

Abstract
Immunocytochemistry showed expression of aquaporin-1 (AQP1) water channels at sites involved in dietary fat processing, including intrahepatic cholangiocytes, gallbladder, pancreatic microvascular endothelium, and intestinal lacteals. To determine whether AQP1 has a role in dietary fat digestion and/or absorption, mice were placed on a diet that contained 50% fat. Whereas wild-type mice (3–3.5 wk of age, 10–12 g) gained 49 ± 5% (SE, n = 50) body weight in 8 days, and heterozygous mice gained 46 ± 4%, AQP1 null mice gained only 4 ± 3%; weights became similar after return to a 6% fat diet after 6 days. The null mice on a high-fat diet acquired an oily appearance, developed steatorrhea with increased stool triglyceride content, and manifested serum hypotriglyceri-demia. Supplementation of the high-fat diet with pancreatic enzymes partially corrected the decreased weight gain in null mice. Absorption of [14C]oleic acid from small intestine was not affected by AQP1 deletion, as determined by blood radio-activity after duodenal infusion. Lipase activity in feces and small intestine was remarkably greater in AQP1 null than wild-type mice on low- and high-fat diets. Fluid collections done in older mice (that are less sensitive to a high-fat diet) by ductal cannulation showed threefold increased pancreatic fluid flow in response to secretin/cholecystokinin, but volumes, pH, and amylase activities were affected little by AQP1 deletion, nor were bile flow rates and bile salt concentrations. Together, these results establish a dietary fat misprocessing defect in AQP1 null mice.
Keywords: water transport, bile, pancreas
LARGE QUANTITIES OF FLUID move across epithelial and endothelial cell barriers in the gastrointestinal tract (36). In humans, 1.5–2.0 liters of bicarbonate-rich fluid are secreted by the pancreas, and 0.5–1.0 liters of bile are secreted by the liver. Seven and one-half liters of fluid are absorbed from small intestine, and 1.3 liters of fluid from colon, to produce dehydrated feces. Various diseases are associated with dysregulation of gastrointestinal fluid balance, such as intestinal hypersecretion in cholera and pancreatic insufficiency and meconium ileus in cystic fibrosis. The gastrointestinal tract is second to kidney in terms of the total amount of transepithelial fluid secretion and absorption.
There is direct evidence that aquaporin-type molecular water channels have an important role in water transport in kidney, lung, and central nervous system (reviewed in Ref. 34). Aquaporins are small, membrane-associated transporting proteins that are expressed in many fluid transporting tissues, including organs of the gastrointestinal tract (reviewed in Ref. 17). Water channel aquaporin-1 (AQP1) is expressed at several sites that are potentially involved in dietary fat processing, including intrahepatic cholangiocytes (bile secretion), gallbladder (bile storage), pancreatic microvascular endothelium (pancreatic fluid/enzyme secretion), and intestinal lacteals (chylomicron absorption) (4, 10, 24). Secretin-regulated trafficking of AQP1 in cholangiocytes has been proposed to occur in bile secretion (19, 20). Water channel AQP4 is expressed in colonic epithelia (8) and gastric parietal cells (8, 21), AQP5 in salivary gland acini (23), and AQP8 and AQP9 in hepatocytes (14, 32). Recent studies in transgenic mice lacking specific aquaporins have implicated the involvement of AQP5 in saliva production (16).
Based on the expression pattern of AQP1 in the proximal gastrointestinal tract, we tested the hypothesis that AQP1 is required for dietary fat processing. AQP1 null mice were generated by targeted gene disruption (18). Except for mild growth retardation, the null mice are grossly phenotypically normal when given unrestricted access to water and standard mouse chow (6% fat). However, they become severely dehydrated when deprived of water because of a urinary concentrating defect resulting from defective proximal tubule absorption (27) and water permeability in thin descending limb of Henle (5) and outer medullary descending vasa recta (25). Fluid transport is also defective in lung (2, 15, 30), but the mice have apparently normal reproductive and neuromuscular function. We report here that AQP1 null mice do not process dietary fat normally, developing steatorrhea and a blunted weight gain response when fed a high-fat diet. To begin to define the mechanism of the defective dietary fat processing, we studied dietary pancreatic enzyme supplementation, analyzed stool fat, measured fecal lipase activity and intestinal oleic acid absorption, and developed a surgical model to collect and analyze pancreatic and biliary fluid secretions.
METHODS
Transgenic mice
AQP1 knockout mice were generated by targeted gene disruption as described previously (18). Heterozygous mice in a CD1 genetic background were bred, and offspring genotype was determined at age 5–7 days. The investigators were blinded to genotype information in dietary studies, stool analysis, oleic acid absorption, and surgical pancreatic/duodenal collection studies. Animal protocols were approved by the University of California, San Francisco (UCSF) and Stanford Committees on Animal Research.
Immunocytochemistry
Liver, pancreas, gallbladder, and small intestine were obtained from mice after systemic perfusion-fixation with 4% paraformaldehyde. Cryostat sections, prepared as described previously (8), were incubated for 30 min with PBS that contained 1% BSA, and then with AQP1 antiserum (1:1,000) for 1 h at 23°C in PBS that contained 1% BSA. Slides were rinsed with 2.7% NaCl and then PBS and incubated with a secondary Cy3-conjugated sheep anti-rabbit F(ab)2 fragment (1:200) for visualization by fluorescence microscopy.
Diet studies
Experiments were carried out on litter-matched wild-type (+/+), AQP1 heterozygous (+/−), and AQP1 knockout (−/−) mice (10–12 g initially, 4–6 days after weaning unless otherwise specified). Older mice were used in some studies. Three different commercial diets were used (Bioserve, Frenchtown, NJ): standard mouse chow (6% fat), high-fat diet containing 50% animal fat (product #F3785), and zero fat diet (product #F3784). In some experiments, the high-fat diet was freshly supplemented with 4 capsules Pancrease (McNeil Pharmaceutical) per 100 g chow, each capsule containing lipase (4,500 USP units), amylase (20,000 USP units), and protease (25,000 USP units). In some experiments, the high-fat diet was supplemented with 45 units of purified bacterial lipase per gram chow (Calbiochem, San Diego, CA). Mice were given free access to water and one of the above diets as their only source of solid food. Body weight was recorded every 2 days. In some experiments, food intake was matched daily by providing the wild-type mice the same amount of food consumed by null mice on the previous day. In some studies, blood was collected for measurement of serum triglyceride and cholesterol concentrations by the UCSF Clinical Laboratory.
Stool fat analysis
Stool samples were collected immediately after spontaneous defecation. Fecal fat content was analyzed qualitatively by Sudan VI staining (29) and semiquantitatively by organic extraction with heptane:diethylether:ethanol (1:1:1 vol/vol) and then twice with heptane: diethylether:ethanol:water (1:1:1:1) (12). Lipids were measured gravimetrically after the organic extractions and solvent evaporation.
Stool lipids were analyzed by thin-layer chromatography (TLC) as described previously (28). Stool samples (5–10 mg) were collected from individual mice and dried in a vacuum oven at 60°C for 1 h. The solid matter was extracted overnight with 0.5 ml of chloroform:methanol (2:1) at room temperature and then centrifuged. The supernatant was back-extracted with 0.5 ml water, and the organic phase was transferred to a preweighed centrifuge tube. The organic solvent was evaporated to dryness and further dried in a vacuum dessicator. Lipids were weighed and then dissolved in chloroform to a final concentration of 5 μg/μl. Two microliters of the sample were spotted onto a prewashed, preactivated silica gel plate (type K6, 20 × 20 cm, 250 μm thick; Whatman, NJ) along with the standards triolein, oleic acid, and cholesterol (Aldrich). The plate was developed by a two-stage one-dimensional chromatographic method in two different solvent mixtures: 1) chloroform (97%):methanol (2%):acetic acid (1%), and 2) hexane (96%):methanol (3%): acetic acid (1%). Lipids were visualized by charring the plate using a 3% cupric acetate: 8% phosphoric acid mixture and heating to 90°C.
Lipase activity
Fresh fecal samples (5–10 mg) obtained after spontaneous defecation or withdrawal from proximal small intestine were collected, immediately weighed, and placed on ice. The samples were mixed with 19 volumes (wt:vol) of ice-cold PBS and pipetted/vortexed to generate a uniform suspension. After centrifugation of the suspensions for 10 min at 15,000 g, 15 μl of each supernatant were assayed for lipase activity using a Lipase-PS kit (Sigma) according to the manufacturer’s instructions. Lipase activity was reported in units/liter original sample.
Intestinal absorption of [14C]oleic acid
Mice (10–12 g) were fasted overnight and anesthetized by halothane inhalation. After a midline incision, the stomach and duodenum were exposed, and 0.1 ml olive oil (ICN Biochemicals) containing 1 μCi [14C]oleic acid (56 mCi/mmol, Amersham) was infused into the duodenum through the pylorus using a 1-cc insulin syringe. After closing the abdomen by suture, the mice were returned to a clean cage with free access to water. All mice recovered within 1 min from anesthesia and did not show signs of distress. Blood samples (10 μl in duplicate) were collected from the tail at 30 min, 1 h, 1.5 h, 2 h, and 4 h. Mice were killed using an overdose of pentobarbital (150 mg/kg) after collection of the final blood sample. Radioactivity was counted using an LS 8000 Beckman scintillation counter.
Surgical collection of biliary and pancreatic secretions
Litter-matched wild-type and AQP1 knockout mice (30–35 g) were fasted overnight in wire-bottom cages and given free access to water containing 10% dextrose. Anesthesia was induced by inhalation of 3% isoflurane (0.5 l/min) in 100% oxygen and initially maintained on 1.0% isoflurane. Mice were warmed with a heating pad and overhead lamps. Rectal temperature (maintained at 37 ± 0.5°C), respiratory rate, and heart rate were monitored and recorded in 10-min intervals. Under high magnification, a low transverse neck incision was made, and the right jugular vein was cannulated with PE-10 tubing (outer diameter 0.6 mm). Lactated Ringer solution containing 5% dextrose was infused at 13 ml·kg−1·h−1. The right common carotid artery was dissected and cannulated with a PE-10 catheter filled with heparinized saline (200 U/ml) for blood pressure monitoring using a standard transducer and pressure monitor (Tektronix, Beaverton, OR). A PE-10 catheter was inserted into the left jugular vein for intravenous administration of lactated Ringer solution as needed to maintain mean arterial pressure >75 mmHg. All catheters were secured in place with 6-0 silk suture. The urinary bladder was also cannulated using PE-10 tubing through a midline abdominal incision.
After exposing the abdominal cavity, the common bile duct adjacent to the duodenum was cannulated for collection of pancreatic secretions. A silk ligature was placed around the common bile duct above the pancreas, and a second catheter was inserted into the proximal common bile duct to collect pure biliary secretions. The cystic duct was ligated to prevent emptying of gallbladder contents. The abdomen was then filled with warmed normal saline and the incision closed. Urine, bile, and pancreatic secretions were collected into preweighed 0.5-ml Eppendorf tubes for gravimetric volume determination. The pH of freshly collected fluid was measured using thin strips of pH-sensitive paper (pHydrion; Micro Essential Laboratory, Brooklyn, NY) viewed under high magnification. A total of six collections (each >30 min) of bile and pancreatic secretions were made: three collections of basal secretion followed by three collections of stimulated secretion during continuous infusion of cholecystokinin octapeptide (105 pmol·kg−1·h−1; Sigma) and secretin (22 pmol·kg−1·h−1; Calbiotech, Redwood City, CA). Samples were frozen at −20°C for subsequent assay of bile acid, amylase, and lipase concentrations. Lipase and amylase activities in pancreatic fluid were measured by the UCSF Clinical Laboratory using standard methods. Bile acid concentration was measured in biliary fluid using a bile acid assay kit (Sigma) according to the manufacturer’s instructions.
RESULTS
AQP1 was localized in mouse liver, gallbladder, pancreas, and small intestine by immunofluorescence using a rabbit anti-AQP1 antibody raised against purified human AQP1. As shown in Fig. 1, AQP1 was expressed strongly in the endothelium of duodenal central lacteals (Fig. 1A). Specific AQP1 fluorescence labeling was also seen in microvessels in gallbladder (Fig. 1B, left) and pancreas (Fig. 1C, left). In liver, AQP1 labeling was mainly present in canaliculi and small bile ducts (Fig. 1D, left). This expression pattern is consistent with the previous localization data in rat (10, 24). No specific staining was found in tissues from AQP1 null mice that were processed in parallel (Fig. 1, B–D, right).
Fig. 1.

Immunofluorescence localization of aquaporin-1 (AQP1) in mouse digestive organs. Duodenal sections (A, longitudinal, left; transverse, right) showing strong AQP1 immunostaining in central lacteals of wild-type mice. In gallbladder, AQP1 is expressed in submucosal microvessels of wild-type mice (B, left); no specific staining in AQP1 null mice (B, right). In pancreas, AQP1 labeling was most apparent in the interlobular vasculature (C, left; knockout control, right). In liver, AQP1 protein was detected in canaliculi and small bile ducts (D, left; knockout control, right).
To test the hypothesis that AQP1 plays a role in dietary fat processing, mice were placed on a diet containing 50% fat. As shown in Fig. 2A, body weight in wild-type and heterozygous mice increased rapidly, increasing by 49% in 8 days, while the AQP1 knockout mice grew little. All mice gained weight when returned to a regular (6%) fat diet on day 8. Whereas the wild-type mice appeared grossly healthy and active, the AQP1 null mice on a high-fat diet were relatively inactive and remarkably smaller than wild-type mice and acquired an oily exterior appearance (Fig. 2B).
Fig. 2.

Weight change and appearance of mice on a 50% fat diet. A: weight curves of wild-type (+/+), heterozygous (+/−), and AQP1 null (−/−) mice on a high-fat diet. A total of 50 mice were studied in each group in 8 independent sets of measurements. Initial mouse weight was 10–12 g. Each point is mean ± SE weight. Where indicated, the mice were switched to normal diet. B: photograph of mice on day 4 of a high-fat diet showing oily-appearing fur and smaller size of AQP1 null mice.
Soft feces were noted in the AQP1 null mice on a high-fat diet, suggesting steatorrhea. Sudan VI staining of feces from AQP1 null mice on a high-fat diet showed numerous fat globules (Fig. 3A, inset), which were rarely seen in wild-type mice on a high-fat diet, or mice of any genotype on a low-fat diet. Semi-quantitative analysis of stool fat content by a lipid extraction method showed elevated stool fat in AQP1 null mice vs. wild-type mice (Fig. 3A). TLC analysis of fecal lipids indicated more triglycerides in the stool of AQP1 null mice (Fig. 3B). The density ratio of triglyceride:free fatty acid was ~2.5-fold higher in AQP1 null than in wild-type mice (0.93 ± 0.13 vs. 0.36 ± 0.12 arbitrary units, means ± SE, P < 0.01), suggesting impaired triglyceride hydrolysis and/or absorption. After 3 days on a high-fat diet, serum triglyceride concentration was remarkably lower in the AQP1 null mice than in the wild-type mice, whereas serum cholesterol concentrations were similar (Fig. 3C). There was no significant difference in triglyceride and cholesterol concentrations in wild-type and null mice on a normal diet.
Fig. 3.

Fecal lipid analysis. A: semi-quantitative analysis of stool lipid content by an organic extraction method (means ± SE, n = 20; *P < 0.05; **P < 0.01). Inset: Sudan IV staining of stool fat from AQP1 null mice after 3 days on a 50% fat diet. B: fecal lipids from 6 wild-type and 6 AQP1 null mice on a high-fat diet analyzed by TLC as described in METHODS. Identical weights of total lipids were run on each lane. The indicated lanes contain the standards triolein (triglyceride), oleic acid (free fatty acid), and cholesterol. C: plasma triglyceride and cholesterol concentrations (means ± SE, n = 10; *P < 0.05) in mice before and after 3 days on a high-fat diet.
In the above studies, despite the development of steatorrhea, the AQP1 null mice consumed approximately one-half of the amount of high-fat food consumed by wild-type mice. To exclude the possibility that differences in food intake accounted for the failure of AQP1 null mice to gain weight, intake-matched diet studies were done as described in METHODS. Figure 4A shows that the differences in mouse weight persisted on a matched diet. As found above, the AQP1 null mice developed steatorrhea, whereas the wild-type mice did not. Figure 4B shows similar weight gains in wild-type and AQP1 null mice placed on a zero fat diet, suggesting that the null mice are able to effectively process dietary proteins and carbohydrates.
Fig. 4.

Time course of body weight (means ± SE) in response to an intake-matched 50% fat diet (A) or a zero fat diet (B). Ten mice were studied in each group. Initial mouse weight was 10–12 g.
To determine whether fat maldigestion contributes to the dietary fat misprocessing in the AQP1 null mice, the high-fat diet was supplemented with high doses of a clinical pancreatic enzyme mixture (Pancrease) used to treat fat maldigestion in diseases of pancreatic insufficiency such as cystic fibrosis. Figure 5 shows that pancreatic enzyme supplementation produced a partial correction of the dietary fat misprocessing in the AQP1 null mice. The weight gain of AQP1 null mice was intermediate between that found in null mice in the absence of supplementation and that in wild-type mice. Addition of Pancrease to the high-fat diet did not affect weight gain in wild-type mice (not shown). A smaller, though significant, correction of the fat misprocessing in AQP1 null mice was found on supplementation of the high-fat diet with bacterial lipase. These results suggest that fat maldigestion may contribute to the fat misprocessing. In an attempt to study fat malabsorption directly, mice were placed on a free fatty acid diet containing a 1:1 mixture of a zero fat diet and a mixture of oleic acid (75%), stearic acid (12.5%), and palmitic acid (12.5%). Unfortunately, both wild-type and AQP1 null mice consumed very little of this diet and lost weight (not shown), rendering the results noninformative.
Fig. 5.
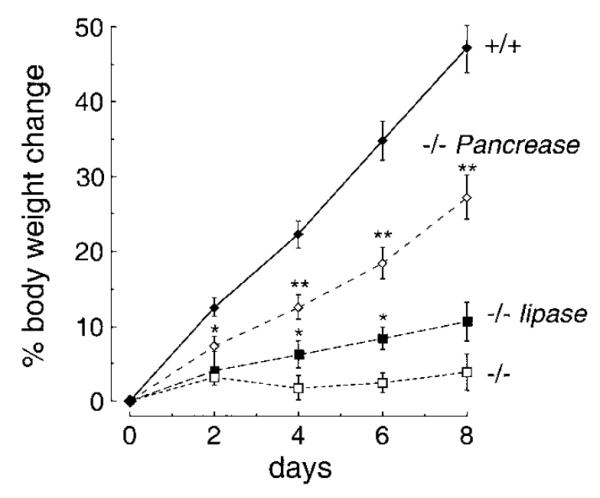
Dietary studies of fat maldigestion and malabsorption. Mice were placed on a high-fat diet as in Fig. 2. Where indicated, the diet was supplemented with Pancrease or purified lipase as described in methods. In each group, 25–30 mice were studied. Each point is mean ± SE body weight. *P < 0.05; **P < 0.01.
Lipase activity in the lumen of small intestine and in feces was measured to evaluate pancreatic exocrine insufficiency. Figure 6A shows that fecal lipase activity in AQP1 null mice was not lower, but nearly 10-fold higher than that in wild-type mice on a low-fat diet. After 3 days on a high-fat diet, fecal lipase concentrations strongly increased; however, fecal lipase concentration remained ~10-fold greater in the null mice. Lipase activity in the lumen of small intestine was also higher in the AQP1 null mice, suggesting that defective pancreatic lipase secretion is not responsible for the steatorrhea in the AQP1 null mice. The increased pancreatic lipase secretion in the AQP1 null mice is probably caused by increased fatty acid concentration in the duodenum, which triggers a neuroendocrine signal (9). To investigate the possibility that duodenal lipase is ineffective because of an abnormal pH, the pH of duodenal contents was measured in a series of mice on a low-fat diet. Figure 6B shows similar duodenal pH (6.5 ± 0.3 in −/− vs. 6.8 ± 0.2 in +/+ mice), suggesting that bicarbonate secretion in the AQP1 null mice is not impaired and that the environment in the duodenal lumen supports normal lipase function. The pH in distal ileum (7.8–8.0) was also not affected by AQP1 deletion.
Fig. 6.

Pancreatic exocrine function. A: lipase activity in feces and small intestinal lumen (means ± SE, 30 mice in each group. *P < 0.01, **P < 0.001). Samples were collected from weight-matched mice (10–12 g) before and after 3 days on a high-fat diet. B: duodenal and ileal pH measurements in mice (10–12 g) on a normal diet. Under pentobarbital anesthesia, the entire small intestine was exposed. The duodenum and distal ileum were transected, and 3 samples from the duodenum and ileum were measured to compute average pH. Data from 5 mice in each group (means ± SE).
Long-chain fatty acids are absorbed mainly by the lymphatic pathway involving the endothelium in intestinal central lacteals (7, 11). [14C]oleic acid absorption was measured as an indicator of long-chain fatty acid absorption. Figure 7 shows the time course of serum [14C]oleic acid radioactivity after a single infusion of olive oil containing [14C]oleic acid, as described in METHODS. No significant difference was found in [14C] oleic acid absorption in wild-type and AQP1 null mice. The apparently normal lipid uptake was a surprising observation, given the increased fecal lipase reported above; however, oleic acid absorption might not be a good model for triglyceride absorption.
Fig. 7.
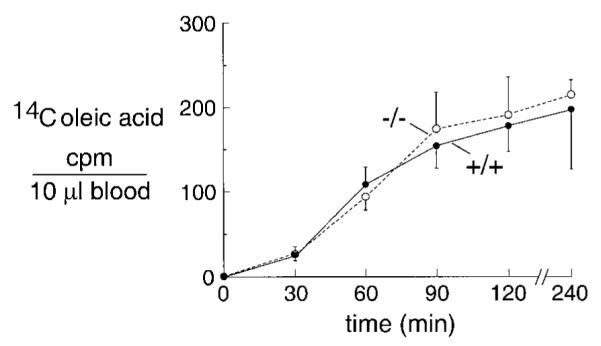
Intestinal oleic acid absorption. Time course of [14C]oleic acid absorption. Weight-matched young mice (10–12 g) were used for the study. Data are means ± SE, 6 mice in each group. cpm, Counts per minute.
As described in discussion, there are caveats in the interpretation of fecal lipase and [14C]oleic acid studies. To directly assess the role of AQP1 in biliary and pancreatic function, bile and pancreatic fluid collections were done in anesthetized adult mice by ductal cannulations as described in METHODS. Because of technical limitations, the bile and pancreatic fluid collections reported in Fig. 8 could be done only on large (35–40 g) mice, which manifest only a very mild fat misprocessing defect. Figure 8A shows that bile flow was not affected by AQP1 deletion, nor was pH (7.8–8.1) and bile salt concentration (7–12 mM). Also, bile flow was not increased in response to cholecystokinin (CCK)/secretin stimulation. In these studies, bile salt depletion was avoided by intravenous administration of taurocholate as reported previously (3), resulting in stable bile salt concentrations for >90 min. These results suggest that defective biliary function does not contribute to the dietary fat misprocessing in AQP1 null mice.
Fig. 8.

Bile and pancreatic fluid secretion. Fluids were collected in anesthetized mice by ductal cannulations (see methods). Where indicated, cholecystokinin (CCK) and secretin were infused intravenously. A: bile fluid analysis showing bile flow rate. B: pancreatic fluid flow rates. C: pancreatic lipase and amylase secretion rate. Data are means ± SE, 20 mice in each group. *P < 0.05.
In collections of pancreatic fluid done at the same time as the bile collections, AQP1 deletion did not affect the rate of pancreatic fluid secretion (Fig. 8B) nor the pH (8.1–8.4) of the pancreatic fluid (not shown). CCK/secretin stimulation significantly increased the pancreatic fluid secretion rate, however, there was no difference in stimulated fluid secretion rates in wild-type vs. AQP1 null mice. Interestingly, there was a small decrease in the secretion of pancreatic lipase and amylase in the AQP1 null mice (Fig. 8C).
Last, we studied the effects of mouse age on dietary fat processing. Older mice were able to tolerate a highfat diet much better than the younger mice (Fig. 9), producing smaller though significant differences in weight gain.
Fig. 9.

Effect of mouse age on dietary fat processing. Mice of indicated starting weights were placed on a 50% fat diet.
DISCUSSION
The principal finding of this study is that AQP1 deletion in mice causes a defect in dietary fat processing, particularly in young mice. The motivation for these studies was the expression of AQP1 at multiple sites in the gastrointestinal tract that participate in fat digestion and absorption. The fat processing machinery was challenged by placing the mice on a high-fat diet. The AQP1-deficient mice given a high-fat diet gained remarkably less weight than matched wild-type mice. The AQP1-deficient mice developed steatorrhea and acquired an oily appearance that may be related to inadequate self grooming. We speculate that dietary fat misprocessing might account for the mild growth retardation found in AQP1 null mice raised on standard (6% fat) mouse chow after weaning (18). We believe it is unlikely that defective urinary concentrating ability (27) or lung water balance (2, 15) could account for the retarded growth of AQP1 null mice; however, other phenotype differences cannot be ruled out, such as defective neuroendocrine or microvascular function.
There are multiple factors that independently or together could account for the dietary fat misprocessing in AQP1 null mice. Intrahepatic cholangiocytes lining the biliary are involved in bile formation, which plays a key role in fat digestion. These cells express AQP1, of which intracellular trafficking has been proposed to be cAMP regulated (19). Fat digestion also requires lipase and colipse that are secreted by the exocrine pancreas, as well as bicarbonate, to produce an appropriate pH in the duodenal lumen. The gallbladder participates in biliary fluid storage and expulsion in response to a fat bolus. The pancreas and gallbladder express AQP1 throughout their microvascular endothelia, and to a lesser extent, in selected epithelia. Endothelial cells in central lacteals of duodenum strongly express AQP1. The lacteals are involved in transport of chylomicrons produced by intestinal digestive processes (7, 31). AQP1 is also expressed in microvascular endothelia in esophagus and stomach, and to a lesser extent, in endothelia throughout the intestine.
The dietary studies and surgical fluid collections provide clues to the factors that might contribute to the fat misprocessing in AQP1 null mice. The partial correction of the fat processing defect by inclusion of pancreatic enzymes in the high-fat diet is consistent with a component of fat maldigestion to the observed dietary fat misprocessing. However, these results must be interpreted cautiously because of the small degree of correction obtained only at very high doses of pancreatic enzymes added to the high-fat diet. Furthermore, we do not know the degree of predigestion of the high-fat diet or whether the high doses of pancreatic enzymes produce other physiological effects in mice.
Several studies addressed the possibility of fat maldigestion in AQP1 null mice. AQP1 deletion was not associated with decreased biliary fluid secretion or bile salt concentration, at least in the larger adult mice. As reported previously in rat (1, 13), secretin did not increase biliary fluid secretion; secretin-stimulated bile secretion in mice appears to require the upregulation of intrahepatic cholangiocytes by chronic bile duct ligation. Our results suggest that defective bile production and secretion do not contribute to the fat processing defect and that AQP1 is not required for biliary function in mice. AQP1 deletion was not associated with decreased production of pancreatic fluid or altered pH in adult mice, suggesting little effect on pancreatic bicarbonate secretion. Alkalinity in the duodenal lumen was not impaired in young AQP1 null mice. There was a small decrease in the concentrations of lipase and amylase in the pancreatic fluid collected from adult mice, which probably does not contribute significantly to the dietary fat misprocessing. Rodents secrete substantial amounts of lingual lipase by the sublingual glands, but less in mice than rats (6); thus pancreatic lipase secretion in mice is probably required for dietary fat processing, especially when stressed by a 50% fat diet. In the young mice that manifested more severe dietary fat misprocessing, fecal and intestinal lipase activities were remarkably greater in AQP1 null mice than wild-type mice, suggesting a significant malabsorptive component to the dietary fat misprocessing. The increased fatty acid content in the duodenal lumen of AQP1 null mice probably provides the signal for increased pancreatic lipase secretion.
Studies were also done to address the possibility of fat maldigestion in AQP1 null mice. Mice were given a free fatty acid diet composed of oleic acid, stearic acid, and palmitic acid to bypass digestion. However, all mice lost weight substantially, so the results were noninformative. In a separate study, mice were infused with [14C]oleic acid into duodenum, and the time course of serum radioactivity was taken as a measure of fat absorption. No differences in the magnitude or the time course of radioactivity were found in the wild-type and AQP1 null mice. However, again, these results should be considered with caution because duodenal oleic acid absorption may not be the rate-limiting step under the experimental conditions, which differ substantially from natural feedings. In addition, there is a difference in oleic acid vs. triglyceride absorption.
What are the possible mechanisms by which genetic deletion of a water-transporting protein might influence dietary fat processing? AQP1 appears to transport water selectively without carrying small solutes such as ions, urea, and sugars (33). Evidence was reported that AQP1 might also transport carbon dioxide (22), and there may be other as yet unidentified functions of AQP1. However, our laboratory was unable to demonstrate CO2 movement through AQP1 in measurements on erythrocytes and lungs of wild-type vs. AQP1 null mice, as well as reconstituted proteoliposomes (35). AQP1 deletion might be associated with developmental or acquired structural abnormalities in gastrointestinal function. Based on studies of proximal tubule absorption in AQP1 null mice (27) and salivary gland fluid secretion in AQP5 null mice (16), the paradigm has emerged that high water permeability and aquaporins are required for active near-isosmolar fluid transport in epithelia (34). Because of AQP1 expression in relevant epithelial and endothelial cells in the gastrointestinal tract, defective secretion of fluid across cells in the biliary tract and pancreatic acini/ ducts could alter the quantity and/or composition of secreted fluids. For example, slowed fluid movement across pancreatic endothelia and acinar cells might alter the convective flow of pancreatic enzymes through the pancreatic duct. AQP1 expression in endothelial cells of intestinal lacteals might be involved in transient bidirectional movements of water in response to osmotic gradients created by entry of fluid from the duodenal lumen. Although these and additional mechanisms are possible, definition of the multifactorial mechanism(s) will be very challenging for a process with the complexity of fat processing.
Three Colton blood group null human females lacking functional AQP1 protein were identified and reported to be phenotypically normal (26). However, neither clinical histories nor laboratory analyses were done to evaluate the possibility of a urinary concentrating defect, dietary fat misprocessing, or other clinical abnormalities. There are many reasons why Colton null humans might not manifest overt fat malabsorption. The highly heterogeneous genetic background in humans would predict considerable variability in a complex phenotype such as dietary fat misprocessing. If dietary fat misprocessing in Colton null humans is age dependent as in AQP1 null mice, then problems may occur only in early life. Also, humans do not generally consume diets containing 50% fat, and dietary modifications would be made if steatorrhea and associated symptoms developed on consumption of a high-fat meal. Finally, it is recognized that there can be significant differences in organ physiology and aquaporin expression among mammalian species. It would be interesting to obtain dietary and gastrointestinal system histories from Colton null humans, as well as to evaluate the possibility of dietary fat misprocessing using established clinical protocols.
In summary, we have identified a dietary fat processing defect in AQP1 null mice that is most overt in young mice. The AQP1 null mice developed steatorrhea and remarkably increased fecal lipase. Several studies on fat malabsorptive vs. maldigestive mechanisms suggest that maldigestion may contribute more than malabsorption; however, the complexity of the system does not permit clear quantitation of the individual factors contributing to this interesting phenotype.
Acknowledgments
We thank Dr. Nicholas Larusso for helpful advice and Liman Qian for breeding and genotyping the mice.
This work was supported by National Institutes of Health Grants DK-35124, HL-59198, HL-60288, DK-43840, and DK-09526 and a National Cystic Fibrosis Foundation grant (award R613).
Information can be found at http://www.ucsf.edu/verklab.
REFERENCES
- 1.Alpini G, Lenzi R, Sarkozi L, Tavoloni N. Biliary physiology in rats with bile ductular cell hyperplasia. Evidence for a secretory function of proliferated bile ductules. J Clin Invest. 1988;81:569–578. doi: 10.1172/JCI113355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bai C, Fukuda N, Song Y, Ma T, Matthay MA, Verkman AS. Lung fluid transport in aquaporin-1 and aquaporin-4 knockout mice. J Clin Invest. 1999;103:555–561. doi: 10.1172/JCI4138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Berr F, Stellaard F, Goetz A, Hammer C, Paumgartner G. Ethinylestradiol stimulates a biliary cholesterol-phospholipid cosecretion mechanism in the hamster. Hepatology. 1988;8:619–624. doi: 10.1002/hep.1840080330. [DOI] [PubMed] [Google Scholar]
- 4.Bondy C, Chin E, Smith BL, Preston GM, Agre P. Developmental gene expression and tissue distribution of the CHIP28 water-channel protein. Proc Natl Acad Sci USA. 1993;90:4500–4504. doi: 10.1073/pnas.90.10.4500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chou CL, Knepper MA, van Hoek AN, Brown D, Yang B, Ma T, Verkman AS. Reduced water permeability and altered ultrastructure in thin descending limb of Henle in aquaporin-1 null mice. J Clin Invest. 1999;103:491–496. doi: 10.1172/JCI5704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.DeNigris SJ, Hamosh M, Kasbekar DK, Lee TC, Hamosh P. Lingual and gastric lipases: species differences in the origin of prepancreatic digestive lipases and in the localization of gastric lipase. Biochim Biophys Acta. 1988;959:38–45. doi: 10.1016/0005-2760(88)90147-6. [DOI] [PubMed] [Google Scholar]
- 7.Friedman HI, Nylund B. Intestinal fat digestion, absorption, and transport. A review. Am J Clin Nutr. 1980;33:1108–1139. doi: 10.1093/ajcn/33.5.1108. [DOI] [PubMed] [Google Scholar]
- 8.Frigeri A, Gropper M, Turck CW, Verkman AS. Immunolocalization of the mercurial-insensitive water channel and glycerol intrinsic protein in epithelial cell plasma membranes. Proc Natl Acad Sci USA. 1995;92:4328–4331. doi: 10.1073/pnas.92.10.4328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Green GM, Taguchi S, Friestman J, Chey WY, Liddle RA. Plasma secretin, CCK, and pancreatic secretion in response to dietary fat in the rat. Am J Physiol Gastrointest Liver Physiol. 1989;256:G1016–G1021. doi: 10.1152/ajpgi.1989.256.6.G1016. [DOI] [PubMed] [Google Scholar]
- 10.Hasegawa H, Lian SC, Finkbeiner WE, Verkman AS. Extrarenal tissue distribution of CHIP28 water channels by in situ hybridization and antibody staining. Am J Physiol Cell Physiol. 1994;266:C893–C903. doi: 10.1152/ajpcell.1994.266.4.C893. [DOI] [PubMed] [Google Scholar]
- 11.Hyun SA, Vahouny V, Treadwell CR. Portal absorption of fatty acids in lymph- and portal vein-cannulated rats. Biochim Biophys Acta. 1967;137:296–305. doi: 10.1016/0005-2760(67)90105-1. [DOI] [PubMed] [Google Scholar]
- 12.Jeejeebhoy KN, Ahmad S, Kozak G. Determination of fecal fats containing both medium and long chain triglycerides and fatty acids. Clin Biochem. 1970;3:157–163. [PubMed] [Google Scholar]
- 13.Kountouras J, McKavanagh S, Burmicky M, Billing BH. The effect of secretin on bile flow and bile acid and bilirubin excretion following relief of prolonged bile duct obstruction in the rat. J Hepatol. 1987;4:198–205. doi: 10.1016/s0168-8278(87)80080-6. [DOI] [PubMed] [Google Scholar]
- 14.Koyama Y, Yamamoto T, Kondo D, Funaki H, Yaoita E, Kawasaki K, Sato N, Hatakeyama K, Kihara I. Molecular cloning of a new aquaporin from rat pancreas and liver. J Biol Chem. 1997;272:30329–30333. doi: 10.1074/jbc.272.48.30329. [DOI] [PubMed] [Google Scholar]
- 15.Ma T, Fukuda N, Song Y, Matthay MA, Verkman AS. Lung fluid transport in aquaporin-5 knockout mice. J Clin Invest. 2000;105:93–100. doi: 10.1172/JCI8258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ma T, Song Y, Gillespie A, Carlson EJ, Epstein CJ, Verkman AS. Defective secretion of saliva in transgenic mice lacking aquaporin-5 water channels. J Biol Chem. 1999;274:20071–20074. doi: 10.1074/jbc.274.29.20071. [DOI] [PubMed] [Google Scholar]
- 17.Ma T, Verkman AS. Aquaporin water channels in gastrointestinal physiology. J Physiol (Lond) 1999;517:317–326. doi: 10.1111/j.1469-7793.1999.0317t.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ma T, Yang B, Gillespie A, Carlson EJ, Epstein CJ, Verkman AS. Severely impaired urinary concentrating ability in transgenic mice lacking aquaporin-1 water channels. J Biol Chem. 1998;273:4296–4299. doi: 10.1074/jbc.273.8.4296. [DOI] [PubMed] [Google Scholar]
- 19.Marinelli RA, Pham L, Agre P, LaRusso NF. Secretin promotes osmotic water transport in rat cholangiocytes by increasing aquaporin-1 water channels in plasma membrane. Evidence for a secretin-induced vesicular translocation of aquaporin-1. J Biol Chem. 1997;272:12984–12988. doi: 10.1074/jbc.272.20.12984. [DOI] [PubMed] [Google Scholar]
- 20.Marinelli RA, Tietz PS, Pham LD, Rueckert L, Agre P, LaRusso NF. Secretin induces the apical insertion of aquaporin-1 water channels in rat cholangiocytes. Am J Physiol Gastrointest Liver Physiol. 1999;276:G280–G286. doi: 10.1152/ajpgi.1999.276.1.G280. [DOI] [PubMed] [Google Scholar]
- 21.Misaka T, Abe K, Iwabuchi K, Kusakabe Y, Ichinose M, Miki K, Emori Y, Arai S. A water channel closely related to rat brain aquaporin 4 is expressed in acid- and pepsinogen-secretory cells of human stomach. FEBS Lett. 1996;381:208–212. doi: 10.1016/0014-5793(96)00092-0. [DOI] [PubMed] [Google Scholar]
- 22.Nakhoul NL, Davis BA, Romero MF, Boron WF. Effect of expressing the water channel aquaporin-1 on the CO2 permeability of Xenopus oocytes. Am J Physiol Cell Physiol. 1998;274:C543–C548. doi: 10.1152/ajpcell.1998.274.2.C543. [DOI] [PubMed] [Google Scholar]
- 23.Nielsen S, King LS, Christensen BM, Agre P. Aquaporins in complex tissues. II. Subcellular distribution in respiratory and glandular tissues of rat. Am J Physiol Cell Physiol. 1997;273:C1549–C1561. doi: 10.1152/ajpcell.1997.273.5.C1549. [DOI] [PubMed] [Google Scholar]
- 24.Nielsen S, Smith BI, Christensen EI, Agre P. Distribution of the aquaporin CHIP in secretory and resorptive epithelia and capillary endothelia. Proc Natl Acad Sci USA. 1993;90:7275–7279. doi: 10.1073/pnas.90.15.7275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pallone TL, Edwards A, Ma T, Silldorff EP, Verkman AS. Requirement of aquaporin-1 for NaCl-driven water transport across descending vasa recta. J Clin Invest. 2000;105:215–222. doi: 10.1172/JCI8214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Preston GM, Smith BL, Zeidel ML, Moulds JJ, Agre P. Mutations in aquaporin-1 in phenotypically normal humans without functional CHIP water channels. Science. 1994;265:1585–1587. doi: 10.1126/science.7521540. [DOI] [PubMed] [Google Scholar]
- 27.Schnermann J, Chou CL, Ma T, Traynor T, Knepper MA, Verkman AS. Defective proximal tubular fluid reabsorption in transgenic aquaporin-1 null mice. Proc Natl Acad Sci USA. 1998;95:9660–9664. doi: 10.1073/pnas.95.16.9660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schwarz M, Lund EG, Setchell KDR, Kayden HJ, Zerwekh JE, Bjorkhem I, Herz J, Russell DW. Disruption of cholesterol 7α-hydroxylase gene in mice. J Biol Chem. 1996;271:18024–18031. doi: 10.1074/jbc.271.30.18024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Simko V. Fecal fat microscopy. Acceptable predictive value in screening for steatorrhea. Am J Gastroenterol. 1981;75:204–208. [PubMed] [Google Scholar]
- 30.Song Y, Ma T, Matthay MA, Verkman AS. Role of aquaporin-4 in airspace-to-capillary water permeability in intact mouse lung measured by a novel gravimetric method. J Gen Physiol. 2000;115:17–27. doi: 10.1085/jgp.115.1.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tso P, Pitts V, Granger DN. Role of lymph flow in intestinal chylomicron transport. Am J Physiol Gastrointest Liver Physiol. 1985;249:G21–G28. doi: 10.1152/ajpgi.1985.249.1.G21. [DOI] [PubMed] [Google Scholar]
- 32.Tsukaguchi H, Shayakul C, Berger UV, Mackenzie B, Devidas S, Guggino WB, van Hoek AN, Hediger MA. Molecular characterization of a broad selectivity neutral solute channel. J Biol Chem. 1998;273:24737–24743. doi: 10.1074/jbc.273.38.24737. [DOI] [PubMed] [Google Scholar]
- 33.Verkman AS, Mitra AK. Structure and function of aquaporin water channels. Am J Physiol Renal Physiol. 2000;278:F13–F28. doi: 10.1152/ajprenal.2000.278.1.F13. [DOI] [PubMed] [Google Scholar]
- 34.Verkman AS, Yang B, Song Y, Manley G, Ma T. Role of water channels in fluid transport studied by phenotype analysis of aquaporin knockout mice. Exp Physiol. 2000;85S:233S–241S. doi: 10.1111/j.1469-445x.2000.tb00028.x. [DOI] [PubMed] [Google Scholar]
- 35.Yang B, Fukuda N, van Hoek AN, Matthay MA, Ma T, Verkman AS. Carbon dioxide permeability of aquaporin-1 measured in erythrocytes and lung of aquaporin-1 null mice and in reconstituted proteoliposomes. J Biol Chem. 2000;275:2686–2692. doi: 10.1074/jbc.275.4.2686. [DOI] [PubMed] [Google Scholar]
- 36.Zhang EB. In: Intestinal water and electrolyte transport. In: Gastrointestinal, Hepatobiliary, and Nutritional Physiology. Zhang EB, Sitrin MD, Black DB, editors. Lippincott-Raven; Philadelphia, PA: 1996. pp. 91–118. [Google Scholar]