

Abstract
The spore-forming bacterium Clostridium difficile represents the principal cause of hospital-acquired diarrhea and pseudomembranous colitis worldwide. C. difficile infection (CDI) is mediated by 2 bacterial toxins, A and B; neutralizing these toxins with monoclonal antibodies (mAbs) provides a potential nonantibiotic strategy for combating the rising prevalence, severity, and recurrence of CDI. Novel antitoxin mAbs were generated in mice and were humanized. The humanized antitoxin A mAb PA-50 and antitoxin B mAb PA-41 have picomolar potencies in vitro and bind to novel regions of the respective toxins. In a hamster model for CDI, 95% of animals treated with a combination of humanized PA-50 and PA-41 showed long-term survival relative to 0% survival of animals treated with standard antibiotics or comparator mAbs. These humanized mAbs provide insight into C. difficile intoxication and hold promise as potential nonantibiotic agents for improving clinical management of CDI.
Clostridium difficile is a gram-positive, spore-forming, anaerobic bacterium that represents the leading cause of hospital-acquired diarrhea in developed countries [1, 2]. C. difficile infection (CDI) results in a spectrum of disease ranging from mild-to-severe diarrhea to fulminant colitis and death. The incidence and severity of CDI have increased markedly over the past decade, due in part to the emergence of unusually virulent, antibiotic-resistant strains. Chief amongst these are strains characterized as group BI by restriction endonuclease analysis, North American pulse-field type 1 (NAP1) by pulse-field gel electrophoresis, and ribotype 027 by polymerase chain reaction. CDI currently affects approximately 500 000 individuals and causes more than 20 000 deaths annually in the United States [1, 3].
CDI is typically precipitated when an individual is exposed to C. difficile spores while receiving antibiotics, which disrupt the normal colonic flora and provide an opportunity for C. difficile to flourish. Current practice for managing CDI involves discontinuing the culpable antibiotic and initiating treatment with metronidazole, vancomycin, or fidaxomicin [4]. Unfortunately, antibiotic therapy is associated with incomplete response or disease recurrence in approximately 30% of patients. The per-patient healthcare costs of CDI have been estimated to be approximately $4000 for primary cases and $16 000 for recurrent cases in the United States [5]. Consequently, the C. difficile bacterium places a significant burden on the healthcare systems of the United States and many other countries.
The main virulence factors of C. difficile are 2 large protein toxins, A and B. The toxins share similar size and domain organization composed of an amino-terminal glucosyltransferase domain followed by a proteolytic domain, a hydrophobic translocation domain, and a carboxy-terminal receptor-binding domain. Both toxins induce cell rounding and death by glucosylating GTPases that are required for cytoskeletal integrity [6, 7]. These toxins have been reported to be overexpressed in hypervirulent strains [8], are absent from nontoxigenic strains [9], and provide targets for novel therapies.
Neutralizing C. difficile toxins with monoclonal antibodies (mAbs) or vaccine-induced antibodies constitutes a nonantibiotic treatment strategy that has shown preclinical promise [10–16]. Initial clinical proof of principle was demonstrated recently with human anti-C. difficile toxin mAbs [17]. When used clinically in combination with antibiotic therapy, the mAbs significantly reduced the rate of CDI recurrence [17]. The results are consistent with prior findings that serum levels of endogenous antitoxin antibodies correlate with protection from primary and recurrent CDI [18, 19]. Although the toxin-encoding genes tcdA and tcdB are variable elements of the C. difficile genome [20, 21], little is known about how their genetic variation influences the activity of neutralizing antibodies.
We have generated novel humanized mAbs, PA-50 and PA-41, which define potent neutralization epitopes on toxins A and B, respectively. This report describes the mAbs’ binding properties and breadth of neutralizing activity. Additionally, combination therapy with PA-50/PA-41 in a well-established animal model of CDI resulted in long-lived protection from lethal disease beyond that observed with standard antibiotic therapy.
MATERIALS AND METHODS
Cell Lines, Purified C. difficile Toxins, and Supernatants
CHO-K1 and T-84 cells were obtained from American Type Culture Collection (ATCC, Rockville, Maryland). CHO-K1 cells were cultured in F-12K medium supplemented with 10% qualified fetal bovine serum (FBS) and l-glutamine, nonessential amino acids, and sodium pyruvate (Invitrogen). T-84 human colonic epithelial cells were cultured in a 1:1 mixture of F-12K and DMEM (Invitrogen) supplemented with 5% FBS, l-glutamine, nonessential amino acids, sodium pyruvate, and HEPES. Purified toxin and toxoid proteins from strain VPI 10463 were obtained from List Biological Laboratories (Campbell, California) or TechLab (Blacksburg, Virginia). C. difficile culture supernatants were produced at TechLab as described elsewhere [22].
Generation of Murine PA-50 and Murine PA-41
Female Balb/c mice (Charles River Labs, Wilmington, Massachusetts) were immunized subcutaneously with 2 or 3 doses of 10 μg of toxin A toxoid (inactivated with formaldehyde) with 10 μg Quil A adjuvant (Accurate Chemical, Westbury, New York) at 3-week intervals prior to boosting with increasing doses of active toxin A or B, also at 3-week intervals. The doses of toxin A were escalated from 20 ng to 2.5 μg, whereas doses of toxin B were escalated from 2 to 12.5 μg. Animals were boosted intraperitoneally with 10 μg toxin A or 20 μg toxin B 3 days before death. Hybridomas were generated by standard methods [23]. Hybridoma supernatants were tested for neutralization of toxin A or B on T-84 or CHO-K1 cells, respectively. Two potently inhibitory mAbs were designated murine PA-50 (mPA-50, antitoxin A) and murine PA-41 (mPA-41, antitoxin B). Hybridomas producing these mAbs were grown in cell culture or in mouse ascites, and murine mAbs were purified to >95% homogeneity by protein A chromatography, dialyzed into phosphate-buffered saline (PBS) and stored at −80°C.
Humanization of PA-50 and PA-41 and Preparation of Comparator mAbs
The variable regions of the heavy and light chain genes of mPA-50 and mPA-41 were cloned from the hybridomas using published methods [24]. Complementarity-determining regions and the relevant framework amino acids from the murine mAbs were grafted into human IgG1,κ framework sequences. The humanized VH and VL regions were cloned into the expression vectors, pCON-gamma1 and pCON-kappa (Lonza Biologics, Berkshire, UK). Humanized PA-50 and PA-41 were produced as full-length IgG1,κ antibodies in stably transfected CHO-K1SV cells (Lonza) and purified to >95% homogeneity by protein A chromatography.
CDA1 and CDB1 [10, 17] were produced for use as comparator mAbs. Published DNA sequences encoding the heavy and light chain variable regions of CDA1/3D8 and CDB1/124 [25] were synthesized (DNA2.0, Menlo Park, California) and cloned into vectors pCON-gamma1 and pCON-kappa (Lonza). Full-length IgG1,κ mAbs were expressed and purified as described above. CDA1 and CDB1 exhibited binding affinities, neutralization potencies, and hemagglutination activities in accordance with published data [25].
In Vitro Neutralization Assays
T-84 and CHO-K1 cells were used to evaluate neutralization activity of mAbs against purified toxin A and toxin B, respectively. T-84 or CHO-K1 cells were added to 96-well flat-bottom cell culture plates (Perkin Elmer) at 2 × 103 or 1.5 × 104 cells per well, respectively, and incubated for 4 hours at 37°C and 5% CO2. Toxin A (240 ng/mL) or toxin B (8 pg/mL) was combined with serially diluted mAbs (Falcon) for 1 hour at 37°C and then added to the cells. After incubation for 72 hours, 20 µL/well CellTiter-Blue (Promega) was added, and the plates were incubated for an additional 4 hours. Plates were read using a fluorescence excitation wavelength of 560 nm and an emission wavelength of 590 nm. Cell survival was compared in treated and untreated cultures, and the mAb concentration required for 50% neutralization (EC50) was calculated.
For C. difficile culture supernatants, toxin titer was determined by performing serial 2-fold dilutions of supernatants in the CHO-K1 and T-84 assays described above. The minimum dilution that caused >90% cytotoxicity was used in neutralization experiments.
Efficacy Studies
Fifty-day-old Golden Syrian hamsters (Charles River Laboratories, Stone Ridge, New York) were pretreated (day −1) with a single subcutaneous dose of clindamycin at 50 mg/kg to disrupt the normal colonic flora. On the following day (day 0), hamsters received an oral dose (3.1 × 106 colony-forming units in 0.5 mL) of a suspension of C. difficile (strain 545, ATCC 43596) [26]. Hamsters (8 or 10 per group) received either no treatment, vancomycin orally at 20 mg/kg twice daily (BID) × 5 days, starting on day 1, or mAbs administered intraperitoneally every other day (QOD) × 4, starting on day −1. Animals were weighed at least weekly and monitored daily for health and survival during the 39-day study. Necropsy was performed at study termination, and C. difficile cecal counts were determined following anaerobic culture at 37°C for 48 hours in selective medium. The limit of detection was 20 CFU/g of cecum contents.
Treatment of the animals was in accordance with regulations outlined in the USDA Animal Welfare Act (9 CFR Parts 1, 2, and 3) and/or the conditions specified in the “Guide for Care and Use of Laboratory Animals” (National Academy Press, Washington, DC, 1996).
Statistical Analyses
Neutralization data were fit to a 4-parameter logistic equation using GraphPad Prism software (v. 4.0; GraphPad, San Diego, California). Two-sided t tests or log-rank tests were used for comparison of means or survival data, respectively.
RESULTS
Epitope Specificity
Immunizations yielded a panel of murine mAbs with toxin-neutralizing activity. Included in this panel were mAbs mPA-50 and mPA-41 to toxins A and B, respectively. Biacore experiments indicated that the mPA-50 epitope is present in multiple copies on toxin A and does not overlap with the epitope for CDA1 (Supplementary Figure 1A). Immobilized mPA-50 specifically bound toxin A with an avidity of 0.16 nM. The mPA-41 was observed to bind a single site on toxin B with an affinity of 0.59 nM. Binding of mPA-41 to toxin B did not block subsequent binding of CDB1 (Supplementary Figure 1B). Neither mPA-50 nor mPA-41 showed measurable cross-reactivity with toxin B or toxin A, respectively, in Biacore or enzyme-linked immunosorbent assay (ELISA) studies (data not shown).
The mAbs’ binding sites were localized to specific regions of the respective toxins by limited proteolysis of the toxins followed by Western blotting (Supplementary Figure 1C and 1D). The mPA-50 bound the carboxy-terminal domain of toxin A, whereas mPA-41 bound the amino-terminal domain of toxin B. Collectively, the data indicate that mPA-50 binds multiple sites within the receptor-binding domain of toxin A, and mPA-41 binds a single site within the enzymatic domain of toxin B.
In Vitro Neutralization Activity of Murine and Humanized mAbs
Murine and humanized mAbs were analyzed for binding avidities and neutralization of purified toxins from strain VPI 10463 in vitro. Comparable binding avidities were observed for murine and humanized mAbs (data not shown). Murine and humanized forms of PA-50 neutralized the cytotoxicity of toxin A with EC50 values of 90 pM (Figure 1A). The mPA-41 and humanized PA-41 neutralized toxin B with EC50 values of 9 and 4 pM, respectively (Figure 1B). Essentially complete neutralization of toxins A and B was obtained at higher mAb concentrations. Humanized PA-50 and PA-41 thus recapitulate the toxin-neutralizing activities of the respective mouse mAbs.
Figure 1.

Neutralization of purified toxins by murine and humanized monoclonal antibodies (mAbs). A, Humanized PA-50 and murine PA-50 (mPA-50) were compared for neutralization of purified toxin A on T-84 cells. B, Humanized PA-41 and mPA-41 were compared for neutralization of toxin B on CHO-K1 cells.
Breadth of In Vitro Neutralization of Humanized mAbs
Because C. difficile exhibits interstrain heterogeneity in the genes encoding toxins A and B, studies were undertaken to examine the breadth of toxin neutralization by PA-50 and PA-41. These studies employed toxin-containing supernatants generated from diverse C. difficile isolates from North America and Europe (Table 1). The panel included ribotypes 001, 002, 003, 012, 014, 017, 027, and 078 in approximate frequency proportion to that observed clinically [27, 28], with the exception of ribotype 017 tcdA−tcdB+ strains, which are overrepresented in the panel. Supernatants from tcdA−tcdB+ strains were used as tools to identify cells that were refractory to killing by supernatants containing toxin B alone and thus would be suitable for examining cytotoxicity mediated by toxin A. Strain VPI 10463 was included in the panel and allowed a comparison of results obtained with purified and unpurified toxins. CDA1 and CDB1 were tested as comparator mAbs.
Table 1.
Neutralization of Toxins From Diverse Clostridium difficile Strains In Vitro
| EC50, pM |
|||||
|---|---|---|---|---|---|
| Antitoxin A mAbs |
Antitoxin B mAbs |
||||
| Ribotype | Strain | PA-50 | CDA1 | PA-41 | CDB1 |
| 001 | CCL14137 | 39 | 611 | 9.7 | 129 |
| 001 | MH5 | 127 | 384 | 18 | 73 |
| 001 | Pitt 2 | 27 | 247 | 13 | 92 |
| 002 | UVA17 | 26 | 825 | 129 | 671 |
| 003 | VPI 10463 | 20 | 1271 | 7.7 | 136 |
| 012 | 545 | 38 | 4552 | 15 | 153 |
| 012 | 630 | 54 | 1019 | 111 | 1782 |
| 014 | UVA30/TL42 | 51 | 625 | 21 | 324 |
| 017 | CCL13820 | N/A | N/A | >105 | 72 |
| 017 | F1470 | N/A | N/A | >105 | 46 |
| 017 | Pitt 102 | N/A | N/A | >105 | 8.1 |
| 027 | CCL678 | 29 | 58 950 | 77 | >105 |
| 027 | CCL14402 | ND | ND | 19 | 4678 |
| 027 | CD196 | 61 | 132 600 | 16 | 9812 |
| 027 | HMC553 | 29 | 109 000 | 24 | 14 730 |
| 027 | Montreal 5 | 29 | 87 090 | 36 | 25 810 |
| 027 | Montreal 7.1 | 31 | 109 400 | 29 | 16 800 |
| 027 | Pitt 45 | 43 | 108 100 | 29 | 26 510 |
| 036 | 8864 | N/A | N/A | 370 | >105 |
| 078 | Pitt 07 | 32 | >105 | 59 | 8415 |
Data are representative of 2–3 replicates for each mAb/strain combination. Abbreviations: N/A, not applicable; ND, not done.
PA-50 neutralized toxin A in a strain-independent manner. The median EC50 was 32 pM (range, 20–127 pM; Table 1), and Hill slopes typically exceeded 2 (Figure 2A). PA-50 was more active than CDA1 against each of the test isolates. The greatest potency differences were observed for hypervirulent ribotype 027 strains; PA-50 was approximately 1000-fold more potent than CDA1 (P = .0002) against these strains, as well as against a ribotype 078 strain included in the panel.
Figure 2.

Neutralization of toxins produced by diverse strains of Clostridium difficile. A, Humanized PA-50 and comparator CDA1 antitoxin A monoclonal antibodies (mAbs) were tested for neutralization of the cytotoxicity of C. difficile culture supernatants against T-84 cells. B, Humanized PA-41 and comparator CDB1 antitoxin B mAbs were tested for neutralization of the cytotoxicity of C. difficile culture supernatants against CHO-K1 cells.
PA-41 inhibited each of the tcdA+tcdB+ strains with a median EC50 of 23 pM (range, 7.7–129 pM; Table 1) (Figure 2B). PA-41 was generally more effective than CDB1 against tcdA+tcdB+ strains and was approximately 500-fold more potent against the ribotype 027 strains (P = .003). CDB1 was more effective than PA-41 against ribotype 017 tcdA− tcdB+ strains; however, the reverse was true for a ribotype 036 tcdA−tcdB+ strain. Finally, PA-41 and PA-50 exhibited comparable activities against crude and purified forms of VPI 10463 toxins (Table 1 and Figure 1).
Efficacy of PA-50 and PA-41 in a Hamster Model of CDI
In vivo efficacy was examined in hamsters. The study examined 20 and 50 mg/kg/mAb doses of PA-50 and PA-41 used in combination, that is, “PA-50/PA-41″, in animals challenged with the ribotype 012 strain 545. Strain 545 represents a heterologous challenge strain that is unrelated to the ribotype 003 strain (VPI 10463) used in generating these mAbs. Strain 545 is similar to VPI 10463 in terms of the susceptibility of its toxins to neutralization by PA-50, PA-41, and the comparator mAbs (Table 1). Control groups in the study received no treatment, vancomycin, or the CDA1/CDB1 combination (Table 2).
Table 2.
Survival Outcomes by Treatment Group in the Hamster Efficacy Study
| Treatment | Dose (route) | Treatment days | No. of animals | Median survival, d | Day 39 survival, % |
|---|---|---|---|---|---|
| No treatment | None | None | 8 | 2 | 0 |
| Vancomycin | 20 mg/kg BID (PO) | 1–5 | 10 | 20 | 0 |
| CDA1/CDB1 combination | 20 mg/kg each mAb QOD (IP) | −1, 1, 3, 5 | 10 | 11 | 0 |
| CDA1/CDB1 combination | 50 mg/kg each mAb QOD (IP) | −1, 1, 3, 5 | 10 | 14 | 0 |
| PA-50/PA-41 combination | 20 mg/kg each mAb QOD (IP) | −1, 1, 3, 5 | 10 | N/A | 100 |
| PA-50/PA-41 combination | 50 mg/kg each mAb QOD (IP) | −1, 1, 3, 5 | 10 | N/a | 90 |
Abbreviations: BID, twice daily; IP, intraperitoneally; mAP, monoclonal antibodies; N/A, not applicable; survival exceeded 50% at the end of the study; QOD, every other day.
Untreated animals had a median survival of 2 days, with the longest survival being 3 days. In contrast, 19 of 20 animals treated with either 20 or 50 mg/kg/mAb of PA-50/PA-41 survived through the 39-day study (Table 2 and Figure 3A). One PA-50/PA-41 animal perished on day 8 with symptoms consistent with CDI.
Figure 3.
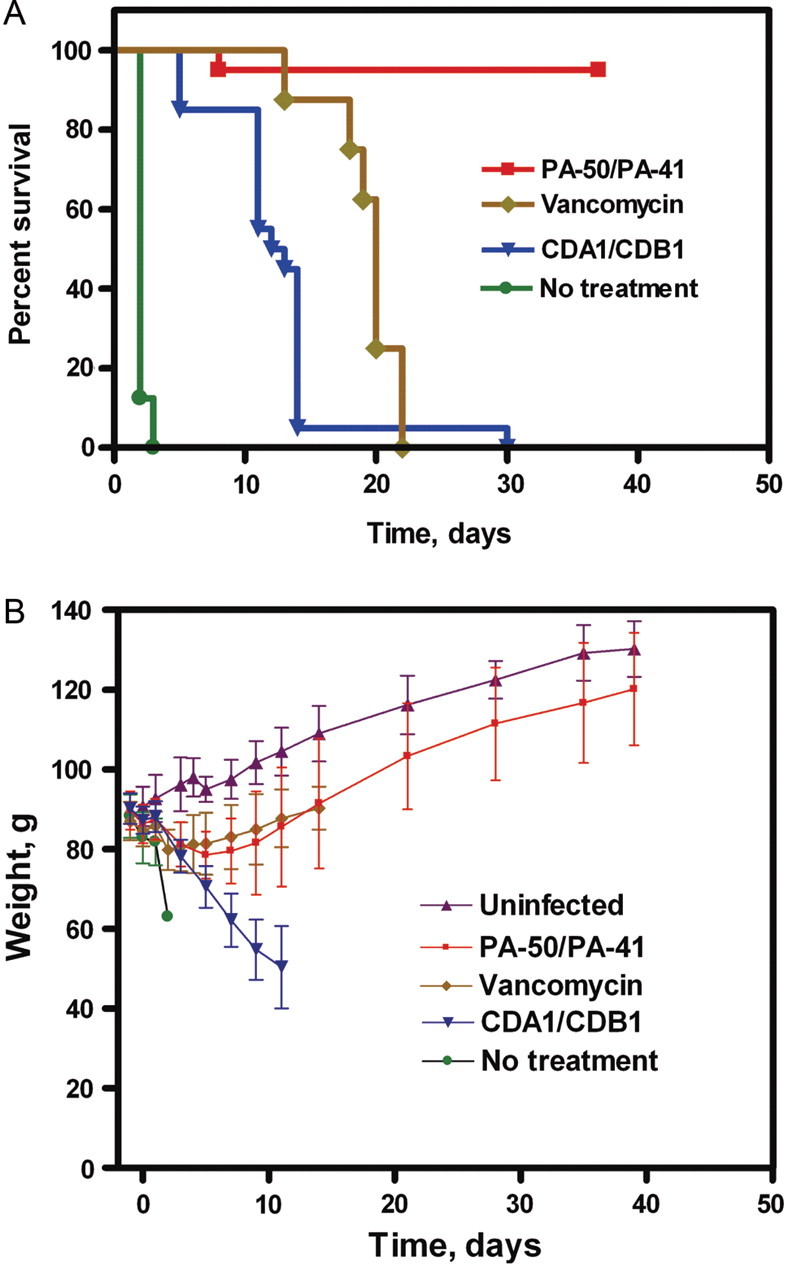
Survival outcomes and weight changes in the hamster efficacy study. A, Kaplan-Meier survival curves of Clostridium difficile-infected animals that received treatment with a 1:1 combination of humanized PA-50/PA-41; vancomycin; a 1:1 combination of comparator monoclonal antibodies (mAbs); or no treatment. B, Mean (±SD) body weights of animals over time in the different treatment groups compared with those of uninfected control animals. Dosing regimens are described in Table 2. Survival outcomes were similar for both the 20 mg/kg and the 50 mg/kg doses of PA-50/PA-41 and of CDA1/CDB1 (Table 2); therefore, combined data are shown for the purpose of illustration.
Animals treated with the CDA1/CDB1 combination, or with vancomycin, displayed intermediate survival. Treatment with CDA1/CDB1 at 20 or 50 mg/kg/mAb resulted in median survival times of 11 and 14 days, respectively. Maximum survival was 30 days. The results observed for CDA1/CDB1 in this study are consistent with the 55% survival at 11 days postinfection observed in a prior hamster study [10]. Vancomycin treatment improved median survival to 20 days, but all vancomycin-treated animals experienced a fatal relapse of disease typical of this model. Given the similar outcomes observed for the 20 and 50 mg/kg/mAb doses in mAb-treated animals, the data for these dose groups were pooled in plotting survival curves (Figure 3A). The survival benefit for treatment with PA-50/PA41 was significant (P < .0001) relative to each of the other treatment groups.
Additional evaluations included weight measurements, gross necropsy, and cecal titer of C. difficile at termination. Mean body weights of animals treated with vancomycin or PA-50/PA-41 decreased during the first week postinfection and then rebounded (Figure 3B). By day 39, the mean body weights of animals treated with PA-50/PA-41 were similar to those of healthy, uninfected animals that were housed in parallel (P > .05). The mean body weights of animals treated with CDA1/CDB1 declined steadily (Figure 3B).
At day 39, the gastrointestinal tracts of the 19 surviving PA-50/PA-41 animals appeared similar to those of uninfected animals (data not shown). Cecal titers of C. difficile were either undetectable (<1.3 log10 CFU, n = 11) or low (4.15 ± 0.76 log10 CFU, n = 8) in the PA-50/PA-41-treated animals. In contrast, inflamed gastrointestinal tracts were observed in some or all of the animals in the other treatment groups at time of death. C. difficile was detected in 4 of 4 untreated animals (mean CFU = 8.96 ± 0.59 log10, P < .0001 relative to PA-50/PA-41) and in 4 of 4 vancomycin-treated animals (mean CFU = 6.01 ± 0.93 log10, P < .017 relative to PA-50/PA-41) for which cecal analyses were performed. Most hamsters treated with CDA1/CDB1 had little to no cecum contents at the time of analysis, which precluded quantitation of C. difficile titers. The empty ceca were an unexpected finding that may relate to prolonged disruption of normal gastrointestinal function in these animals.
A second study compared the efficacies of PA-50 and PA-41 used alone or in combination (see Supplementary Data). Treatment groups included vehicle, vancomycin, 40 mg/kg PA-50 alone, 40 mg/kg PA-41 alone, and PA-50/PA-41 at 20 mg/kg/mAb or 40 mg/kg/mAb. All animals that received either dose of the PA-50/PA-41 combination survived until the end of the study (42 days, Supplementary Figure 2). Median survival times for the other treatment groups were 2 days for vehicle, 2 days for PA-41, 3 days for PA-50, and 16 days for vancomycin.
DISCUSSION
This study describes novel humanized mAbs, PA-50 and PA-41, that broadly neutralize toxins from conventional and hypervirulent strains of C. difficile in vitro and define important neutralization epitopes on toxins A and B. When used in combination in a stringent hamster model of CDI, the mAbs provided long-lived protection against lethal disease beyond that observed for standard antibiotic therapy. The gastrointestinal tracts of the animals treated with the PA-50/PA-41 combination were characterized by a healthy appearance and low or undetectable levels of C. difficile, indicating that therapy led to normalization of the colon. PA-50 and PA-41 potently neutralized toxins from genetically diverse C. difficile strains representative of the current epidemic.
The mAbs’ breadth of activity is notable in light of the considerable genotypic and phenotypic variation within the toxins [20, 29, 30]. PA-50 and PA-41 neutralized toxins produced by the hypervirulent ribotype 027 strains with picomolar activity, whereas comparator mAbs exhibited nanomolar activity. This result is consistent with reduced binding of CDA1 to toxin A from ribotype 027 strains [31]. Toxin B from ribotype 027 strains exhibits marked sequence variation associated with increased cytotoxicity in vitro [21, 29]. However, this sequence divergence did not affect the activity of PA-41, which binds an epitope within the amino-terminal domain. Overall, the findings indicate that the epitopes for PA-50 and PA-41 are broadly conserved through the ribotype 027 lineage. Further delineation of the epitopes for PA-50 and PA-41 may provide insight into structure-function relationships of C. difficile toxins.
CDI is typically caused by C. difficile strains that produce both toxins A and B. However, tcdA−tcdB+ strains, predominantly ribotype 017, can cause disease [32, 33]. Ribotype 017 strains exhibit reduced pathogenicity in hamsters [34] and encode an atypical tcdB whose amino-terminal region bears 70%–80% sequence identity with both tcdB from VPI 10463 and lethal toxin (tcsL) from C. sordellii [35]. Phenotypically, ribotype 017 tcdB has hybrid characteristics and exhibits the receptor-binding properties and glucosylating specificities of typical tcdB and tcsL toxins, respectively [35]. The atypical amino-terminal region of ribotype 017 tcdB provides a likely explanation for why it was not neutralized by PA-41. Although ribotype 017 strains can be regionally prevalent [36–39], overall they comprise <2% of the strains encountered in recent international phase 3 clinical studies [27, 28]. In contrast to the findings for ribotype 017 strains, PA-41 effectively neutralized toxin from a ribotype 036 tcdA−tcdB+ strain.
PA-50 exhibited a steep dose-response neutralization curve with Hill coefficients >2, indicating cooperative inhibition. Cooperative interactions are common in nature and are characterized by Hill coefficients of >1 [40]. PA-50 binds toxin A in a multivalent fashion, a condition that is often necessary, but not sufficient for cooperativity. We are not aware of another mAb that shows cooperative neutralization of C. difficile toxin. Studies to ascertain if cooperativity occurs at the level of PA-50 binding to toxin A and to determine the valency of the PA-50 epitope on toxin A are ongoing.
Combination treatment with PA-50/PA-41 was highly efficacious in the well-established hamster model of CDI. A short course of treatment with PA-50/PA-41 resulted in 95% survival of animals at 39 days postinfection, compared with 0% survival of animals that received no treatment, standard antibiotic therapy, or comparator mAbs. At 39 days postinfection, animals treated with PA-50/PA-41 had normal weights and showed no obvious gastrointestinal lesions. C. difficile bacterial counts could not be recovered from most animals, reflecting a >6-log10 clearance relative to untreated animals. One likely explanation for these findings is that mAb neutralization of toxins in the absence of antibiotics enabled reestablishment of protective microbial flora in the gastrointestinal tracts of the animals.
Either toxin A or toxin B alone can cause fatal disease in hamsters [34, 41, 42], and mAbs to both toxins are generally required for maximum efficacy [10, 43]. Consistent with these findings, treatment with PA-50 or PA-41 alone exhibited minimal activity (Supplementary Data), underscoring the requirement for combination treatment. The QOD mAb treatment regimen used here was selected on the basis of a prior study of antitoxin mAbs [10]. Given the high-level efficacy observed for PA-50/PA-41, further exploration of dose and schedule is warranted. Examination of additional mAb combinations (eg, PA-50/CDB1, CDA1/PA-41, and 3 or 4 mAb combinations) is also of future interest.
In summary, humanized anti-C. difficile toxin mAbs PA-50 and PA-41 define novel neutralization epitopes that are broadly conserved across contemporary and historical strains of C. difficile. Combination treatment of animals with PA-50/PA-41 provided durable protection against lethal disease in hamsters. The properties of these mAbs make them attractive development candidates as a potential nonantibiotic therapy for CDI.
Supplementary Data
Supplementary materials are available at The Journal of Infectious Diseases online (http://jid.oxfordjournals.org/). Supplementary materials consist of data provided by the author that are published to benefit the reader. The posted materials are not copyedited. The contents of all supplementary data are the sole responsibility of the authors. Questions or messages regarding errors should be addressed to the author.
Notes
Acknowledgments. We thank Raymond N. Ogawa for technical assistance and Patty Schneider for graphical assistance.
Financial support. This work was supported by grant number R01AI094424 from the National Institute of Allergy and Infectious Diseases (NIAID) and by Progenics Pharmaceuticals, Inc. Its contents are solely the responsibility of the authors and do not necessarily represent the official views of NIAID.
Potential conflicts of interest. All authors are all current or former employees of Progenics Pharmaceuticals, Inc. The information contained in this article was partially presented at the 51st Interscience Conference on Antimicrobial Agents and Chemotherapy.
All authors have submitted the ICMJE Form for Disclosure of Potential Conflicts of Interest. Conflicts that the editors consider relevant to the content of the manuscript have been disclosed.
References
- 1.Redelings MD, Sorvillo F, Mascola L. Increase in Clostridium difficile-related mortality rates, United States, 1999–2004. Emerg Infect Dis. 2007;13:1417–9. doi: 10.3201/eid1309.061116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rupnik M, Wilcox MH, Gerding DN. Clostridium difficile infection: new developments in epidemiology and pathogenesis. Nat Rev Microbiol. 2009;7:526–36. doi: 10.1038/nrmicro2164. [DOI] [PubMed] [Google Scholar]
- 3.McDonald LC, Killgore GE, Thompson A, et al. An epidemic, toxin gene-variant strain of Clostridium difficile. N Engl J Med. 2005;353:2433–41. doi: 10.1056/NEJMoa051590. [DOI] [PubMed] [Google Scholar]
- 4.Cohen SH, Gerding DN, Johnson S, et al. Clinical practice guidelines for Clostridium difficile infection in adults: 2010 update by the Society for Healthcare Epidemiology of America (SHEA) and the Infectious Diseases Society of America (IDSA) Infect Control Hosp Epidemiol. 2010;31:431–55. doi: 10.1086/651706. [DOI] [PubMed] [Google Scholar]
- 5.Ghantoji SS, Sail K, Lairson DR, DuPont HL, Garey KW. Economic healthcare costs of Clostridium difficile infection: a systematic review. J Hosp Infect. 2010;74:309–18. doi: 10.1016/j.jhin.2009.10.016. [DOI] [PubMed] [Google Scholar]
- 6.Lyerly DM, Krivan HC, Wilkins TD. Clostridium difficile: its disease and toxins. Clin Microbiol Rev. 1988;1:1–18. doi: 10.1128/cmr.1.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Voth DE, Ballard JD. Clostridium difficile toxins: mechanism of action and role in disease. Clin Microbiol Rev. 2005;18:247–63. doi: 10.1128/CMR.18.2.247-263.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Warny M, Pepin J, Fang A, et al. Toxin production by an emerging strain of Clostridium difficile associated with outbreaks of severe disease in North America and Europe. Lancet. 2005;366:1079–84. doi: 10.1016/S0140-6736(05)67420-X. [DOI] [PubMed] [Google Scholar]
- 9.Cohen SH, Tang YJ, Silva J., Jr Analysis of the pathogenicity locus in Clostridium difficile strains. J Infect Dis. 2000;181:659–63. doi: 10.1086/315248. [DOI] [PubMed] [Google Scholar]
- 10.Babcock GJ, Broering TJ, Hernandez HJ, et al. Human monoclonal antibodies directed against toxins A and B prevent Clostridium difficile–induced mortality in hamsters. Infect Immun. 2006;74:6339–47. doi: 10.1128/IAI.00982-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hussack A, Arbabi-ghahroudi M, van Faassen H, et al. Neutralization of Clostridium difficile toxin A with single-domain antibodies targeting the cell receptor binding-domain. J Bio Chem. 2011;286:8961–76. doi: 10.1074/jbc.M110.198754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Corthier G, Muller MC, Wilkins TD, Lyerly D, L'Haridon R. Protection against experimental pseudomembranous colitis in gnotobiotic mice by use of monoclonal antibodies against Clostridium difficile toxin A. Infect Immun. 1991;59:1192–5. doi: 10.1128/iai.59.3.1192-1195.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Demarest SJ, Hariharan M, Elia M, et al. Neutralization of Clostridium difficile toxin A using antibody combinations. MAbs. 2010;2:190–8. doi: 10.4161/mabs.2.2.11220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Permpoonpattana P, Hong HA, Phetcharaburanin J, et al. Immunization with Bacillus spores expressing toxin A peptide repeats protects against infection with Clostridium difficile strains producing toxins A and B. Infect Immun. 2011;79:2295–302. doi: 10.1128/IAI.00130-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Torres JF, Lyerly DM, Hill JE, Monath TP. Evaluation of formalin-inactivated Clostridium difficile vaccines administered by parenteral and mucosal routes of immunization in hamsters. Infect Immun. 1995;63:4619–27. doi: 10.1128/iai.63.12.4619-4627.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Giannasca PJ, Zhang ZX, Lei WD, et al. Serum antitoxin antibodies mediate systemic and mucosal protection from Clostridium difficile disease in hamsters. Infect Immun. 1999;67:527–38. doi: 10.1128/iai.67.2.527-538.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lowy I, Molrine DC, Leav BA, et al. Treatment with monoclonal antibodies against Clostridium difficile toxins. N Engl J Med. 2010;362:197–205. doi: 10.1056/NEJMoa0907635. [DOI] [PubMed] [Google Scholar]
- 18.Kyne L, Warny M, Qamar A, Kelly CP. Asymptomatic carriage of Clostridium difficile and serum levels of IgG antibody against toxin A. N Engl J Med. 2000;342:390–7. doi: 10.1056/NEJM200002103420604. [DOI] [PubMed] [Google Scholar]
- 19.Kyne L, Warny M, Qamar A, Kelly CP. Association between antibody response to toxin A and protection against recurrent Clostridium difficile diarrhoea. Lancet. 2001;357:189–93. doi: 10.1016/S0140-6736(00)03592-3. [DOI] [PubMed] [Google Scholar]
- 20.Rupnik M. Heterogeneity of large clostridial toxins: importance of Clostridium difficile toxinotypes. FEMS Microbiol Rev. 2008;32:541–55. doi: 10.1111/j.1574-6976.2008.00110.x. [DOI] [PubMed] [Google Scholar]
- 21.Stabler RA, Dawson LF, Phua LT, Wren BW. Comparative analysis of BI/NAP1/027 hypervirulent strains reveals novel toxin B-encoding gene (tcdB) sequences. J Med Microbiol. 2008;57(Pt 6):771–5. doi: 10.1099/jmm.0.47743-0. [DOI] [PubMed] [Google Scholar]
- 22.Lyerly DM, Phelps CJ, Toth J, Wilkins TD. Characterization of toxins A and B of Clostridium difficile with monoclonal antibodies. Infect Immun. 1986;54:70–6. doi: 10.1128/iai.54.1.70-76.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Harlow E, Lane D. Antibodies, A Laboratory Manual. Cold Spring Harbor: Cold Spring Harbor Laboratory Press; 1988. [Google Scholar]
- 24.Co MS, Avdalovic NM, Caron PC, Avdalovic MV, Scheinberg DA, Queen C. Chimeric and humanized antibodies with specificity for the CD33 antigen. J Immunol. 1992;148:1149–54. [PubMed] [Google Scholar]
- 25.Ambrosino D, Babcock GJ, Broering T, et al. Antibodies against Clostridium difficile toxins and uses thereof. US patent 7,625,559. [Google Scholar]
- 26.Delmee M, Avesani V, Delferriere N, Burtonboy G. Characterization of flagella of Clostridium difficile and their role in serogrouping reactions. J Clin Microbiol. 1990;28:2210–4. doi: 10.1128/jcm.28.10.2210-2214.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gerding DN, Sambol SP, Nagaro K, et al. Restriction endonuclease analysis (REA) typing of Clostridium difficile in a phase 3 treatment trial of fidaxomicin vs vancomycin: decreased cure rate for epidemic BI/NAP1/027 strain. 49th Interscience Conference on Antimicrobial Agents and Chemotherapy. Abstract L1-1642. San Francisco, CA, 12–15 September 2009. [Google Scholar]
- 28.Cheknis AK, Sambol SP, Davidson DM, et al. Distribution of Clostridium difficile strains from a North American, European and Australian trial of treatment for C. difficile infections: 2005–2007. Anaerobe. 2009;15:230–3. doi: 10.1016/j.anaerobe.2009.09.001. [DOI] [PubMed] [Google Scholar]
- 29.Lanis JM, Barua S, Ballard JD. Variations in TcdB activity and the hypervirulence of emerging strains of Clostridium difficile. PLoS Pathog. 2010;6:e1001061. doi: 10.1371/journal.ppat.1001061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Scaria J, Ponnala L, Janvilisri T, Yan W, Mueller LA, Chang YF. Analysis of ultra-low genome conservation in Clostridium difficile. PLoS ONE. 2010;5:e15147. doi: 10.1371/journal.pone.0015147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Babcock GJ, Thomas WD, Broering TJ, et al. Human monoclonal antibodies neutralize toxins produced by epidemic strains of Clostridium difficile. 43rd Annual Meeting of the Infectious Diseases Society of America. Abstract 164. San Francisco, CA, 6–9 October 2005. [Google Scholar]
- 32.van den Berg RJ, Claas EC, Oyib DH, et al. Characterization of toxin A-negative, toxin B-positive Clostridium difficile isolates from outbreaks in different countries by amplified fragment length polymorphism and PCR ribotyping. J Clin Microbiol. 2004;42:1035–41. doi: 10.1128/JCM.42.3.1035-1041.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Johnson S, Sambol SP, Brazier JS, et al. International typing study of toxin A-negative, toxin B-positive Clostridium difficile variants. J Clin Microbiol. 2003;41:1543–7. doi: 10.1128/JCM.41.4.1543-1547.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sambol SP, Tang JK, Merrigan MM, Johnson S, Gerding DN. Infection of hamsters with epidemiologically important strains of Clostridium difficile. J Infect Dis. 2001;183:1760–6. doi: 10.1086/320736. [DOI] [PubMed] [Google Scholar]
- 35.Chaves-Olarte E, Low P, Freer E, et al. A novel cytotoxin from Clostridium difficile serogroup F is a functional hybrid between two other large clostridial cytotoxins. J Biol Chem. 1999;274:11046–52. doi: 10.1074/jbc.274.16.11046. [DOI] [PubMed] [Google Scholar]
- 36.Drudy D, Quinn T, O'Mahony R, Kyne L, O'Gaora P, Fanning S. High-level resistance to moxifloxacin and gatifloxacin associated with a novel mutation in gyrB in toxin-A-negative, toxin-B-positive Clostridium difficile. J Antimicrob Chemother. 2006;58:1264–7. doi: 10.1093/jac/dkl398. [DOI] [PubMed] [Google Scholar]
- 37.Goorhuis A, Legaria MC, van den Berg RJ, et al. Application of multiple-locus variable-number tandem-repeat analysis to determine clonal spread of toxin A-negative Clostridium difficile in a general hospital in Buenos Aires, Argentina. Clin Microbiol Infect. 2009;15:1080–6. doi: 10.1111/j.1469-0691.2009.02759.x. [DOI] [PubMed] [Google Scholar]
- 38.Huang H, Fang H, Weintraub A, Nord CE. Distinct ribotypes and rates of antimicrobial drug resistance in Clostridium difficile from Shanghai and Stockholm. Clin Microbiol Infect. 2009;15:1170–3. doi: 10.1111/j.1469-0691.2009.02992.x. [DOI] [PubMed] [Google Scholar]
- 39.Pituch H, Brazier JS, Obuch-Woszczatynski P, Wultanska D, Meisel-Mikolajczyk F, Luczak M. Prevalence and association of PCR ribotypes of Clostridium difficile isolated from symptomatic patients from Warsaw with macrolide-lincosamide-streptogramin B (MLSB) type resistance. J Med Microbiol. 2006;55(Pt 2):207–13. doi: 10.1099/jmm.0.46213-0. [DOI] [PubMed] [Google Scholar]
- 40.Chou TC. Derivation and properties of Michaelis-Menten type and Hill type equations for reference ligands. J Theor Biol. 1976;59:253–76. doi: 10.1016/0022-5193(76)90169-7. [DOI] [PubMed] [Google Scholar]
- 41.Lyras D, O'Connor JR, Howarth PM, et al. Toxin B is essential for virulence of Clostridium difficile. Nature. 2009;458:1176–9. doi: 10.1038/nature07822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kuehne SA, Cartman ST, Heap JT, Kelly ML, Cockayne A, Minton NP. The role of toxin A and toxin B in Clostridium difficile infection. Nature. 2010;467:711–3. doi: 10.1038/nature09397. [DOI] [PubMed] [Google Scholar]
- 43.Kink JA, Williams JA. Antibodies to recombinant Clostridium difficile toxins A and B are an effective treatment and prevent relapse of C. difficile-associated disease in a hamster model of infection. Infect Immun. 1998;66:2018–25. doi: 10.1128/iai.66.5.2018-2025.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.