystrophin N-ABD is a dimer in solution; 2), whether it is in an open conformation in solution, similar to that in the crystal structure (Fig. 1 a); and 3), whether it is in a closed conformation in its actin-bound form, as predicted by the low-resolution cryo-EM studies (27).
Materials and Methods
Cloning, expression, and purification
The cDNA coding for dystrophin N-ABD (residues 1–246; a kind gift from Steve Winder, University of Sheffield, UK) was amplified using polymerase chain reaction, subcloned into pET28a expression vector using NdeI and HindIII restriction sites, and transformed into DH5α by heat-shock method. The plasmid was amplified using the Qiagen miniprep kit (Valencia, CA), and single cysteine mutants (C10S, C188S) were generated using the Qiaquick mutagenesis protocol. Amplified DNA sequences of dystrophin N-ABD and its mutants were confirmed by DNA sequencing. All three plasmids were transformed into BL21-DE3 by the heat-shock method, and glycerol stocks were made for further expression and purification. A 2-L culture of transformed bacteria in LB broth was induced with 0.5 mM IPTG for 3 h at 37°C, and the cells were harvested as a pellet. For the purification of dystrophin N-ABD, the cell pellet was suspended in a buffer containing 50 mM Tris, 200 mM NaCl, pH 7, 1 mM PMSF, and lysed by sonication (15 cycles at 20-watt power and 45% amplitude, each of 20 s duration with 20 s gaps between cycles, and with samples kept on ice). The lysed cell suspension was centrifuged at 30,000 × g using a SS-34 rotor (Sorvall, Newtown, CT) and Oak Ridge centrifuge tubes (Nalgene, Penfield, NY). The supernatant was centrifuged again and the soluble protein from clear supernatant was purified using Ni-Sepharose Fast Flow (GE Healthcare, Buckinghamshire, UK) metal affinity chromatography. The purity of the protein was evaluated using SDS-PAGE (Fig. 2 a). Pure elutes were pooled and dialyzed against phosphate buffered saline (PBS) buffer (0.1 M NaH2PO4, 0.15 M NaCl, pH 7). Single cysteine mutants were purified using the same protocol as described above for the WT protein.
Figure 2.
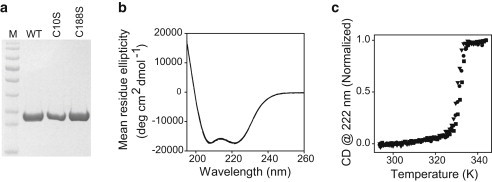
(a) SDS-PAGE of the purified WT dystrophin N-ABD and its two single-cysteine mutants C10S and C188S. Lane M represents the molecular mass markers (bottom to top: 17, 26, 34, 43, 56, 72, 95, 130, and 170 kDa, respectively). (b) CD spectrum of the WT and two mutants (5 μM). Note that all three spectra essentially overlap. (c) Thermal melts of the WT and two mutants (5 μM). As shown, cysteine mutations did not affect the protein structure (panel b) or stability (panel c).
Circular dichroism and fluorescence measurements
Circular dichroism (CD) spectra of pure proteins (5 μM) were recorded in PBS buffer using a Chirascan Plus spectrometer (Applied Photophysics, Leatherhead, UK). Thermal melts were recorded using a continuous temperature ramp. Urea (Nacalai Tesque, Kyoto, Japan; Code No. 35940-81) was used to measure the stability of dystrophin N-ABD in PBS buffer. For this experiment, buffer samples with varying urea concentration were initially prepared and the protein was added from a stock solution. The samples were equilibrated for 1 h before recording the CD or fluorescence signals. Fluorescence measurements were made using a PTI Fluorometer (South Brunswick, NJ). The data were normalized from 0 to 1 before plotting.
Pyrene labeling
WT and mutant N-ABDs were labeled with pyrene using the reagent N-(1-pyrene)maleimide (Molecular Probes, Eugene, OR). A stock of 5 mM N-(1-pyrene)maleimide was made in acetonitrile. Single cysteine mutants (20 μM) were reacted with 20 μM N-(1-pyrene)maleimide, whereas the WT (20 μM) was reacted with 40 μM N-(1-pyrene)maleimide because it contains two cysteine reactive groups. The reaction was carried out at room temperature overnight (12 h). Proteins were exchanged into PBS using PD-10 prepacked columns with Sephadex G25 (GE Lifesciences). Percentage labeling was estimated using a PM-Cysteine molar extinction coefficient of 37,500 M−1cm−1 at 343 nm (Molecular Probes Handbook) and micro-BCA protein estimation kit (Pierce, Franklin, MA).
Actin binding
Rabbit skeletal muscle actin (Cytoskeleton, Denver, CO) was polymerized into F-actin by incubating in polymerization buffer (10×, 0.5 M KCl, 20 mM MgCl2, 10 mM ATP) for 1 h at room temperature. Fluorescence spectra of pyrene-labeled proteins (10 μM) were recorded in the absence and presence of F-actin (10 μM) on a PTI fluorometer with excitation at 345 nm. Spectra were normalized to the intensity of the pyrene monomer fluorescence peak at 375 nm.
Results and Discussion
Cysteine mutations did not affect the structure and stability of dystrophin N-ABD
When expressed in Escherichia coli and purified using affinity chromatography, we obtained high yields of highly pure WT dystrophin N-ABD and its single-cysteine mutants (Fig. 2 a). We first confirmed that the mutations did not significantly alter the structure or stability of the N-ABD. Mutants had CD spectra similar to that of the WT protein, with minima at 208 and 222 nm characteristic of α-helical secondary structure (Fig. 2 b). When the proteins were subjected to thermal denaturation, the mutants unfolded with a midpoint temperature (Tm = 331.3 ± 0.9 K) similar to that of the WT protein (Fig. 2 c). Although these temperature melts are not reversible, they do indicate that the protein stability does not change upon mutations.
Dystrophin N-ABD is a monomer in solution
We monitored excimer fluorescence of the pyrene-labeled C188S mutant, which has a single cysteine residue at position 10 in the CH1 domain (Fig. 1 a). We expected to observe excimer fluorescence if the protein formed a parallel dimer in which the CH1 domain of one monomer is close to that of the adjacent monomer, allowing the pyrenes from the two monomers to stack against one another. Alternatively, nonspecific labeling of residues other than C10 within the CH1 domain during the labeling reaction might result in stacking of the two pyrene molecules from the same monomer resulting in excimer fluorescence. Under our experimental conditions, the fraction of the labeled protein was ∼0.55 ± 0.05, and we did not observe any excimer fluorescence (C10-Py in Fig. 3 a), indicating the absence of parallel dimers or nonspecific labeling. We similarly investigated the presence of excimer fluorescence in C10S single-cysteine mutant, which has a single cysteine residue at position 188 in the CH2 domain (Fig. 1 a). Under the labeling conditions we used, the fraction of labeled protein was ∼0.43 ± 0.05, and we did not observe any excimer fluorescence (C188-Py in Fig. 3 a), indicating the absence of parallel dimers or nonspecific labeling within the CH2 domain.
Figure 3.
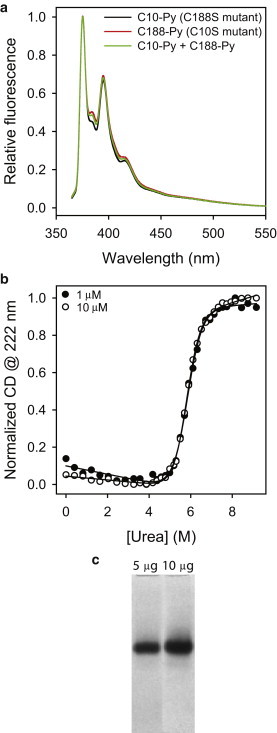
Dystrophin N-ABD is a monomer in solution. (a) Pyrene fluorescence of single-cysteine mutants (C188S (C10-Py) and C10S (C188-Py); 10 μM), and when mixed at equimolar ratio (C10-Py + C188-Py; 10 μM total protein concentration). The absence of excimer fluorescence indicates the absence of dimers or higher oligomers in solution. (b) Thermodynamic stability of dystrophin N-ABD measured at 1 and 10 μM. Urea was used to unfold the protein, and the CD at 222 nm was used to monitor unfolding of the α-helical secondary structure of the protein. The stability is independent of the protein concentration confirming the absence of stable dimers at or below 10 μM protein concentration. (c) Native PAGE of dystrophin N-ABD with 5 and 10 μg loading protein amounts. A similar result was observed even when the gel was overloaded with 30 μg of protein (7).
Next, we investigated excimer fluorescence in an equimolar (1:1) mixture of the pyrene-labeled single-cysteine mutants C188S and C10S (C10-Py + C188-Py in Fig. 3 a). If the protein formed an antiparallel dimer as observed in the x-ray crystal structure (Fig. 1 a) in which C10 in one monomer is close to the C188 residue in the other monomer, or if there had been nonspecific labeling anywhere in the two CH domains, excimer fluorescence might be expected. However, we observed no excimer fluorescence (Fig. 3 a), indicating the absence of antiparallel dimers or nonspecific labeling.
The dimer seen in x-ray crystal studies (Fig. 1 a) has been proposed to form with a dimerization constant, Kd of 4 μM (5). To exclude such possibility, we monitored the melting of the protein secondary structure upon the addition of urea using the CD signal at 222 nm at two different protein concentrations 1 and 10 μM (Fig. 3 b). If there is a dimer formation, the relative dimer concentration would be higher at 10 μM compared to 1 μM; therefore, an increase in protein stability would be expected at higher protein concentration. The protein melting curves revealed an overlapping sigmoidal transition at the two concentrations (Fig. 3 b), indicating that the protein stability is independent of the protein concentration, suggesting the absence of dimers under our experimental conditions.
Results from our earlier NMR relaxation experiments also support the previous conclusion that dystrophin’s tandem CH domain is a monomer in solution (7). The rotational correlation time (τc), calculated from the longitudinal (T1) and transverse (T2) relaxation times, is close to that expected for a monomer of the same size as that of dystrophin’s N-ABD. Furthermore, we ran native PAGE at different protein concentrations (Fig. 3 c), to show that the protein exists as a single species in solution. A single band was observed on the native gel when the gel was loaded with 5 and 10 μg of the protein. We also found the same result with 30 μg of protein (7), indicating that the protein sample contains a single species at all the concentrations.
Taken together, results from all the previous experiments (absence of excimer fluorescence (Fig. 3 a), denaturant melt as a function of protein concentration (Fig. 3 b), NMR relaxation (7), and native gel (Fig. 3 c)) indicate that the dystrophin N-ABD is a monomer in solution, and if dimer population is present, it should be minimal (at the most 5%) such that it does not significantly affect the measured signals within the experimental errors.
Dystrophin N-ABD is in a closed conformation in solution
Now that we have shown that the N-ABD exists primarily as a monomer in solution, we use the doubly labeled WT protein to determine whether the protein exists in a closed conformation similar to that observed in the x-ray structures of ABDs of fimbrin (15), plectin (16), and α-actinin (17) (Fig. 1 b), or if it exists in an open conformation observed in its domain-swapped dimer crystal structure (5) (Fig. 1 a). We monitored excimer fluorescence in the pyrene-labeled WT protein, which has two cysteine residues, i.e., at positions 10 and 188. If the molecule exists in an open conformation as indicated by the x-ray structure, the distance between the two cysteines would be 56 Å (Fig. 1 a). In such a conformation, no excimer fluorescence would be expected because the pyrene aromatic rings would be too far apart to form an excited-state dimer. However, excimer fluorescence might be observed if the protein is in a closed conformation with significant interactions between the two CH domains, allowing the pyrenes labeled at C10 and C188 to stack against each other. Indeed, we observed an additional fluorescence band at 467 nm (WT-Py in Fig. 4 a) consistent with excimer formation, in addition to the pyrene monomer fluorescence observed earlier with pyrene-labeled single-cysteines mutants (C10-Py, C188-Py, and C10-Py + C188-Py in Fig. 4 a). This observation clearly indicates that the protein exists in a closed conformation in solution, contrary to the open conformation observed in the earlier x-ray crystal studies (Fig. 1 a). Because the labeling reaction was performed on the WT protein having two cysteines, three species would be expected in solution: those having the pyrene label at C10 alone, C188 alone, and at both C10 and C188. In this case, the fraction of the doubly labeled protein would be the product of the probabilities of labeling individual cysteines, C10 and C188, i.e., 0.55 × 0.43 = 0.24. Under the labeling conditions we used, the fraction of the pyrene label per one protein molecule was ∼0.68 ± 0.05. Because the pyrene monomer fluorescence seen in Fig. 4 a originates from all three species (doubly labeled, and two single-labeled proteins), whereas excimer fluorescence originates only from the doubly labeled protein, the excimer fluorescence intensity would be expected to be higher than what was observed if the sample contained 100% of the doubly labeled WT. However, these experiments do not exclude the presence of an open conformation that might not contribute to the excimer fluorescence signal. The denaturant melting experiments described below and in Fig. 4 b indicate that the population of such an open conformation, if it exists, would be minimal.
Figure 4.
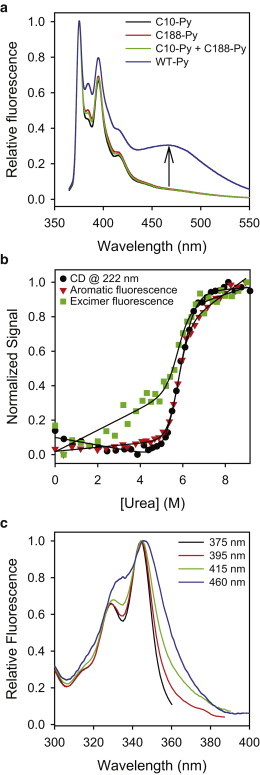
Dystrophin N-ABD is in a closed conformation in solution. (a) Pyrene fluorescence of the WT protein (WT-Py; 10 μM) in comparison with that of the single-cysteine mutants, C188S (C10-Py; 10 μM), C10S (C188-Py; 10 μM), and 1:1 C188S + C10S (C10-Py + C188-Py; 10 μM total protein concentration). The arrow denotes the excimer fluorescence of the WT protein, indicating that the protein is in a closed conformation with two cysteines close in space. (b) Urea melting of pyrene excimer fluorescence, in comparison with that measured by CD at 222 nm and aromatic fluorescence (λex = 280 nm) of the unlabeled protein. The protein concentration used in these experiments was 5 μM. All three signals measured the same protein stability (ΔG = 10.8 ± 0.8 kcal/mol, m = −1.9 ± 0.1 kcal/mol/M [urea]) when fit to a 2-state Santoro-Bolen equation (48,49), indicating that the pyrene labeling did not affect the protein structure. (c) Excitation spectra at different emission wavelengths. The spectrum corresponding to the excimer fluorescence (λem = 460 nm) is much broader than that of the monomer fluorescence (λem = 375 nm), indicating that the excimer fluorescence originates from both ground-state pyrene monomers as well as dimers.
To exclude the possibility that the covalent labeling with pyrene is changing the structure or dynamics of the protein, we measured protein stability using urea as the denaturant and pyrene excimer fluorescence as the signal, and we made a comparison with the stability measured from the melting of the secondary structure (CD at 222 nm) and the tertiary structure (fluorescence of aromatic amino acids with λex = 280 nm) of the unlabeled protein (Fig. 4 b). All three probes exhibited an overlapping sigmoidal, cooperative melting transition with the same ΔG value (10.8 ± 0.8 kcal/mol), indicating that the covalent labeling did not alter the protein conformation. The denaturant melt monitored by pyrene fluorescence has a native baseline with a higher slope compared to that of CD and fluorescence, which was expected because of the strong solvent dependence of pyrene fluorescence demonstrated earlier (43). However, the observation of a ΔG similar to that of fluorescence and CD indicates that the solution conformation detected by pyrene excimer fluorescence represents the average conformation of the entire protein ensemble. This implies that if any open conformation exists in solution that does not contribute to the excimer fluorescence signal, it should be minimal; otherwise, the ΔG of the closed conformation measured by pyrene excimer would differ from the ΔG of the protein ensemble measured by CD and fluorescence. In addition, labeled WT protein having the same conformational stability as that of the unlabeled monomeric protein excludes two other possibilities: 1), covalent labeling of the WT protein with pyrenes is forcing the protein to adopt a new conformation, and 2), covalent labeling with pyrenes might favor dimer formation in the doubly labeled WT protein.
Our previously published results (7) also support the above conclusion that the dystrophin N-ABD is in a closed conformation solution. The rotational correlation time (τc) calculated from NMR relaxation experiments was 18 ns, close to 16 ns if dystrophin N-ABD is a perfectly spherical protein (7). Further evidence for the closed conformation of the dystrophin N-ABD was obtained from the excitation spectra of the pyrene monomer and excimer fluorescence. If the dystrophin N-ABD is in a closed conformation, it is quite likely that the pyrene dimers might be present in the ground state. In such a case, the excitation spectrum corresponding to the excimer fluorescence would be much broader (33,34), because the excimer fluorescence originates from both ground-state pyrene monomer as well as ground-state pyrene dimers. Our results (Fig. 4 c) confirmed this scenario, i.e., the excimer fluorescence (λem = 460 nm) has much broader excitation spectrum than that of the monomer fluorescence (λem = 375 nm), supporting the above conclusion that the dystrophin N-ABD is in a closed conformation.
Dystrophin N-ABD is in a closed conformation upon binding to F-actin
Dystrophin N-ABD binds to F-actin with a Kd of 13 μM and with 1:1 stoichiometry where one N-ABD molecule binds to one actin monomer (11). In F-actin, actin monomers are separated by a translation of 27.5 Å and a rotation of 166° (44). Depending on the exact binding geometry, CH domains from neighboring N-ABD molecules might come close in space, as observed in an earlier cryo-EM study (27). This would result in intermolecular pyrene excimer formation, which does not provide information about the conformation of individual protein molecules. To exclude this probability, we performed the actin-binding experiments under limited labeling conditions in which the WT protein was labeled such that the fraction of the doubly labeled protein was 10%, and correspondingly single-cysteine mutants were labeled by 30%. We confirmed the absence of intermolecular excimer fluorescence with the pyrene-labeled single-cysteine mutants, C188S and C10S. Upon adding F-actin, we did not observe an excimer fluorescence band (Fig. 5 a), indicating no intermolecular excimer formation. Under identical conditions, the intensity of the excimer fluorescence of the WT protein increased upon binding to F-actin (Fig. 5 b). As stated previously, because the WT contains two cysteines, the sample contained three species, two single-labeled and one doubly labeled; therefore, the true increase in excimer fluorescence upon actin binding would be higher than that observed in Fig. 5 b if both the cysteines in the WT protein are labeled with pyrene. Two possible scenarios could explain such an increase in excimer fluorescence: the dystrophin N-ABD transitions to a new conformation, bringing the two labeled cysteines much closer in 3D space compared to its unbound form, or the quantum yield of pyrenes increases upon actin binding because of their inaccessibility to the solvent. Irrespective of the reason, the presence of excimer fluorescence clearly indicates that the dystrophin N-ABD is in a closed conformation when bound to actin.
Figure 5.
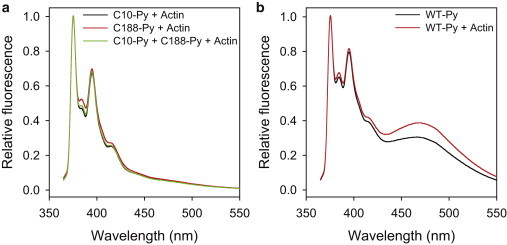
Dystrophin N-ABD is in a closed conformation when bound to F-actin. Panel (a) shows the pyrene fluorescence of the two single-cysteine mutants, C188S (C10-Py; 10 μM), C10S (C188-Py; 10 μM), and 1:1 C10-Py + C188-Py (10 μM protein concentration), whereas panel (b) shows the pyrene fluorescence of the WT N-ABD (10 μM), respectively, upon binding to F-actin (10 μM). WT protein shows excimer fluorescence, indicating that it is in a closed conformation when bound to F-actin. For these experiments, the single-cysteine mutants were labeled by 30%, whereas the WT protein was labeled such that the fraction of the doubly labeled protein was 10%.
Comparison of the solution conformation of dystrophin N-ABD with other tandem CH domains
Our results indicate that dystrophin N-ABD is a monomer in solution, rather than a domain-swapped dimer observed in crystallography studies. Similar examples exist in the literature where soluble monomeric proteins swap their domains to form oligomers under appropriate solution conditions (45,46), which implies that observing a domain-swapped dimer for a protein in its crystal state does not always guarantee that the protein is a dimer in solution. For the dystrophin N-ABD, dimer formation might have resulted from solvent conditions used during crystallization, as suspected earlier (5,26).
Our results on the dystrophin N-ABD, along with that of utrophin (13,14), fimbrin (15), plectin (16), and α-actinin (17), indicate that tandem CH domains in general may be monomeric in solution. In addition, the closed conformations of the tandem CH domains of dystrophin (shown here), fimbrin (15), plectin (16), α-actinin (17), and utrophin (24) indicate that it may be a common conformation for tandem CH domains. However, the actin-bound conformations may differ between tandem CH domains. We show here that the dystrophin N-ABD binds to F-actin in a closed conformation, whereas recent studies indicated that utrophin N-ABD binds to F-actin in an open conformation (24,25). This difference might explain why dystrophin and utrophin have different modes of binding to actin, and do not compete with one another for actin binding (11). Similar solution structural information is needed for other tandem CH domains to make general conclusions about how they interact with F-actin.
Conclusions
In summary (Fig. 6), we used the very simple and elegant technique of pyrene excimer fluorescence to resolve the three long-standing controversies about the conformation of the dystrophin N-ABD: 1), it is a monomer in solution; 2), it exists in a closed conformation; and 3) it binds to F-actin in a closed conformation.
Figure 6.
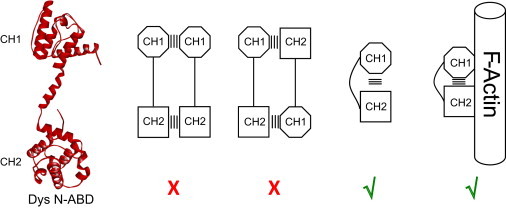
Summary of the results presented in this study indicate that dystrophin’s tandem CH domain is a monomer in solution, exists in a closed conformation, and binds to F-actin in a closed conformation.
Acknowledgments
We acknowledge the help of the Biophysics Core, University of Colorado Anschutz Medical Campus, during the course of this work. We thank Steve Winder, Emil Reisler, and Edward Egelman for helpful discussions, and Steve Winder for a critical reading of the manuscript.
This project was funded by the American Heart Association, a Jane and Charlie Butcher grant in Genomics and Biotechnology, and the ALSAM Foundation through the Skaggs Scholars Program.
References
- 1.Emery A.E.H. The muscular dystrophies. Lancet. 2002;359:687–695. doi: 10.1016/S0140-6736(02)07815-7. [DOI] [PubMed] [Google Scholar]
- 2.Blake D.J., Weir A., Davies K.E. Function and genetics of dystrophin and dystrophin-related proteins in muscle. Physiol. Rev. 2002;82:291–329. doi: 10.1152/physrev.00028.2001. [DOI] [PubMed] [Google Scholar]
- 3.Ervasti J.M. Dystrophin, its interactions with other proteins, and implications for muscular dystrophy. Biochim. Biophys. Acta. 2007;1772:108–117. doi: 10.1016/j.bbadis.2006.05.010. [DOI] [PubMed] [Google Scholar]
- 4.Koenig M., Monaco A.P., Kunkel L.M. The complete sequence of dystrophin predicts a rod-shaped cytoskeletal protein. Cell. 1988;53:219–228. doi: 10.1016/0092-8674(88)90383-2. [DOI] [PubMed] [Google Scholar]
- 5.Norwood F.L., Sutherland-Smith A.J., Kendrick-Jones J. The structure of the N-terminal actin-binding domain of human dystrophin and how mutations in this domain may cause Duchenne or Becker muscular dystrophy. Structure. 2000;8:481–491. doi: 10.1016/s0969-2126(00)00132-5. [DOI] [PubMed] [Google Scholar]
- 6.Huang X., Poy F., Eck M.J. Structure of a WW domain containing fragment of dystrophin in complex with β-dystroglycan. Nat. Struct. Biol. 2000;7:634–638. doi: 10.1038/77923. [DOI] [PubMed] [Google Scholar]
- 7.Singh S.M., Kongari N., Mallela K.M.G. Missense mutations in dystrophin that trigger muscular dystrophy decrease protein stability and lead to cross-β aggregates. Proc. Natl. Acad. Sci. USA. 2010;107:15069–15074. doi: 10.1073/pnas.1008818107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Henderson D.M., Lee A., Ervasti J.M. Disease-causing missense mutations in actin binding domain 1 of dystrophin induce thermodynamic instability and protein aggregation. Proc. Natl. Acad. Sci. USA. 2010;107:9632–9637. doi: 10.1073/pnas.1001517107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rybakova I.N., Ervasti J.M. Dystrophin-glycoprotein complex is monomeric and stabilizes actin filaments in vitro through a lateral association. J. Biol. Chem. 1997;272:28771–28778. doi: 10.1074/jbc.272.45.28771. [DOI] [PubMed] [Google Scholar]
- 10.Gimona M., Winder S.J. The calponin homology (CH) domain. In: Cesareni G., Gimona M., Sudol M., Yaffe M., editors. Modular Protein Domains. Wiley-VCH Verlag GmbH & Co. KGaA; Weinheim: 2005. [Google Scholar]
- 11.Rybakova I.N., Humston J.L., Ervasti J.M. Dystrophin and utrophin bind actin through distinct modes of contact. J. Biol. Chem. 2006;281:9996–10001. doi: 10.1074/jbc.M513121200. [DOI] [PubMed] [Google Scholar]
- 12.Singh S.M., Molas J.F., Mallela K.M.G. Thermodynamic stability, unfolding kinetics, and aggregation of the N-terminal actin binding domains of utrophin and dystrophin. Proteins. 2012;80:1377–1392. doi: 10.1002/prot.24033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Winder S.J., Hemmings L., Kendrick-Jones J. Utrophin actin binding domain: analysis of actin binding and cellular targeting. J. Cell Sci. 1995;108:63–71. doi: 10.1242/jcs.108.1.63. [DOI] [PubMed] [Google Scholar]
- 14.Keep N.H., Winder S.J., Kendrick-Jones J. Crystal structure of the actin-binding region of utrophin reveals a head-to-tail dimer. Structure. 1999;7:1539–1546. doi: 10.1016/s0969-2126(00)88344-6. [DOI] [PubMed] [Google Scholar]
- 15.Goldsmith S.C., Pokala N., Almo S.C. The structure of an actin-cross-linking domain from human fimbrin. Nat. Struct. Biol. 1997;4:708–712. doi: 10.1038/nsb0997-708. [DOI] [PubMed] [Google Scholar]
- 16.García-Alvarez B., Bobkov A., de Pereda J.M. Structural and functional analysis of the actin binding domain of plectin suggests alternative mechanisms for binding to F-actin and integrin β4. Structure. 2003;11:615–625. doi: 10.1016/s0969-2126(03)00090-x. [DOI] [PubMed] [Google Scholar]
- 17.Franzot G., Sjöblom B., Djinović Carugo K. The crystal structure of the actin binding domain from α-actinin in its closed conformation: structural insight into phospholipid regulation of α-actinin. J. Mol. Biol. 2005;348:151–165. doi: 10.1016/j.jmb.2005.01.002. [DOI] [PubMed] [Google Scholar]
- 18.Muir L.A., Chamberlain J.S. Emerging strategies for cell and gene therapy of the muscular dystrophies. Expert Rev. Mol. Med. 2009;11:e18. doi: 10.1017/S1462399409001100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fairclough R.J., Bareja A., Davies K.E. Progress in therapy for Duchenne muscular dystrophy. Exp. Physiol. 2011;96:1101–1113. doi: 10.1113/expphysiol.2010.053025. [DOI] [PubMed] [Google Scholar]
- 20.Ahmad A., Brinson M., Amalfitano A. Mdx mice inducibly expressing dystrophin provide insights into the potential of gene therapy for duchenne muscular dystrophy. Hum. Mol. Genet. 2000;9:2507–2515. doi: 10.1093/hmg/9.17.2507. [DOI] [PubMed] [Google Scholar]
- 21.Le Rumeur E., Winder S.J., Hubert J.-F. Dystrophin: more than just the sum of its parts. Biochim. Biophys. Acta. 2010;1804:1713–1722. doi: 10.1016/j.bbapap.2010.05.001. [DOI] [PubMed] [Google Scholar]
- 22.Henderson D.M., Belanto J.J., Ervasti J.M. Internal deletion compromises the stability of dystrophin. Hum. Mol. Genet. 2011;20:2955–2963. doi: 10.1093/hmg/ddr199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sjöblom B., Ylänne J., Djinović-Carugo K. Novel structural insights into F-actin-binding and novel functions of calponin homology domains. Curr. Opin. Struct. Biol. 2008;18:702–708. doi: 10.1016/j.sbi.2008.10.003. [DOI] [PubMed] [Google Scholar]
- 24.Lin A.Y., Prochniewicz E., Thomas D.D. Large-scale opening of utrophin’s tandem calponin homology (CH) domains upon actin binding by an induced-fit mechanism. Proc. Natl. Acad. Sci. USA. 2011;108:12729–12733. doi: 10.1073/pnas.1106453108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Broderick M.J.F., Bobkov A., Winder S.J. Utrophin ABD binds to F-actin in an open conformation. FEBS Open Bio. 2012;2:6–11. doi: 10.1016/j.fob.2012.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lehman W., Craig R., Sutherland-Smith A.J. An open or closed case for the conformation of calponin homology domains on F-actin? J. Muscle Res. Cell Motil. 2004;25:351–358. doi: 10.1007/s10974-004-0690-7. [DOI] [PubMed] [Google Scholar]
- 27.Sutherland-Smith A.J., Moores C.A., Lehman W. An atomic model for actin binding by the CH domains and spectrin-repeat modules of utrophin and dystrophin. J. Mol. Biol. 2003;329:15–33. doi: 10.1016/s0022-2836(03)00422-4. [DOI] [PubMed] [Google Scholar]
- 28.Galkin V.E., Orlova A., Egelman E.H. The utrophin actin-binding domain binds F-actin in two different modes: implications for the spectrin superfamily of proteins. J. Cell Biol. 2002;157:243–251. doi: 10.1083/jcb.200111097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Moores C.A., Keep N.H., Kendrick-Jones J. Structure of the utrophin actin-binding domain bound to F-actin reveals binding by an induced fit mechanism. J. Mol. Biol. 2000;297:465–480. doi: 10.1006/jmbi.2000.3583. [DOI] [PubMed] [Google Scholar]
- 30.Hanein D., Volkmann N., Matsudaira P. An atomic model of fimbrin binding to F-actin and its implications for filament cross-linking and regulation. Nat. Struct. Biol. 1998;5:787–792. doi: 10.1038/1828. [DOI] [PubMed] [Google Scholar]
- 31.Galkin V.E., Orlova A., Egelman E.H. High-resolution cryo-EM structure of the F-actin-fimbrin/plastin ABD2 complex. Proc. Natl. Acad. Sci. USA. 2008;105:1494–1498. doi: 10.1073/pnas.0708667105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Galkin V.E., Orlova A., Egelman E.H. Opening of tandem calponin homology domains regulates their affinity for F-actin. Nat. Struct. Mol. Biol. 2010;17:614–616. doi: 10.1038/nsmb.1789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lehrer S.S. Intramolecular pyrene excimer fluorescence: a probe of proximity and protein conformational change. Methods Enzymol. 1997;278:286–295. doi: 10.1016/s0076-6879(97)78015-7. [DOI] [PubMed] [Google Scholar]
- 34.Conibear P.B., Bagshaw C.R., Málnási-Csizmadia A. Myosin cleft movement and its coupling to actomyosin dissociation. Nat. Struct. Biol. 2003;10:831–835. doi: 10.1038/nsb986. [DOI] [PubMed] [Google Scholar]
- 35.Bains G., Patel A.B., Narayanaswami V. Pyrene: a probe to study protein conformation and conformational changes. Molecules. 2011;16:7909–7935. doi: 10.3390/molecules16097909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bains G.K., Kim S.H., Narayanaswami V. The extent of pyrene excimer fluorescence emission is a reflector of distance and flexibility: analysis of the segment linking the LDL receptor-binding and tetramerization domains of apolipoprotein E3. Biochemistry. 2012;51:6207–6219. doi: 10.1021/bi3005285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sahoo D., Narayanaswami V., Ryan R.O. Pyrene excimer fluorescence: a spatially sensitive probe to monitor lipid-induced helical rearrangement of apolipophorin III. Biochemistry. 2000;39:6594–6601. doi: 10.1021/bi992609m. [DOI] [PubMed] [Google Scholar]
- 38.Patel A.B., Khumsupan P., Narayanaswami V. Pyrene fluorescence analysis offers new insights into the conformation of the lipoprotein-binding domain of human apolipoprotein E. Biochemistry. 2010;49:1766–1775. doi: 10.1021/bi901902e. [DOI] [PubMed] [Google Scholar]
- 39.Jain N., Bhattacharya M., Mukhopadhyay S. Chain collapse of an amyloidogenic intrinsically disordered protein. Biophys. J. 2011;101:1720–1729. doi: 10.1016/j.bpj.2011.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Feng L., Kim E., Rubenstein P.A. Fluorescence probing of yeast actin subdomain 3/4 hydrophobic loop 262–274. Actin-actin and actin-myosin interactions in actin filaments. J. Biol. Chem. 1997;272:16829–16837. doi: 10.1074/jbc.272.27.16829. [DOI] [PubMed] [Google Scholar]
- 41.Liou Y.-M., Chao H.-L. Fluorescence spectroscopic analysis of the proximity changes between the central helix of troponin C and the C-terminus of troponin T from chicken skeletal muscle. Biochim. Biophys. Acta. 2007;1774:466–473. doi: 10.1016/j.bbapap.2007.01.004. [DOI] [PubMed] [Google Scholar]
- 42.Ansari S., Alahyan M., El-Mezgueldi M. Role of caldesmon in the Ca2+ regulation of smooth muscle thin filaments: evidence for a cooperative switching mechanism. J. Biol. Chem. 2008;283:47–56. doi: 10.1074/jbc.M706771200. [DOI] [PubMed] [Google Scholar]
- 43.Kalyanasundaram K., Thomas J.K. Solvent-dependent fluorescence of pyrene-3-carboxaldehyde and its applications in the estimation of polarity at micelle-water interfaces. J. Phys. Chem. 1977;81:2176–2180. [Google Scholar]
- 44.Berg J.M., Tymoczko J.L., Stryer L. 6th Edition. W. H. Freeman & Company; New York: 2007. Biochemistry. [Google Scholar]
- 45.Liu Y., Eisenberg D. 3D domain swapping: as domains continue to swap. Protein Sci. 2002;11:1285–1299. doi: 10.1110/ps.0201402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Carey J., Lindman S., Linse S. Protein reconstitution and three-dimensional domain swapping: benefits and constraints of covalency. Protein Sci. 2007;16:2317–2333. doi: 10.1110/ps.072985007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Shatsky M., Nussinov R., Wolfson H.J. A method for simultaneous alignment of multiple protein structures. Proteins. 2004;56:143–156. doi: 10.1002/prot.10628. [DOI] [PubMed] [Google Scholar]
- 48.Santoro M.M., Bolen D.W. Unfolding free energy changes determined by the linear extrapolation method. 1. Unfolding of phenylmethanesulfonyl alpha-chymotrypsin using different denaturants. Biochemistry. 1988;27:8063–8068. doi: 10.1021/bi00421a014. [DOI] [PubMed] [Google Scholar]
- 49.Santoro M.M., Bolen D.W. A test of the linear extrapolation of unfolding free energy changes over an extended denaturant concentration range. Biochemistry. 1992;31:4901–4907. doi: 10.1021/bi00135a022. [DOI] [PubMed] [Google Scholar]