

Abstract
Background.The impact of pre-exposure prophylaxis (PrEP) with antiretrovirals on breakthrough HIV or SHIV infection is not fully documented. We addressed the hypothesis that SHIVSF162P3 infection despite active PrEP results in altered early immune parameters, compared with untreated infection.
Methods.Eleven rhesus macaques were infected during repeated, rectal, low-dose SHIVSF162P3 exposures while receiving concurrent oral PrEP (Truvada [n = 2] or GS7340 [n = 4]) or as untreated controls (n = 5). We measured SHIV RNA, inflammatory cytokines, CD4 cells, and SHIV-specific and memory T cells until 20 weeks after peak viremia.
Results.SHIV infection during PrEP resulted in 100-fold lower peak viremia and lower IL-15, IL-18, and IL-1Ra levels, compared with controls (P < .05; Wilcoxon rank-sum test). Unlike controls, PrEP-treated macaques showed no significant CD4 cell count reduction during acute infection and developed more SHIV-specific central memory T cells, relative to controls. After in vivo CD8 cell depletion, viral load increased to similar levels, indicating that CD8 cells were critical for viral control in both groups.
Conclusions.PrEP with antiretrovirals has beneficial effects on early SHIV infection even when infection is not prevented. Although long-term immune control could not be examined in this SHIV infection model, our results suggest that PrEP results in improved early disease parameters in breakthrough infections.
Pre-exposure prophylaxis (PrEP) with oral or topical antiretrovirals (ARVs) for the prevention of HIV infection is currently undergoing efficacy evaluation in clinical trials. Four trials have reported efficacy among men who have sex with men and heterosexual couples [1–4]; one trial in women was abandoned because of unlikely efficacy at interim analysis [5], and others are at different stages of completion [6]. Although overall results are encouraging or are pending, it is clear that 100% effectiveness is unlikely to be achievable because of imperfect adherence or efficacy. This raises the question of the consequences of breakthrough infection acquired during PrEP.
To address this, we studied innate immune parameters and adaptive T cell responses during acute infection and PrEP in macaques. Adaptive B cell responses have been addressed elsewhere [7]. Breakthrough SHIV infection during PrEP in macaque models can result in blunted peak and set-point viremia [8–10]. This is most likely because ARV drugs can partially inhibit virus replication even though they did not completely block virus transmission. Because of an expected relationship between viremia and the strength of innate and adaptive immune responses (eg, as in elite controllers) [11], we hypothesized that attenuated PrEP breakthrough infection would be characterized by limited inflammation, limited CD4 cell loss, and alterations in adaptive T cell responses. Such immune parameters during acute HIV infection [12] can further influence infection course and are of interest during preclinical and clinical PrEP evaluation.
We used longitudinal specimens generated during efficacy testing of new intermittent PrEP regimens in macaques [8–10], allowing us to maximize insights from ongoing animal studies. The PrEP efficacy studies were geared toward modeling realistic conditions of sexual HIV transmission during intermittent PrEP. SHIV transmission occurred after repeated, low-dose, rectal virus exposures, allowing establishment of infection and initiation of immune responses under physiologic circumstances. ARVs were given only once per week to model intermittent PrEP use; sporadic nonadherence was not modeled. Drugs were continued with the same regimen after documented infection to study the emergence of drug resistance and to model ARV use intended as PrEP by persons who will not immediately know their HIV status. We used SHIVSF162P3, a R5-tropic virus with mucosal transmissibility. It causes initial viremia similar to acute HIV infection. There is a characteristic, transient, but moderate decrease in blood CD4 T cell count at peak viremia [13, 14]. Unlike HIV infection, however, set-point SHIVSF162P3 viremia near undetectable levels (approximately 50 copies/mL of blood plasma) is reached within 12 weeks, and the infection course is generally nonpathogenic. CD8 cells contribute significantly to SHIVSF162P3 control, as demonstrated by in vivo CD8 cell depletion [15]. Thus, we examined the acute phase, when adaptive immune responses develop to effectively control SHIVSF162P3 [14–16].
We analyzed parameters previously measured in S(H)IV+ rhesus macaques. IFNγ, IFNα, IL-1Ra, MCP-1, IL-15, and IL-18 levels increase during acute SIV infection, in contrast to other inflammatory factors (eg, TNFα and IL-6) [17]. Immune markers HLA-DR and CD38 were not measured, because they fluctuate little during SHIVSF162P3 infection [16]. In contrast, central memory T cells (TCM) differ in SIV+ macaques with varying immune activation [17]. CD4 cell count measurements can be inconclusive in SHIVSF162P3 infection because of the moderate decrease and rebound within one week [14]. Because plasma IL-15 level and CD4 cell count decreases are inversely correlated in early SIV infection of rhesus macaques [18], we determined IL-15 as a marker related to CD4 cell count, to further substantiate subtle changes in CD4 cell counts during SHIV infection.
MATERIALS AND METHODS
Macaques, Virus, PrEP regimens, and Repeat Low Dose (RLD) Virus Challenges
The Animal Care and Use Committee (IACUC) of the Centers for Disease Control and Prevention (CDC) approved all described macaque procedures. These were in accordance with standards established in the Guide for the Care and Use of Laboratory Animals [19]. Eleven male Indian rhesus macaques were housed at the CDC (Atlanta, GA) and underwent efficacy testing of intermittent PrEP (Table 1, and as described [8, 9]). In brief, 2 macaques received oral Truvada [9] (emtricitabine [FTC] [20 mg/kg] and tenofovir disoproxyl fumarate [TDF] [22 mg/kg]), administered 7 days before the first rectal viral challenge and then once weekly 2 hours after each challenge. Four macaques received the novel tenofovir pro-drug GS7340 in 1 weekly dose (13.7 mg/kg) 3 days before SHIV exposure [8]. The genetic identity of 8 MHC alleles with potential impact on SHIV infection course is also shown in Table 1. Mamu-B08 and -B17, associated with superior control of SIVmac239 infection [20, 21], were either not present in the PrEP-group, or were present once in both groups, respectively. Trim5 genotypes were not determined, because SHIVSF162P3 replication is not restricted by Trim5 gene products in Indian rhesus macaques (E. Kersh, unpublished data, and as reported for SIVmac239 replication [22]).
Table 1.
Study Animal Description
| Infection After Exposure # | PrEP Continuation After Peak Viremia (weeks) | Mamu-Allele |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Animal ID | Drug Treatment | A01 | A02 | A08 | A11 | B01 | B03 | B04 | B08 | B17 | ||
| PrEP group | ||||||||||||
| 35032 | Truvada | 4 | 18 | − | − | − | − | − | − | − | − | − |
| 34912 | Truvada | 14 | 0 | − | − | + | − | − | − | − | − | + |
| DL2L | GS7340 | 3 | 11 | − | − | + | + | − | − | − | − | − |
| 1784 | GS7340 | 2 | 12 | + | + | − | − | + | − | − | − | − |
| 4292 | GS7340 | 2 | 12 | − | − | + | − | − | − | − | − | − |
| DK43 | GS7340 | 4 | 9 | − | − | − | − | + | − | − | − | − |
| Control group | ||||||||||||
| AG94 | – | 3 | N/A | − | − | + | − | − | − | − | − | − |
| AI22 | – | 5 | N/A | − | − | − | − | + | − | − | − | − |
| 35720 | – | 1 | N/A | + | − | + | − | − | − | − | − | + |
| 19-V | – | 1 | N/A | + | − | − | − | − | − | − | − | − |
| 62-P | – | 1 | N/A | − | − | − | − | − | − | − | + | − |
SHIVSF162P3 ([13], provided by the National Institutes of Health [NIH] AIDS Research and Reference Reagent Program) was administered during repeated, low dose, rectal virus exposures at 10 TCID-50 [8–10] [number of exposures indicated in Table 1]). Viral load was assessed by polymerase chain reaction with a detection limit of 50 copies/mL, as previously described [23].
Blood Sample Collection, Flow Cytometry, IFNg Enzyme-Linked Immunospot Assay (ELISPOT), Inflammatory Factors
We collected blood samples once per week with use of BD Vacutainer CPT tubes (BD Biosciences). PBMCs were counted using an automated Guava cell counter (Millipore). SHIV-specific T cells were determined by IFNγ-ELISPOT, incubating fresh cells with 15-mer peptide pools with 11 amino acid overlaps as described [24].
T cells, gag-specific T cells, intracellular cytokine/chemokine production, and effector and central memory T cell determination (TEM and TCM) based on cytokine expression [25], were determined by flow cytometry as described [24]. In brief, freeze-thawed cells were washed, stimulated with gag1/2 peptide pools, and analyzed with antibodies to CD3, CD4, CD8, CD69, CCR7, CD28, IL-2, TNFα, IFNγ, and MIP-1β. Samples were examined on a LSRII flow cytometer (BD Biosciences) and analyzed using FlowJo software, version 7.2.1 (Tree Star). TCM cells were defined as CD3+ CD69+ (IFNγ, TNFα, MIP-1β, and/or IL-2)+, CD28high, or CCR7+; whereas TEM cells were CD3+ CD69+ (IFNγ, TNFα, MIP-1β, and/or IL-2)+, CD28high/low, or CCR7−.
After freeze-thawing of plasma, we measured IL-15 using the Quantikine IL-15 Immunoassay (R&D Systems) and INFγ, IL-18, MCP-1, and IL-1Ra (a = antagonist) with nonhuman primate-specific reagents (Millipore), according to the instructions by the manufacturer, and using a Luminex-100 system (Invitrogen) with Bio-Plex Manager software, version 4.0 (Bio-Rad). IFNα levels in plasma were measured using a human IFNα enzyme-linked immunosorbent assay (ELISA) kit (PBL Interferon Source).
CD8 Cell Depletion
Anti-CD8 antibody cM-T807 and control IgG antibodies (IgIV, a polyclonal IgG preparation derived from human plasma) were obtained through the NIH Non-Human Primate Resource and from Centocor. Antibodies were injected subcutaneously at 10 mg/kg at day 0, followed by intravenous injections on days 3, 7, and 10 at a dose of 5 mg/kg. We isolated fresh PBMCs and counted T cells by flow cytometry using antibodies to CD8 (clone DK25, DAKO, Denmark), CD3, and CD4 (clones SP34 and RM4-5, respectively; BD Biosciences). Viral RNA was also monitored at each time point.
Statistical Methods
We tested differences between control and PrEP-treated groups for select time points or between time points of a group with Wilcoxon rank-sum tests using GraphPadPrism, version 4.0, or SAS, version 9.2 (SAS Institute). When data from multiple time points per subject were analyzed to compare differences by treatment arm, we used a generalized estimation equations method to estimate robust variances to account for within subject correlation [26]. Temporal decrease in viral load (log-10) in primary infection was examined using mixed effects regression analysis and with left-censoring of undetectable viral load values at <50 copies/mL.
RESULTS
Infection and Viremia
Immune parameters during acute SHIVSF162P3 infection were compared among 6 PrEP-treated and 5 untreated control macaques after repeated, low-dose, rectal virus exposures. The intermittent PrEP regimens with Truvada or the tenofovir prodrug GS7340 are described elsewhere [8–10]; they were either partially or not effective in preventing infection. Only infected macaques were studied here, whereas immune responses in PrEP-protected macaques were previously examined [24].
Macaques, their treatment and its duration, and the number of virus exposures required for their infection are listed in Table 1.
Figure 1 shows plasma viral load up to 20 weeks after peak viremia. ARV treatment was continued on the same intermittent, once weekly regimen as during PrEP after documented infection for a median of 11.5 weeks after peak viremia, except for animal 34912 (Table 1), to monitor for drug resistance development. No resistance mutations developed in this period, as reported elsewhere [8, 9]. Peak viremia was 100-fold lower in PrEP/ARV-treated macaques, relative to control macaques, because treated macaques had a median peak viral load of 105.5 RNA copies/mL of plasma, compared with 107.5 in untreated macaques (P = .02, Wilcoxon rank-sum test). Peak viremia was also low in animal 34912 (105.3 plasma RNA copies/mL, data not shown), the only animal that had ARVs discontinued at the time of infection (Table 1). Temporal decrease in virus load (log-10) after peak viremia differed between the 2 groups (mixed effects regression analysis, P = .04); the estimated decrease was 0.14 log10 copies/mL per week for controls and 0.24 per week for PrEP/ARV-treated animals.
Figure 1.

Peak viremia was 100-fold lower in PrEP/ARV-treated macaques relative to control macaques (P = .02, Wilcoxon rank-sum test). Plasma viral load was measured by quantitative reverse-transcriptase polymerase chain reaction. The detection limit was 50 copies/mL (dotted line). Shown are mean values and standard error of the mean (SEM) for control (grey) and PrEP-treated animals (black). Temporal decrease in virus load (log-10) after peak viremia differed between PrEP/ARV-treated macaques relative to control macaques (mixed effects regression analysis, P = .04).
Inflammatory Immune Factors During Acute Infection
To characterize innate and adaptive immune responses during acute infection, we measured key inflammatory plasma cytokines and chemokines (Figure 2A–2D) [17]. We tested whether IFNγ, IL-1Ra, IL-18, INFα, and MCP-1 levels differed at peak viremia during maximal viral replication and inflammation. Median values of IL-1Ra and IL-18 were lower in PrEP-breakthrough–infected macaques, compared with controls (IL-1Ra: 1.9 pg/mL vs. 48.9 pg/mL [P < .05]; IL-18: 0.3 pg/mL vs. 58.0 pg/mL [P < .05]; Wilcoxon rank-sum tests). Median IFNγ plasma concentrations were 0 and 3.6 pg/mL, and median MCP-1 concentrations were 176 and 257 pg/mL in breakthrough PrEP, compared with control-infected macaques, respectively; these estimated median differences were not statistically significant. IFNα level was not measurable in the samples.
Figure 2.

Inflammatory factors during acute infection. The indicated factors were measured in blood plasma using Luminex technology (A–D). Mean values and standard error of the mean (SEM) are shown. Median values of IL-1Ra and IL-18 were lower at peak viremia in PrEP-breakthrough–infected macaques, compared with controls (*P < .05, Wilcoxon rank-sum tests). Differences in IFNγ and MCP-1 plasma concentrations did not reach statistical significance in breakthrough PrEP-, compared with control-infected macaques.
T cell Immunity
Peripheral blood T cell counts are shown in Figure 3A in samples taken before virus exposures, at peak viremia, and 2, 6, 12, and 20 weeks thereafter. In control animals, there was a temporal drop in overall CD3+ T cell counts at peak viremia, compared with immediately before infection (P = .03, Wilcoxon rank-sum test) (Figure 3A), which is typical for SHIVSF162P3 infection at peak viremia [14] and is attributable to a decrease in CD4 T cells occurring sharply at this time. This decrease was not seen in PrEP breakthrough infections. At peak viremia, the median number of CD4 T cells was higher in breakthrough PrEP–infected macaques, compared with control macaques (P = .02, Wilcoxon rank-sum test) (Figure 3B). Median CD8 T cell counts did not differ between the groups and did not expand significantly, as in previous studies of SHIVSF162P3 infection (data not shown) [14]. During the viral ramp-up phase in SIV infection, IL-15 production precedes the infection of CD4 T cells and their subsequent death [18]. IL-15 concentrations and CD4 cell count decrease have been shown to be inversely correlated [18]. We examined plasma IL-15 level 2 weeks before peak viremia (Figure 3C). Control-infected macaques had higher IL-15 levels, compared with breakthrough-infected macaques (P < .05, Wilcoxon rank-sum test) (Figure 3C). Thus, we observed both higher IL-15 concentrations and lower CD4 cell counts in controls than in breakthrough PrEP–infected animals (Figure 3C).
Figure 3.

Blood lymphocytes were enumerated by flow cytometry. Mean numbers of CD3+ (A), or CD4+ (at peak viremia only, B) T cells are shown. Error bars in A depict standard error of the mean (SEM), the asterisk indicates statistical differences comparing cell numbers between controls and PrEP-animals at peak viremia (P = .03; Wilcoxon rank-sum test). The characteristic decrease in CD4+ T cell count at the peak of acute infection was not observed in breakthrough PrEP-infected macaques. In (C), plasma IL-15 levels were measured by ELISA 1 and 2 weeks prior to peak viremia. The P values in B and C were obtained by Wilcoxon rank sum test (P = .02, P = .009, respectively).
Antigen (SHIV)–specific T cells were further analyzed by flow cytometry (Figure 4A–4C) and IFNγ-ELISPOT (Figure 4D and 4E). T cells were stimulated with gag-derived peptide pools and identified as antigen specific if this prompted an increase in intracellular IFNγ, IL-2, TNFα, and/or MIP-1β. Infection led to similar increases in the number and percentage of gag-specific T cells in both groups (Figure 4A). Gag-specific central memory T cells (TCM) were analyzed because they can correlate well with restoration of gut-resident CD4 cells [27], an important immunological marker for disease attenuation [28]. More gag-specific T cells had a TCM phenotype in breakthrough PrEP-infected macaques than in controls when combined T cells from peak, and 6, 12, and 20 weeks thereafter were analyzed (P = .04, generalized estimation equations method) (Figure 4B), whereas TEM cells had an inverse distribution in the 2 groups (not shown). We examined the ability of T cells to simultaneously produce multiple cytokines or chemokines (referred to as “factors” in Figure 4C), because this is associated with superb antimicrobial potency [29]. At peak viremia, more gag-specific T cells simultaneously produced 4 factors in breakthrough PrEP-infected monkeys, compared with controls (P = .02, Wilcoxon rank-sum test) (Figure 4C), despite a similar number and percentage of gag-specific T cells (Figure 4A), indicating enhanced potency of gag-specific T cells. Functional differences resolved at later time points (Figure 4C and data not shown).
Figure 4.
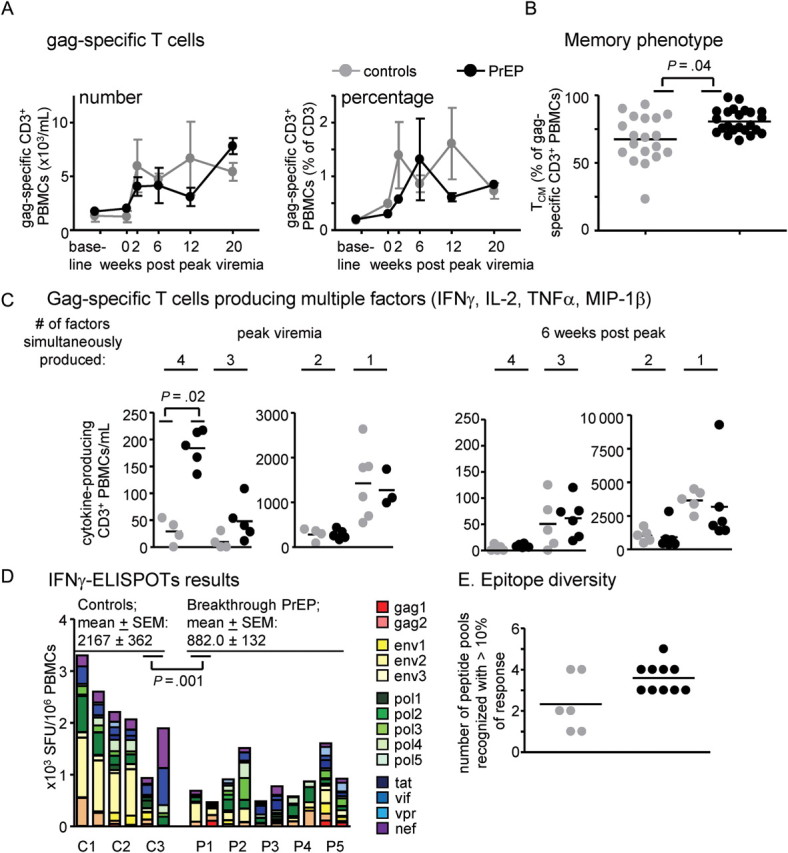
Enumeration and characterization of SHIV-specific T cells (gag-specific [A–C], or specific for gag, env, pol, tat, vif, vpr, and nef products [D and E]). A. Induction of gag-specific T cells. We identified gag-specific T cells by flow cytometry, after in-vitro incubation of freeze-thawed cells with gag1/gag2 peptide pools, then enumeration of CD3+ cells with intracellular production of IFNγ, IL-2, MIP-1β, and/or TNFα. The left panel shows numbers, the right panel shows percentages of gag-specific PBMCs. B. gag-specific T cells were further analyzed for central memory TCM (shown) or effector memory TEM (not shown) phenotype. TCM cells were defined as CD3+ CD69+ (IFNγ, TNFα, MIP-1β, and/or IL-2)+, CD28high, CCR7+. The asterisk indicates statistical significance (P = .04, generalized estimation equations method) between control and PrEP-infected groups when analyzing T cells from peak, and 6, 12, and 20 weeks thereafter, combined. (C). Gag-specific cells were divided by their ability to produce cytokines/ chemokines IFNγ, TNFα, MIP-1β, and/or IL-2 (referred to as “factors”) alone, or to produce them simultaneously in any combination. The P value was obtained by Wilcoxon rank-sum test. Differences in cells producing one to three factors at peak viremia, or differences in cells producing any number of factors after peak viremia, were not statistically significant. D, E. SHIV-specific, IFNγ+ cells in fresh blood samples were analyzed by IFNγ-ELISPOT. We analyzed blood collected on two independent occasions between weeks 10 and 12 post peak viremia from 3 control macaques (C1–C3), and 5 PrEP-breakthrough macaques (P1–P5). Responses to 14 peptide pools derived from the indicated gene products were combined to calculate the indicated spot forming units (SFUs; D). Mean values and standard error of the mean (SEM) are given; the P value was calculated with the generalized estimation equations method. E. The graph displays the number of peptide pools recognized with ≥10% of the response; differences between the groups were not statistically significant.
To study T cell responses to more epitopes, we performed IFNγ ELISPOT analyses using peptide pools with gag, env, pol, vif, vpr, nef, and tat sequences (Figure 4D and 4E). Fresh blood samples were collected on 2 separate occasions 10–12 weeks after peak viremia. When responses to all peptide pools were added, controls showed greater combined T cell responses than did breakthrough PrEP macaques (mean, 2167 SFU/106 PBMCs vs. 882 SFU/106 PBMCs; P = .001, generalized estimation equations method). Epitope diversity was not significantly different between the 2 groups, however, because a mean of 3.6 peptide pools were recognized with >10% of the response in PrEP-treated macaques, compared with a mean of 2.3 peptide pools in controls (Figure 4E).
Effect of In Vivo CD8 Cell Depletion on Viremia
To further address whether CD8 cells play a critical role in controlling SHIVSF162P3 after breakthrough PrEP and control infections, we depleted CD8 cells in vivo by administering anti-CD8 antibodies to 3 controls and 3 PrEP-breakthrough infected macaques [30] approximately 28 weeks after peak viremia and after PrEP discontinuation. One macaque received control IgG antibodies. Figure 5A shows that the antibody regimen led to a temporal disappearance of blood CD8 cells, except in the control IgG-treated macaque. Simultaneously, plasma viral load increased to similar levels in both control and PrEP-infected macaques (Figure 5B). It peaked at a median 105.8 RNA copies/mL in control infected and 105.5 in untreated macaques, indicating no statistically significant difference between the groups (P = .7, Wilcoxon rank-sum test). The rate of viral load decrease was also similar between the 2 groups. This indicates that CD8 cells similarly suppress viremia in this phase of breakthrough and control infection. In controls, median viral load after CD8 cell depletion was lower than during primary viremia (P = .04, Wilcoxon rank-sum test). This could be because non-CD8 cell-mediated adaptive immunity (eg, antibodies) developed at this time point of SHIVSF162P3 infection [7]. There were no statistically significant differences between CD4 cell counts in the control and breakthrough-infected groups after CD8 cell depletion (data not shown). In summary, CD8 cell depletion affected the 2 groups similarly, indicating that CD8 cells efficiently control viremia in both groups.
Figure 5.

In vivo depletion of CD8+ cells. Approximately 28 weeks after peak viremia, CD8+ cells were deleted by anti-CD8 antibody injection (indicated by arrows) in 3 control and 3 breakthrough-infected macaques; 1 macaque received mock IgG antibodies. (A): The disappearance of CD8+ cells from blood was analyzed by flow cytometry on various time points after first antibody administration. Monkey IDs were controls (open symbols): AG94, AI22, 35720; PrEP-treated (filled symbols): 35032, 1784, 4292, and DK43 (IgG control, dotted line). B. Plasma viral loads increased to similar levels in both controls and PrEP-treated macaques after CD8+ cell depletion.
DISCUSSION
Our studies show that PrEP and continued ARV therapy during breakthrough infection can noticeably and beneficially impact early disease parameters in an animal model with relevance for human PrEP. Early systemic inflammatory parameters were lowered during acute infection acquired on active but nonprotective PrEP, compared with untreated infection, and CD4 T cells were spared from the temporal decrease seen in untreated SHIVSF162P3 infection. These data can inform follow-up studies of ongoing or recently completed PrEP clinical studies. If attenuated acute HIV infection is indeed found after human PrEP, this could result in an overall attenuated HIV disease course. Clinical outcomes warrant further study and could include delayed clinical end points, reduced need for ARV therapy, and lowered transmission rates. These parameters could have a beneficial impact on the HIV/AIDS epidemic.
This study focused on evaluation of immunity during acute infection. At later time points and after PrEP was discontinued, there were subtle effects on T cell maturation, such as central memory development. A more comprehensive evaluation of later immunological parameters was not possible in our experimental system of nonpathogenic SHIV infection. Using more pathogenic SIVmac251 or 239 would permit immune analyses during chronic infection that more closely mimics HIV infection. For example, CD4 cell counts, a major predictor of human HIV disease course, could be differentially followed during their steady decrease, allowing a better understanding of long-term immunological control of the infection. This would also permit further analysis of drug resistance development in the context of ongoing vigorous viral replication. Although no resistance was seen in the PrEP breakthroughs in this study, it remains a major concern for PrEP breakthrough HIV infections, particularly if ARVs are continued in a PrEP regimen after infection. Therefore, any potential benefit in long-term immune control may be offset by increased drug resistance development.
The present study was conducted with small animal groups. Powering future studies with larger group sizes will allow a more in-depth analysis of immunological parameters. Further studies should also discontinue PrEP earlier, simultaneously, and at controlled time points and should enumerate CD4 cells in mucosal tissues (eg, gut). The latter could provide a clearer picture of whether gut CD4 counts are spared from destruction, a parameter that has great influence on disease progression [28].
Our observation of altered immune parameters after PrEP is perhaps not surprising, because ARV therapy initiation very early after transmission has similar effects in macaques and humans [27, 31–39]. Our study design included ARV therapy before and after infection and is therefore highly similar, but not identical, to giving ARV therapy very soon after infection. ARV therapy was discontinued in one macaque, 34 912, immediately after the last infecting virus exposure. This macaque was indistinguishable from the other PrEP-breakthrough macaques in terms of viremia and immunological parameters (data not shown), suggesting that continued ARV therapy was not necessary for the altered infection course. We suspect that the main mechanism of action is the effect of ARVs on reducing viral replication immediately after infection, which in turn reduces priming of the immune system. Continued ARV therapy can also limit virus diversification and suppress emergence of immunological escape mutations, thus enhancing immune control [40]. In addition, it is possible that ARVs modulate specific immune-regulatory factors in PBMCs or monocytes independent of virus, as has been reported for tenofovir and its impact on IL-8, IL-10, and IL-12 production [41]. Lastly, chemo-vaccination before the transmission event [24, 42] may impact the subsequent infection through priming of specific arms of the adaptive immune system before infection.
Because it has previously been reported that there is only a transient (limited to one week) and partial decrease in blood CD4 cell count in SHIVSF162P3 infection at peak viremia [14], it was expected that differences in CD4 cell counts are only present at this time point. Differences in SHIV-specific T cell counts were not clearly measurable. Similar numbers of gag-specific T cells were found by flow cytometry, but SHIV-specific T cell numbers differed when examined by IFNγ-ELISPOT using wider epitope representation. The latter could be explained by greater antigenic stimulation in control infections with higher viremia. It is unclear whether these findings represent real differences or are attributable to individual animals in small study groups. To address the functional role of CD8 cell immunity more definitively, we performed an in vivo depletion of such cells. The ensuing similar viremia levels clearly showed that CD8+ cells controlled viremia efficiently in both contexts. Thus, the early ARV treatment had not induced a CD8-independent mechanism to suppress viremia. This experiment could not address whether CD8+ CD3+ (presumably SHIV-specific T cells) or CD3− cells (presumably mostly NK cells) are responsible for the effect. Of note, viral load did not increase to the level seen in acute control infection, possibly because of antibody-mediated immunity at this time point in both control and PrEP breakthrough–infected macaques [7]. Additional studies might shed more definitive light on long-lasting effects of PrEP on T cell maturation (eg, by analyzing the degree of viral divergence in known T cell epitopes from founder virus stocks in control and PrEP breakthroughs) [43].
A recently presented analysis of breakthrough infections from the CAPRISA 004 tenofovir-gel study reports preservation of virus-specific CD4 T cells in intercurrent infections [44], suggesting an attenuated disease course in HIV-infected persons after PrEP. This was not seen in the small number and limited follow-up of breakthrough infections in the iPrEX trial [2], possibly because of low drug adherence in the study. Of note, one patient receiving a prophylactic ARV regimen experienced an attenuated clinical HIV infection characterized by high CD4 cell counts, as reported by a case study [45]. A comprehensive evaluation of HIV disease attenuation after oral and topical PrEP and after vaginal, rectal, and penile infection will be of interest and could even uncover selective benefits of systemic versus topical PrEP use.
In conclusion, our study provides a first characterization of inflammatory markers, T cell memory and epitope specificity, and CD4 cell counts in acute PrEP breakthrough infections of macaques that are acquired during active but only partially effective oral PrEP. These results indicate a potentially beneficial effect of PrEP. Further study is needed to determine whether PrEP during virus acquisition has longer-term effects on immune preservation and control of HIV infection.
Notes
Acknowledgments. We thank Dr. Gil Kersh for his support; CDC veterinary staff (Drs. Katherine Paul and Gregg Langham), animal care providers, and technicians, who made the PrEP studies possible from which specimens for this follow-up study were obtained; Dr. Keith Reimann (provision of cM-T807 antibodies through the NIH Nonhuman Primate Reagent Resource); the MHC Typing Core AIDS Vaccine Research Laboratory at the University of Wisconsin; and the NIH AIDS Research and Reference Reagent Program. The findings and conclusions in this report are those of the authors and do not necessarily represent the views of the Centers for Disease Control and Prevention.
Financial support. This work was supported by the CDC and by an Interagency Agreement (Y1-AI-0681-02) between the CDC and NIH.
Potential conflicts of interest. All authors: No reported conflicts.
All authors have submitted the ICMJE Form for Disclosure of Potential Conflicts of Interest. Conflicts that the editors consider relevant to the content of the manuscript have been disclosed.
References
- 1.Abdool Karim Q, Abdool Karim SS, Frohlich JA, et al. Effectiveness and safety of tenofovir gel, an antiretroviral microbicide, for the prevention of HIV infection in women. Science. 2010;329:1168–74. doi: 10.1126/science.1193748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Grant RM, Lama JR, Anderson PL, et al. Preexposure chemoprophylaxis for HIV prevention in men who have sex with men. New Engl J Med. 2010;363:2587–99. doi: 10.1056/NEJMoa1011205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Thigpen MC, Kebaabetswe PM, Smith DK, et al. Daily oral antiretroviral use for the prevention of HIV infection in heterosexually active young adults in Botswana: results from the TDF2 study [abstract WELBC0] In: 16th IAS Conference on HIV Pathogenesis, Treatment and Prevention (Rome) [Google Scholar]
- 4.Baeten J, Celum C Team oboTPPS. Antiretroviral pre-exposure prophylaxis for HIV-1 prevention among heterosexual African men and women: the Partners PrEP study [abstract MOAX0106] In: 6th IAS Conference on HIV Pathogenesis, Treatment and Prevention. (Rome) [Google Scholar]
- 5.Roehr B. HIV prevention trial in women is abandoned after drugs show no impact on infection rates. BMJ. 2011;342:d2613. doi: 10.1136/bmj.d2613. [DOI] [PubMed] [Google Scholar]
- 6.Veronese F, Anton P, Fletcher CV, et al. Implications of HIV PrEP trials results. AIDS Res Hum Retrov. 2011;27:81–90. doi: 10.1089/aid.2010.0226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Curtis KA, Kennedy MS, Luckay A, et al. Delayed maturation of antibody avidity but not seroconversion in rhesus macaques infected with simian HIV during oral pre-exposure prophylaxis. J Acquir Immun Defic Syndr. 2011;57:355–62. doi: 10.1097/QAI.0b013e3182234a51. [DOI] [PubMed] [Google Scholar]
- 8.Garcia-Lerma JG, Aung W, Cong ME, et al. Natural substrate concentrations can modulate the prophylactic efficacy of nucleotide HIV reverse transcriptase inhibitors. J Virol. 2011;85:6610–7. doi: 10.1128/JVI.00311-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Garcia-Lerma JG, Cong ME, Mitchell J, et al. Intermittent prophylaxis with oral truvada protects macaques from rectal SHIV infection. Sci Transl Med. 2010;2:14ra4. doi: 10.1126/scitranslmed.3000391. [DOI] [PubMed] [Google Scholar]
- 10.Garcia-Lerma JG, Otten RA, Qari SH, et al. Prevention of rectal SHIV transmission in macaques by daily or intermittent prophylaxis with emtricitabine and tenofovir. PLoS Med. 2008;5:e28. doi: 10.1371/journal.pmed.0050028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hunt PW, Brenchley J, Sinclair E, et al. Relationship between T cell activation and CD4+ T cell count in HIV-seropositive individuals with undetectable plasma HIV RNA levels in the absence of therapy. J Infect Dis. 2008;197:126–33. doi: 10.1086/524143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Streeck H, Nixon DF. T cell immunity in acute HIV-1 infection. J Infect Dis. 2010;202(Suppl 2):S302–8. doi: 10.1086/655652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Harouse JM, Gettie A, Eshetu T, et al. Mucosal transmission and induction of simian AIDS by CCR5-specific simian/human immunodeficiency virus SHIV(SF162P3) J Virol. 2001;75:1990–5. doi: 10.1128/JVI.75.4.1990-1995.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pahar B, Wang X, Dufour J, Lackner AA, Veazey RS. Virus-specific T cell responses in macaques acutely infected with SHIV(sf162p3) Virology. 2007;363:36–47. doi: 10.1016/j.virol.2007.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kersh EN, Luo W, Adams DR, et al. Short communication: no evidence of occult SHIV infection as demonstrated by CD8(+) cell depletion after chemoprophylaxis-induced protection from mucosal infection in rhesus macaques. AIDS Res Hum Retro. 2008;24:543–6. doi: 10.1089/aid.2007.0222. [DOI] [PubMed] [Google Scholar]
- 16.Kersh EN, Luo W, Adams DR, et al. Repeated rectal SHIVSF162P3 exposures do not consistently induce sustained T cell responses prior to systemic infection in the repeat-low dose preclinical macaque model. AIDS Res Hum Retro. 2009;25:905–17. doi: 10.1089/aid.2008.0287. [DOI] [PubMed] [Google Scholar]
- 17.Liu J, Keele BF, Li H, et al. Low-dose mucosal simian immunodeficiency virus infection restricts early replication kinetics and transmitted virus variants in rhesus monkeys. J Virol. 2010;84:10406–12. doi: 10.1128/JVI.01155-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Eberly MD, Kader M, Hassan W, et al. Increased IL-15 production is associated with higher susceptibility of memory CD4 T cells to simian immunodeficiency virus during acute infection. J Immunol. 2009;182:1439–48. doi: 10.4049/jimmunol.182.3.1439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.National Research Council. Guide for the Care and Use of Laboratory Animals. Washington, DC: National Academy Press; 1996. [Google Scholar]
- 20.Yant LJ, Friedrich TC, Johnson RC, et al. The high-frequency major histocompatibility complex class I allele Mamu-B*17 is associated with control of simian immunodeficiency virus SIVmac239 replication. J Virol. 2006;80:5074–7. doi: 10.1128/JVI.80.10.5074-5077.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Loffredo JT, Sidney J, Bean AT, et al. Two MHC class I molecules associated with elite control of immunodeficiency virus replication, Mamu-B*08 and HLA-B*2705, bind peptides with sequence similarity. J Immunol. 2009;182:7763–75. doi: 10.4049/jimmunol.0900111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kirmaier A, Wu F, Newman RM, et al. TRIM5 suppresses cross-species transmission of a primate immunodeficiency virus and selects for emergence of resistant variants in the new species. PLoS Biol. 2010;8:e1000462. doi: 10.1371/journal.pbio.1000462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Subbarao S, Otten RA, Ramos A, et al. Chemoprophylaxis with tenofovir disoproxil fumarate provided partial protection against infection with simian human immunodeficiency virus in macaques given multiple virus challenges. J Infect Dis. 2006;194:904–11. doi: 10.1086/507306. [DOI] [PubMed] [Google Scholar]
- 24.Kersh EN, Adams DR, Youngpairoj AS, et al. T cell chemo-vaccination effects after repeated mucosal SHIV exposures and oral pre-exposure prophylaxis. PLoS One. 2011;6:e19295. doi: 10.1371/journal.pone.0019295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hansen SG, Vieville C, Whizin N, et al. Effector memory T cell responses are associated with protection of rhesus monkeys from mucosal simian immunodeficiency virus challenge. Nat Med. 2009;15:293–9. doi: 10.1038/nm.1935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zeger SL, Liang KY, Albert PS. Models for longitudinal data: a generalized estimating equation approach. Biometrics. 1988;44:1049–60. [PubMed] [Google Scholar]
- 27.Verhoeven D, Sankaran S, Silvey M, Dandekar S. Antiviral therapy during primary simian immunodeficiency virus infection fails to prevent acute loss of CD4+ T cells in gut mucosa but enhances their rapid restoration through central memory T cells. J Virol. 2008;82:4016–27. doi: 10.1128/JVI.02164-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mehandru S, Dandekar S. Role of the gastrointestinal tract in establishing infection in primates and humans. Curr Opin HIV AIDS. 2008;3:22–7. doi: 10.1097/COH.0b013e3282f331b0. [DOI] [PubMed] [Google Scholar]
- 29.Owen RE, Heitman JW, Hirschkorn DF, et al. HIV+ elite controllers have low HIV-specific T-cell activation yet maintain strong, polyfunctional T-cell responses. AIDS. 2010;24:1095–105. doi: 10.1097/QAD.0b013e3283377a1e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schmitz JE, Kuroda MJ, Santra S, et al. Control of viremia in simian immunodeficiency virus infection by CD8+ lymphocytes. Science. 1999;283:857–60. doi: 10.1126/science.283.5403.857. [DOI] [PubMed] [Google Scholar]
- 31.Rosenwirth B, ten Haaft P, Bogers WM, et al. Antiretroviral therapy during primary immunodeficiency virus infection can induce persistent suppression of virus load and protection from heterologous challenge in rhesus macaques. J Virol. 2000;74:1704–11. doi: 10.1128/jvi.74.4.1704-1711.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.van Rompay KK, Dailey PJ, Tarara RP, et al. Early short-term 9-[2-(R)-(phosphonomethoxy)propyl]adenine treatment favorably alters the subsequent disease course in simian immunodeficiency virus-infected newborn rhesus macaques. J Virol. 1999;73:2947–55. doi: 10.1128/jvi.73.4.2947-2955.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Watson A, McClure J, Ranchalis J, et al. Early postinfection antiviral treatment reduces viral load and prevents CD4+ cell decline in HIV type 2-infected macaques. AIDS Res Hum Retro. 1997;13:1375–81. doi: 10.1089/aid.1997.13.1375. [DOI] [PubMed] [Google Scholar]
- 34.Streeck H, Jessen H, Alter G, et al. Immunological and virological impact of highly active antiretroviral therapy initiated during acute HIV-1 infection. J Infect Dis. 2006;194:734–9. doi: 10.1086/503811. [DOI] [PubMed] [Google Scholar]
- 35.Hecht FM, Wang L, Collier A, et al. A multicenter observational study of the potential benefits of initiating combination antiretroviral therapy during acute HIV infection. J Infect Dis. 2006;194:725–33. doi: 10.1086/506616. [DOI] [PubMed] [Google Scholar]
- 36.Kubo M, Nishimura Y, Shingai M, et al. Initiation of antiretroviral therapy 48 hours after infection with simian immunodeficiency virus potently suppresses acute-phase viremia and blocks the massive loss of memory CD4+ T cells but fails to prevent disease. J Virol. 2009;83:7099–108. doi: 10.1128/JVI.02522-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kader M, Hassan WM, Eberly M, et al. Antiretroviral therapy prior to acute viral replication preserves CD4 T cells in the periphery but not in rectal mucosa during acute simian immunodeficiency virus infection. J Virol. 2008;82:11467–71. doi: 10.1128/JVI.01143-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lifson JD, Piatak M, Jr, Cline AN, et al. Transient early post-inoculation anti-retroviral treatment facilitates controlled infection with sparing of CD4+ T cells in gut-associated lymphoid tissues in SIVmac239-infected rhesus macaques, but not resistance to rechallenge. J Med Primatol. 2003;32:201–10. doi: 10.1034/j.1600-0684.2003.00026.x. [DOI] [PubMed] [Google Scholar]
- 39.George MD, Reay E, Sankaran S, Dandekar S. Early antiretroviral therapy for simian immunodeficiency virus infection leads to mucosal CD4+ T-cell restoration and enhanced gene expression regulating mucosal repair and regeneration. J Virol. 2005;79:2709–19. doi: 10.1128/JVI.79.5.2709-2719.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zheng Q, Ruone S, Switzer WM, Heneine W, Garcia-Lerma JG. Limited SHIV env diversification in macaques failing oral antiretroviral pre-exposure prophylaxis. Retrovirology. 2012;9:40. doi: 10.1186/1742-4690-9-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Melchjorsen J, Risor MW, Sogaard OS, et al. Tenofovir selectively regulates production of inflammatory cytokines and shifts the IL-12/IL-10 balance in human primary cells. J Acquir Immun Defic Syndr. 2011;57:265–75. doi: 10.1097/QAI.0b013e3182185276. [DOI] [PubMed] [Google Scholar]
- 42.Cranage M, Sharpe S, Herrera C, et al. Prevention of SIV rectal transmission and priming of T cell responses in macaques after local pre-exposure application of tenofovir gel. PLoS Med. 2008;5:e157. doi: 10.1371/journal.pmed.0050157. discussion e157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rolland M, Tovanabutra S, deCamp AC, et al. Genetic impact of vaccination on breakthrough HIV-1 sequences from the STEP trial. Nat Med. 2011;17:366–71. doi: 10.1038/nm.2316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mureithi MW, Poole D, Naranbhai V, et al. Preservation HIV-1-specific IFNgamma+ CD4+ T-cell responses in breakthrough infections after exposure to tenofovir gel in the CAPRISA 004 Microbicide Trial. J Acquir Immun Defic Syndr. 2012;60:124–27. doi: 10.1097/QAI.0b013e31824f53a9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Prada N, Davis B, Jean-Pierre P, et al. Drug-susceptible HIV-1 infection despite intermittent fixed-dose combination tenofovir/emtricitabine as prophylaxis is associated with low-level viremia, delayed seroconversion, and an attenuated clinical course. J Acquir Immun Defic Syndr. 2008;49:117–22. doi: 10.1097/QAI.0b013e3181869a9b. [DOI] [PMC free article] [PubMed] [Google Scholar]