

Abstract
Human leukocyte antigen-homozygous parthenogenetic stem cells (pSC) could provide a source of progenitors for regenerative medicine, lowering the need for immune suppression in patients. However, the high level of homozygosis and the lack of a paternal genome might pose a safety challenge for their therapeutic use, and no study so far has evaluated the spread and significance of gene expression changes across serial potency changes in these cells. We performed serial rounds of differentiation and reprogramming to assess pSC gene expression stability, likely of epigenetic source. We first derived pSC from activated MII oocytes, and differentiated them to parthenogenetic mesenchymal stem cells (pMSC). We then proceeded to induce pluripotency in pMSC by over expression of the four transcription factors Oct4, Sox2, Klf4 and c-Myc. pMSC-derived iPS (piPS) were further differentiated into secondary pMSC (pMSC-II). At every potency change, we characterized the obtained lines both molecularly and by functional differentiation, and performed an extensive genome-wide expression study by microarray analysis. Although overall gene expression of parthenogenetic cells resembled that of potency-matched biparental lines, significantly broader changes were brought about upon secondary differentiation of piPS to pMSC-II compared with matched biparental controls; our results highlight the effect of the interplay of epigenetic reprogramming on a monoparental background, as well as the importance of heterozygosis and biparental imprinting for stable epigenetic reprogramming.
INTRODUCTION
Parthenogenesis is a mode of reproduction that dispenses from the contribution of the paternal genome rarely used in vertebrates (1). Mammalian oocytes can develop parthenogenetically to implantation or beyond (2,3) and stem cell lines can be derived from them (3,4). Reprogramming by over expression of the transcription factors Oct4, Sox2, c-Myc and Klf4 resets the cell to an embryonic stem cell (ESC) like state. The induced pluripotent stem cells (iPS) thus obtained have characteristics similar to those of ESC. Some of the differences between ESC and iPS are found in discreet imprinted loci (5). Imprinting is an epigenetic mechanism by which a gene is expressed in a parent of origin way. Imprinted gene functions are related to growth and development, and imprinting alterations cause complex developmental syndromes, such as Angelman and Prader–Willi (6). Parthenogenetic stem cells (pSC) could be a welcome source of progenitors for regenerative medicine because of their mostly homozygous human leukocyte antigen (HLA) phenotype. However, the high level of homozygosis and the lack of a paternal genome might pose a safety challenge for their use, and no study so far has evaluated the spread and significance of gene expression variation across serial potency changes in these cells.
RESULTS
Cell lines generation and characterization
To derive the pSC line used in this study, 18 oocytes were activated. Of those, six (33%) reached the blastocyst stage and were seeded after removal of the zona pellucida. Four parthenotes attached to the feeder layer; one of them gave an outgrowth, which gave rise to a pSC line (derivation efficiency of 5.5%). The pSC line we derived was homozygotic at HLA loci, and maintained a stable immunological makeup during further differentiation and reprogramming rounds (Supplementary Material, Fig. S1).
To generate parthenogenetic iPS (piPS), we first differentiated the obtained pSC to a multipotent cell type. We elected parthenogenetic mesenchymal stem cells (pMSC) because the combination of immunophenotype and in vitro differentiation tests provides a clear assay to determine the cells identity.
Two different pMSC clonal lines were then reprogrammed by retroviral mediated delivery of four monocystronic constructs carrying the open reading frame of Oct4, Klf4, Sox2 and c-Myc. All experiments concerning differentiation and reprogramming parthenogenetic cells were carried out in parallel with a previously reported 46XX biparental line (ES[10], ES1 and hESC hereafter) (7). Moreover, in order to exclude that leftover pluripotent cells in the differentiated cultures gave rise to putative iPS, we cultured pMSC and MSC under ES condition in all experiment. None of these cultures gave rise to undifferentiated colonies. All pluripotent lines expressed pluripotency-related mRNA and proteins, underwent in vitro and in vivo differentiation and telomerase assay, and were karyotypically stable (Figs 1A, D and E, 2 and 3, and Supplementary Material, Figs S2–S4). We then proceeded to differentiate piPS to secondary pMSC (pMSC-II), and to analyze their expression pattern across the genome. Both pMSC and pMSC-II expressed to almost purity the surface markers CD90, CD73, CD105 and CD106, had a low percentage of CD34 and CD45 cells and were negative for the pluripotency-associated markers SSEA4 and TRA1-60, and could differentiate towards the chondro-, osteo- and adipogenic lineages (Fig. 1B and C, and Supplementary Material, Fig. S5).
Figure 1.
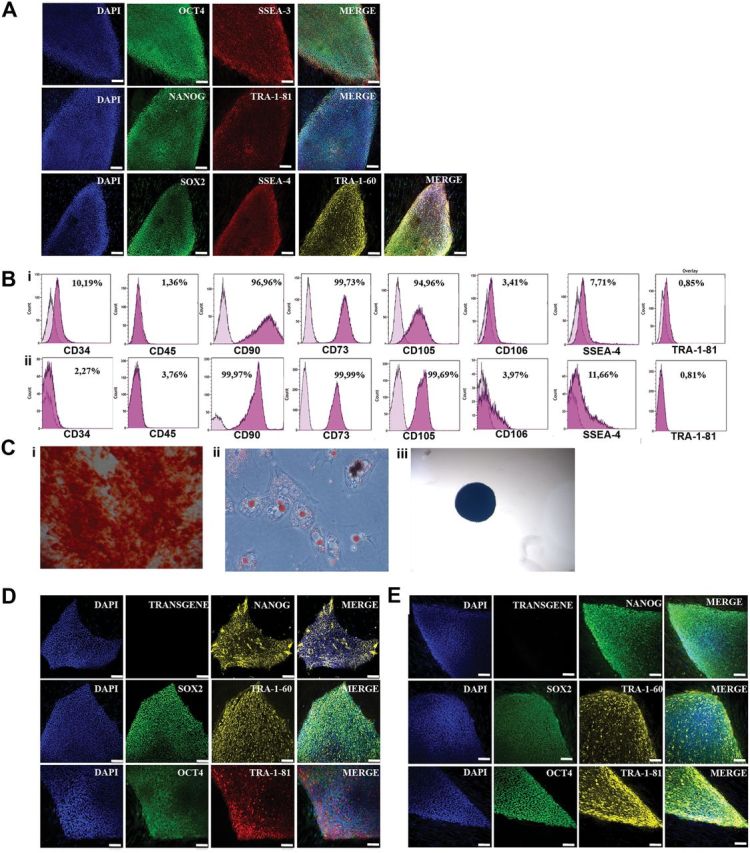
Characterization of parthenogenetic pluripotent cell lines. Expression of the pluripotency markers OCT4, SSEA3, NANOG, Tra-1-81, SOX2, SSEA4 and TRA-1-60 in hSC cells (A), piPS (D) and control biparental iPS (E). Expression of the surface markers CD34, CD45, CD90, CD73, CD105, CD106, SSEA4 and TRA-1-81 in differentiated mesenchymal progenitor pMSC (Bi) and pMSC-II (Bii), and their ability to differentiate towards osteogenic (Ci), adipogenic (Cii) and chondrogenic fate (Ciii). (F) HCA of array data. There is a clear first division in clustering between pluripotent lines (either parthenogenetic or biparental, either primary or induced pluripotent) and MSC progenitors (either parthenogenetic or biparental, either primary or secondary differentiations). Furthermore, within potency groups, the cell lines cluster along round of pluripotency (primary or secondary differentiation, and primary or induced pluripotency). (G) Methylation state of H19 and SNRPN promoters in representative pluripotent and mesenchymal progenitor cells of different parentality and origin.
Figure 2.
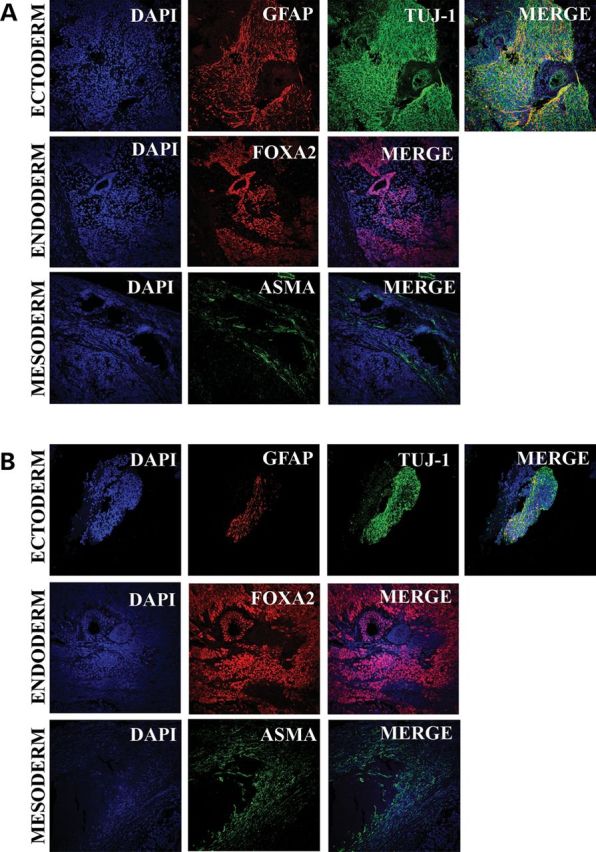
In vitro differentiation through EB formation towards ectoderm, endoderm and mesoderm of a representative biparental iPS line (A) and a representative piPS line (B).
Figure 3.
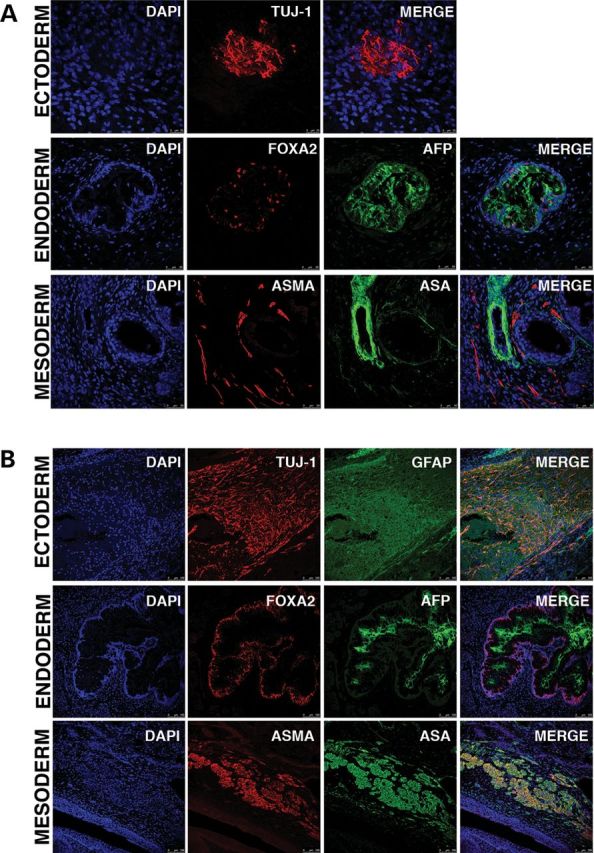
In vivo differentiation through teratoma formation towards ectoderm, endoderm and mesoderm of a representative biparental iPS line (A) and a representative piPS line (B).
Genome-wide changes in gene expression
After array normalization, all lines clustered correctly with similar samples by hierarchical cluster heat-map (HCA; Fig. 1F, Supplementary Material, Fig. S2 and Table S1). Moreover, pluripotent cells, of any origin, clustered more closely together than MSC, again of any origin. HCA identified correctly MSC derived from either hESC or iPS, and clustered them separately. Principal component analysis indicated that iPSs of different parental origin are closer among themselves than iPSs and hESC are; however, secondary MSC, of any parental origin, presented a more scattered pattern than primary MSC. We found small variations in the number of differentially expressed transcripts between parthenogenetic and biparental lines during the passage from pluripotent cells to MSC, to iPS, and to secondary MSC (Supplementary Material, Table S2); analysis of the transcriptional changes across successive time points indicated that 70–80% of genes expressed at each potency changes were shared among mono- and biparental lines. The array results were confirmed by real-time polymerase chain reaction (PCR) on 27 genes (Supplementary Material, Table S3).
During differentiation of pSC to pMSC (n = 2383), the most significant Gene Ontology (GO) Biological Process categories were second-level chromatin organization and DNA packaging (Supplementary Material, Table S1). In contrast, no specific GO enrichment was found for genes specifically expressed by biparental lines (n = 920).
Comparing genes differentially expressed in the analogous steps from piPS to pMSC-II and from biparental iPS to biparental MSC-II, a different picture emerges; first, genes specifically expressed in parthenogenetic lines (n = 2142) are mostly involved in transcription, regulation of transcription, patterning and regionalization. Secondly, GO enrichments are also found in biparental lines (n = 1745), representing functions related to DNA binding and transmembrane transport (Supplementary Material, Table S2). These data suggest that the effects of differentiation after reprogramming are stronger when acting on a parthenogenetic background.
To further dissect this issue, we evaluated the genes that are differentially expressed between biparental MSC and MSC-II, and found that the degree of overlap is very high, with just 165 transcript differentially expressed between the two cell populations. In contrast, the amount of difference between pMSC and pMSC-II was much wider, with 3013 transcripts differentially expressed. Of those 3013, just 92 (3%) overlapped with those seen in the biparental line. The remaining 2921 transcripts revealed an overrepresentation of transcription and transcriptional regulation (Supplementary Material, Table S2). This discrepancy between pMSC and pMSC-II further suggests that when parthenogenetic cells undergo reprogramming they accumulate epigenetic instability, which becomes apparent later during differentiation.
Imprinted gene expression and regulatory region methylation
Ninety-one (54%) of all imprinted genes expressed on the array (n = 169) were differentially expressed between hESC and pSC; 59 more highly expressed in hESC and 32 in pSC. Most of the confirmed imprinted genes over-expressed in hESC are expressed from the paternally inherited chromosome (14 of 19), with the highest fold changes seen in the Prader–Willi/Angelman Syndrome cluster on Ch15 (SNRPN, SNORD64, SNORD109A, MAGEL2, NDN). Interestingly, 5 of the 19 imprinted genes over-expressed in biparental hESC are paternally imprinted, and their expression should therefore not be down-regulated in parthenogenetic cells (TFPI2, CPA4, OSBPL5, ATP10A, ZNF264). Chromosomal location analysis identified that, apart for OSBPL5, all paternally imprinted genes in this group lay in imprinting clusters with other, maternally imprinted, components that are down-regulated in pSC, suggesting that local DNA cues might override parent of origin directions. Of the seven imprinted genes that are more highly expressed in pSC, six are, as expected, paternally imprinted (MEG3, H19, DLX5, PPP1R9A, SLC22A18, PHLDA2).
Next, we compared the methylation status of the differentially methylated region (Fig. 1G) across potency states of the imprinted genes SNRPN and H19. We found that both genes present a remarkably stable methylation pattern, which directly correlates with changes in expression seen among these two cell sources.
DISCUSSION
Reprogramming by induced pluripotency has been unfeasible with parthenogenetic mouse embryonic fibroblasts, and is in general less efficient in parthenogenetic versus biparental progenitors (8), possibly due to the precocious senescence of cells brought about by the lack of expression of the maternally imprinted, paternally expressed gene IGF2 (9). We therefore attempted to differentiate pSC to a progenitor cell type, rather than to a fully differentiated one, before attempting reprogramming. We have found that reprogramming efficiency and speed of pMSC is comparable to that of biparental MSC, confirming a recent result (10) in which fibroblast-like cells from ovarian teratomas were reprogrammed to pluripotency.
As many imprinted genes are involved in embryonic development, it is reasonable to assume that the monoparental imprinting plays a role in the observed difference. Although some paternally expressed gene clusters are completely abolished in all parthenogenetic lines tested, our results confirm the observation that in parthenogenetic iPS (10), maternally imprinted gene expression can be either down-regulated or be expressed at similar levels as in biparental cells. We found that this holds true for both pSC and piPS.
Superficially, parthenogenetic cells both reprogram and differentiate normally, however, mesenchymal progenitors derived from these cells present a significant amount of genes differentially expressed compared with biparental controls. Moreover, the accumulation of reprogramming and differentiation increases this aberration, as pMSC and pMSC-II have significantly fewer common genes than MCS and MSC-II do. Our results extend the observation that pluripotent parthenogenetic cells resemble their biparental counterparts, but a higher degree of difference is seen when the cells are differentiated (10). In summary, a significant amount of homozygosis and maternal only imprinting in human cells does not prevent reprogramming to pluripotency. However, differentiation after direct reprogramming of parthenogenetic cells brings about a wide array of transcriptional changes across the genome. Although said changes are compatible with grossly normal differentiation ability, our results serve as a caveat for the use of monoparental lines in cell therapy and regenerative medicine, offer insights into the efficiency of specific reprogramming strategies and aid in understanding the process of imprinted genes expression during development and differentiation.
MATERIALS AND METHODS
Generation of human parthenotes and pSC
All oocytes and embryos used in this study were obtained after signed informed consent of the donors or the couples, the approval of the Ethics Committee of the CMRB and the approval of the Comisión de Seguimiento y Control de la Donación de Células y Tejidos Humanos del Instituto de Salud Carlos III (project number: PI052847).
The oocytes used in this study were obtained after informed consent from healthy female donors already participating in a gamete donor program for reproductive purposes. The average oocyte donor age was 28 (range: 20–33). The oocytes were collected from preovulatory follicle by intravaginal ultrasound guided follicular punction after controlled ovarian hyperstimulation, and the cumulus oocytes complexes were allowed to recover for 2 h in collection medium G.1 (Vitrolife AB, Goteborg, Sweden) at 37°C and 6% CO2. Following this period, the cumulus cells were removed by exposure to a hyaluronidase solution (80 Ui/ml; Hyase, Vitrolife) and mild pipetting. Once denuded, the oocytes were visually evaluated, and only those arrested in the metaphase of the second meiotic division (MII) were allocated to this study. Immediately after inclusion in the study, MII oocytes were activated by exposure to the Ca+ ionophore Ionomycin (5 µm) for 5 min. The oocytes were washed extensively in the G.1 medium and exposed to 6-dymethyl amino purine (2 mm) for 4 h (11). At the end of the activation period, putative parthenotes were washed extensively and cultured in the G1/G2 media system. Eighteen hours after exposure to Ionomycin and in keeping with standard IVF practices, the activated oocytes were scored for the presence of pronuclei. Oocytes presenting 1 PN and 1 PB were considered successfully activated.
Five days from activation, the zona pellucida of the parthenotes that reached the blastocyst stage was removed by exposure to Pronase. Dezoned whole embryos were seeded over a feeder layer of gamma irradiated human foreskin fibroblasts (HFFs), and exposed to the SC derivation media. SC derivation media consisted of knock-out dulbecco's modified eagle medium (KO-DMEM) (Invitrogen, Carlsbad, CA, USA) supplemented with KO serum replacement (10%; Invitrogen), and basic fibroblast growth factor (bFGF) (10 ng/ml). Before using it to derive pSC, the SC derivation media were left overnight to condition by mitotically inactivated HFFs (CCD1112Sk ATCC, Manassas, VA, USA) and human ESC. This medium was changed every other day for the first 10 days of culture. Once the first colony of pES cells was passaged to a new plate and the cell line established, the medium was switched to the hES medium at 37°C and 5% CO2. The composition of the hES medium is: KO-DMEM (Invitrogen), 20% KO serum replacement (Invitrogen), Glutamax (2 mmol/l; Invitrogen), penicillin–streptomycin, non-essential amino acids (Lonza, Verviers, Belgium), 2-mercaptoethanol (0.05 mmol/l, Invitrogen) and bFGF (10 ng/m; Invitrogen) and was changed every 24 h. This system was also used for the culture of biparental hESC lines used as controls in all experiments. Stem cells were passaged mechanically onto new HFF layers every 7 days.
Retrovirus production for iPS generation
For retrovirus production, 4.3 × 106 Phoenix Amphotropic 293 cells (ATCC) were seeded in the DMEM (Invitrogen) medium containing 10% fetal bovine serum (FBS) (Invitrogen), Glutamax 2 mm, penicillin/streptomycin (100 U ml−1, 100 µg ml−1) in 100 mm culture dishes and placed in a 37°C 5% CO2 incubator. Next day, FuGENE6:DNA complexes were prepared according to the manufacturer’s instructions (Roche Applied Science). A ratio of 3 µg FuGENE to 1 µg plasmid DNA was used (pMSCV-based retroviral vectors are commercially available for OCT4, SOX2, KLF4 and c-Myc in Addgene, reference numbers: 20072, 20073, 20074 and 20075, respectively). Infection efficiency was monitored with pMSCV-based retroviral vector expressing green fluorescent protein (Addgene). Next day, the medium was changed and viral producing cells were cultured overnight at 32°C in a 5% CO2 incubator. Viral supernatants were collected every 12 h through a 0.45 µm polyvinylidene fluoride filter to remove any cells. Finally, 1 µl polybrene (10 mg ml−1; Chemicon) was added for each ml of viral supernatant.
Transduction of mesenchymal stem cells derived from human embryonic stem cells (sMSC) and parthenogenetic mesenchymal stem cells (pMSC)
Pools of 80 000 sMSC and pMSC were infected three times at 12 h intervals with retroviruses carrying polycistronic vectors and centrifuged at 750g at 32°C for 45 min. One day after the last infection cells were trypsinized and plated onto irradiated mouse embryonic fibroblasts or gelatin in hES medium. The hES medium: KO-DMEM, Glutamax (1 mm), penicillin–streptomycin, non-essential amino acids (100 μm), 2-mercaptoethanol (100 μm) and bFGF (10 ng ml−1).
Isolation and subculture of sMCS- and pMSC-derived iPS
Fourteen to 15 days after injection, small iPS colonies become visible from both MSC and pMSC sources. Twenty days after infection, large fast-growing reprogrammed colonies were identifiable. At least 10 iPS lines were established from both MSCs and pMSC cell sources. iPSs were picked mechanically, sub-cloned and cultured on irradiated human fibroblasts in the hES media.
Characterization of cell lines
The self-renewing ability of all cell lines described in this study has been characterized by immunohistochemistry and by real-time PCR. Cells were fixed in 4% paraformaldehyde and the following antibodies were used: OCT4 (Santa-Cruz Biotechnologies), sex-determining region Y-box 2 (Sox2; ABR), Nanog (Everest Biotech), Tumor Rejection Antigen 1 (TRA-1) TRA-1-60 (MAB), TRA-1-81 (MAB), SSEA-4 (Hybridoma Bank) and Stage Specific Embryonic Antigen 3 (SSEA-3; Hybridoma Bank). Direct alkaline phosphatase (AP) activity was analyzed using AP Blue/Red Membrane substrate solution kit (Sigma) according to the manufacturer's specification.
For the molecular characterization and microarray processing of all cell lines, total RNA was isolated using All Prep DNA/RNA columns (Qiagen), following manufacturer's guidelines. All RNA samples were treated with TURBO DNase inhibitor (Ambion, Foster City, CA, USA) to remove any residual genomic DNA. For real-time PCR, 1 µg of RNA was used to synthesize cDNA using the Invitrogen SuperScript II Reverse Transcriptase kit. Twenty-five nanograms of cDNA were used to quantify gene expression by using Platinum Syber Green qPCR Super Mix (Invitrogen) in an ABI Prism 7000 thermocycler (Applied Biosystems, Foster City, CA, USA) and primers as previously described (12), or designed and validated in house (Primer 3 Software). Thermal cycling was carried out with the following conditions: 2 min at 50°C, 10 min at 95°C, 15 s at 95°C and 1 min at 60°C for 40 cycles, plus dissociation step. Gene expression was normalized to the housekeeping gene glyceraldehyde 3-phosphate dehydrogenase, and fold change determined using the comparative Ct method.
HLA typing and microsatellite analysis
Molecular typing of cell lines was performed by Banc de Sang i Teixits (Barcelona, Spain). Microsatellite DNA analysis was performed using multiplex PCR of nine microsatellites and short tandem repeats plus amelogenin gene using AmplFlSTR Profiler Plus Kit (Applied Biosystems).
In vitro differentiation
For unguided in vitro differentiation, colonies of pluripotent cells were collected with a handmade glass instrument and left in suspension in low attachment plastic plates (Nunc) for 3 to 7 days. During this time, the media were gradually changed to EB media. EB media consist of KO-DMEM (Invitrogen) supplemented with 15% FBS (Hyclone), and 2 mm Glutamax (Invitrogen), 50 μm 2-b-mercaptoethanol (Invitrogen), 0.1 mm non-essential amino acids (Cambrex) and 100 U/ml penicillin/streptomycin (Invitrogen). After EBs formed, they were seeded on gelatin-coated slide flasks (Nunc) in the presence of either EB media (endoderm differentiation), EB medium supplemented with 0.5 mm L-ascorbic acid (Sigma) (mesoderm differentiation) or EB medium supplemented with 1% B2 and 0.5% N27 (ectoderm differentiation). The medium was replaced every 2 days. Two to three weeks after EB formation, slide flasks were washed in PBS, fixed at 4°C in 4% paraformaldehyde and processed for immunohistochemistry. The following antibodies were used: Alpha 1 fetoprotein (AFP; Dako), Forkhead Box Protein A2 (FOXA2; R&D System), neuron-specific class III beta-tubulin (Tuj1; Covance), Nestin (Hybridoma Bank), Glial fibrillary acidic protein (glial fibrillary acidic protein; Dako), Smooth Muscle Actin (Sigma), Sex-determining region Y-box 9 (Sox9; R&D Systems, Minneapolis, MN, USA) and Vimentin (Sigma).
Generation and characterization of MSCs
In order to guide the differentiation to mesenchymal stem cells (MSC) of any parentality, groups of 6–8 EBs were plated after formation on gelatine coated six-well plates in the EB medium. The plates were left to differentiate for 3 weeks, until a fibroblast-like population appeared. At this time, leftover EBs were aspirated from the plate, and adherent cells of fibroblast-like aspect were trypsinized and MSC status assessed by fluorescence activated cell sorting. Cells were stained with antibodies against CD34, CD45, CD73, and CD90, CD105, CD106 (BD). Moreover, cells were stained for the pluripotency-associated markers SSEA4 and TRA-1-60 to determine definitive exit from the pluripotent cell pool. Further, differentiation towards bone, cartilage and adipose cells was performed to further assess MSC functionality; MSCs derived from embryonic stem cells (sMSC) or pMSC were seeded at 50 000 cells/cm2 in αMEM (Invitrogen) with 10% fetal calf serum (FCS), 1% penicillin/streptomycin and 10 ng ml−1 bFGF (Sigma). Sub-confluent MSC were obtained within a week and passed at 5000 cells/cm2. Cells were used for generation of iPSs after a maximum of three passages.
In order to evaluate the differentiation capacity to osteogenic fate, cells were plated at 1000 cells/cm2 in osteogenic media [αMEM with 10% FCS, 1% antibiotics, 10 mm β-glicerolphosphate (Sigma), 0.2 mm ascorbate-2-phosphate (Sigma) and 0.01 µm dexamethasone (Sigma)]. Chondrogenic differentiation was performed with 200 000 pelleted cells suspended in the chondrogenic media [DMEM-high glucose (Invitrogen) with 1% antibiotics, 10 ng/ml TGF-β3 (R&D Systems), 50 mg ml−1 ITS+Premix (BD), 50 µg ml−1 proline (Sigma), 500 ng ml−1 BMP6 (R&D Systems), 50 µg ml−1 ascorbate-2-P and 0.1 µm dexamethasone]. For adipogenic differentiation, once cells reached 95–100% confluence, expansion media were replaced with adipogenic media [αMEM with 10% FCS, 1% antibiotics, 50 µM indomethacin (Sigma), 0.5 mm IBMX (Sigma) and 1 µm dexamethasone]. All differentiation protocols were maintained for ∼21 days. Analysis of cell differentiation was performed by histochemical staining by Oil red O (adipogenic), Alizarin red (osteogenic) and toluidine blue (chondrogenic) following previously described protocols.
In vivo differentiation
Severe combined immune deficient-Beige male mice, ∼8 weeks old, were injected with cell pellets of each pluripotent line described of 0.5–1 × 106 cells each in a volume not exceeding 150 µl. The injection sites were intratesticular and subcutaneous. Mice were sacrificed 8–10 weeks after the injections, or when a tumor became apparent, whichever came first. The tumor was fixed overnight at 4°C in 4% paraformaldehyde, and processed for immunohistochemistry as described for in vitro differentiation.
Promoter methylation assay
Genomic DNA was extracted from ∼1 000 000 cells using QIA AMP DNA Mini Kit (Qiagen). Purified DNA was mutagenized with Epitect (Qiagen), according to the manufacturers’ specifications. The promoter sequences for H19, SNRPN, MEST and NESPAS were amplified by either a nested PCR or two subsequent PCRs using previously described primers (13).
The resulting amplified products were cloned into pGEM T Easy plasmids, amplified in TOP10 chemically competent cells, purified and sequenced. Mutagenesis rates of >98%, excluding putative methylation sites, were considered acceptable.
Microarray processing and analysis
RNA integrity was assessed using Agilent 2100 Bioanalyzer (Agilent Technologies, Palo Alto, CA, USA). All RNA samples had high integrity (RNA integrity number ≥7.3) and were subsequently used in microarray experiments.
Amplification, labeling and hybridizations were performed according to the protocols from Ambion and Affymetrix. Briefly, 200 ng of total RNA was amplified using the Ambion® WT Expression Kit (Ambion/Applied Biosystems), labeled using the WT Terminal Labeling Kit (Affymetrix Inc., Santa Clara, CA, USA) and then hybridized to Human Gene 1.0 ST Array (Affymetrix; GEO accession number GSE37711) in a GeneChip® Hybridization Oven 640. Washing and scanning were performed using the Hybridization Wash and Stain Kit and the GeneChip® System of Affymetrix (GeneChip® Fluidics Station 450 and GeneChip® Scanner 3000 7G). Microarray data analysis was performed as follows: after quality control of raw data, data were background corrected, quartile-normalized and summarized to a gene level using the robust multi-chip average (14) obtaining a total of 28832 transcript clusters, excluding controls, which roughly correspond to genes. Only transcripts with an intensity signal of more than a 10% of all intensities of the mean of the studied groups and then over 50% of variance from total resting variance were considered for further analysis, which lead to 13 250 transcript clusters. Core annotations (version NetAffx 31, human genome 19) were used to summarize data into transcript clusters and to annotate analyzed data. Linear models for microarray (LIMMA) (15), a moderated t-statistics model, was used for detecting differentially expressed genes between the conditions in study and among different steps/times. Correction for multiple comparisons was performed using false discovery rate. Genes with an adjusted P-value <0.05 were selected as significant.
Hierarchical cluster analysis (HCA) was also performed to see how data aggregate and linear model for regression purposes. K-means was used for cluster analysis. All data analysis was performed in R (version 2.11.1) with packages aroma.affymetrix, Biobase, Affy, LIMMA and genefilter. Ingenuity Pathway Analysis v 9.0, (Ingenuity® Systems, www.ingenuity.com) was used to perform functional analysis of the results.
SUPPLEMENTARY MATERIAL
Supplementary Material is available at HMG online.
ACKNOWLEDGEMENTS
The authors wish to thank Ida Paramonov for technical assistance with data analysis, José Miguel Andrés Vaquero for assistance with flow cytometry, Lola Mulero Pérez, Cristina Pardo, Cristina Morera and Mercé Gaudes Martí for bioimaging assistance and Gustavo Tiscornia for helpful discussion.
Conflict of Interest statement. None declared.
FUNDING
N.M. was partially supported by Juan de la Cierva Program. This work was supported by grants to J.C.I.B. from MICINN, Tercel, Fundacion Cellex, Sanofi-Aventis and the G. Harold and Leila Y. Mathers Charitable Foundation.
REFERENCES
- 1.Feldheim K.A., Chapman D.D., Sweet D., Fitzpatrick S., Prodöhl P.A., Shivji M.S., Snowden B. Shark virgin birth produces multiple, viable offspring. J. Hered. 2010;101:374–377. doi: 10.1093/jhered/esp129. [DOI] [PubMed] [Google Scholar]
- 2.de Fried E.P., Ross P., Zang G., Divita A., Cunniff K., Denaday F., Salamone D., Kiessling A., Cibelli J. Human parthenogenetic blastocysts derived from noninseminated cryopreserved human oocytes. Fertil. Steril. 2008;89:943–947. doi: 10.1016/j.fertnstert.2007.04.045. [DOI] [PubMed] [Google Scholar]
- 3.Cibelli J.B., Grant K.A., Chapman K.B., Cunniff K., Worst T., Green H.L., Walker S.J., Gutin P.H., Vilner L., Tabar V., et al. Parthenogenetic stem cells in nonhuman primates. Science. 2002;295:819. doi: 10.1126/science.1065637. [DOI] [PubMed] [Google Scholar]
- 4.Lin G., OuYang Q., Zhou X., Gu Y., Yuan D., Li W., Liu G., Liu T., Lu G. A highly homozygous and parthenogenetic human embryonic stem cell line derived from a one-pronuclear oocyte following in vitro fertilization procedure. Cell Res. 2007;17:999–1007. doi: 10.1038/cr.2007.97. [DOI] [PubMed] [Google Scholar]
- 5.Stadtfeld M., Apostolou E., Akutsu H., Fukuda A., Follett P., Natesan S., Kono T., Shioda T., Hochedlinger K. Aberrant silencing of imprinted genes on chromosome 12qF1 in mouse induced pluripotent stem cells. Nature. 2010;465:175–181. doi: 10.1038/nature09017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Buiting K. Prader-Willi syndrome and Angelman syndrome. Am. J. Med. Genet. 2010;154C:365–376. doi: 10.1002/ajmg.c.30273. [DOI] [PubMed] [Google Scholar]
- 7.Aran B., Rodriguez-Piza I., Raya A., Consiglio A., Muñoz Y., Barri P.N., Izpisúa J.C., Veiga A. Derivation of human embryonic stem cells at the Center of Regenerative Medicine in Barcelona. In Vitro Cell Dev. 2010;46:356–366. doi: 10.1007/s11626-010-9288-0. [DOI] [PubMed] [Google Scholar]
- 8.Do J.T., Joo J.Y., Han D.W., Araúzo-Bravo M.J., Kim M.J., Greber B., Zaehres H., Sobek-Klocke I., Chung H.M., Schöler H.R. Generation of parthenogenetic induced pluripotent stem cells from parthenogenetic neural stem cells. Stem Cells. 2009;27:2962–2968. doi: 10.1002/stem.233. [DOI] [PubMed] [Google Scholar]
- 9.Hernandez L., Kozlov S., Piras G., Stewart C.L. Paternal and maternal genomes confer opposite effects on proliferation, cell-cycle length, senescence, and tumor formation. Proc. Natl Acad. Sci. 2003;100:13344–13349. doi: 10.1073/pnas.2234026100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Stelzer Y., Yanuka O., Benvenisty N. Global analysis of parental imprinting in human parthenogenetic induced pluripotent stem cells. Nat. Struct. Mol. Biol. 2011;18:735–741. doi: 10.1038/nsmb.2050. [DOI] [PubMed] [Google Scholar]
- 11.Paffoni A., Brevini T.A., Somigliana E., Restelli L., Gandolfi F., Ragni G. In vitro development of human oocytes after parthenogenetic activation or intracytoplasmic sperm injection. Fertil. Steril. 2007;87:77–82. doi: 10.1016/j.fertnstert.2006.05.063. [DOI] [PubMed] [Google Scholar]
- 12.Aasen T., Raya A., Barrero M.J., Garreta E., Consiglio A., Gonzalez F., Vassena R., Bilić J., Pekarik V., Tiscornia G., et al. Efficient and rapid generation of induced pluripotent stem cells from human keratinocytes. Nat. Biotechnol. 2008;26:1276–1284. doi: 10.1038/nbt.1503. [DOI] [PubMed] [Google Scholar]
- 13.Park T.S., Galic Z., Conway A.E., Lindgren A., van Handel B.J., Magnusson M., Rcihter L., Teitell M.A., Mikkola H.K., Lowry W.E., Plath K., Clark A.T. Derivation of primordial germ cells from human embryonic and induced plurpotent stem cells is significantly improved by coculture with human fetal gonadal cells. Stem Cells. 2009;27:783–795. doi: 10.1002/stem.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Irizarry R.A., Bolstad B.M., Collin F., Cope L.M., Hobbs B., Speed T.P. Summaries of Affymetrix GeneChip probe level data. Nucleic Acids Res. 2003;31:e15. doi: 10.1093/nar/gng015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Smyth G.K. Linear models and empirical Bayes methods for assessing differential expression in microarray experiments. Stat. Appl. Genet. Mol. 2004;3 doi: 10.2202/1544-6115.1027. Article3. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.