

Abstract
Background
The marginal seas of northwestern Pacific are characterized by unique topography and intricate hydrology. Two hypotheses have been proposed to explain genetic patterns of marine species inhabiting the region: the historical glaciations hypothesis suggests population genetic divergence between sea basins, whereas the Changjiang River outflow hypothesis suggests genetic break in line with the Changjiang Estuary. Here the phylogeography of bivalve Cyclina sinensis was investigated to test the validity of these two hypotheses for intertidal species in three marginal seas—the East China Sea (ECS), the South China Sea (SCS), and the Japan Sea (JPS).
Methodology/Principal Findings
Fragments of two markers (mitochondrial COI and nuclear ITS-1) were sequenced for 335 individuals collected from 21 populations. Significant pairwise ΦST were observed between different marginal sea populations. Network analyses illustrated restricted distribution of haplogroups/sub-haplogroups to sea basins, with a narrow secondary contact zone between the ECS and SCS. Demographic expansion was inferred for ECS and SCS lineages using mismatch distributions, neutral tests, and extended Bayesian Skyline Plots. Based on a molecular clock method, the divergence times among COI lineages were estimated dating from the Pleistocene.
Conclusions
The phylogeographical break revealed for C. sinensis populations is congruent with the historical isolation of sea basins rather than the putative Changjiang River outflow barrier. The large land bridges extending between seas during glaciation allowed accumulation of mutations and subsequently gave rise to deep divergent lineages. The low-dispersal capacity of the clam and coastal oceanography may facilitate the maintenance of the historical patterns as barriers shift. Our study supports the historical glaciations hypothesis for interpreting present-day phylogeographical patterns of C. sinensis, and highlights the importance of historical glaciations for generating genetic structure of marine coastal species especially those with low-dispersal abilities in northwestern Pacific.
Introduction
A series of marginal seas that has historically connected and isolated Asia from the Pacific Ocean contributes to the intricate geography of the northwestern Pacific [1]. When the last Pleistocene glaciers advanced, the sea level in the East China Sea (ECS) declined about 130–150 m than the present level while the South China Sea (SCS) was 100–120 m lower [2]. The ECS was reduced to an elongated trough (the Okinawa Trough) and the SCS became an enclosed inland sea connected to the Pacific through the Bashi Strait (Fig. 1). The ECS and SCS were thus isolated due to a large bridge extending from eastern China to Taiwan Island [3]. The Japan Sea (JPS) and ECS were also separated from each other when the Tsushima Strait was almost exposed (Fig. 1).
Figure 1. Map of East Asia showing the sampling sites of Cyclina sinensis and the coastal currents in summer.

Populations are labelled with numbers as shown in Table 1. Different symbols are used to represent populations from three marginal seas: circles, East China Sea populations; triangles, South China Sea populations; square, Japan Sea population. Shaded sea areas indicate regions < 120 m depth that would have been exposed during periods of low sea level. WKCC, West Korea Coastal Current; SCC, Subei Coastal Current; CCC, China Coastal Current; CDW, Changjiang diluted water.
The historical separation of the sea basins was reported to have dramatically influenced the current genetic distribution of various marine species [4]–[7]. A general phylogeographical pattern resulting from the historical glaciation hypothesis is concluded [5]: three marginal seas had served as separate refugia and accumulated substantial genetic differentiation during glacial periods; two secondary contact zones were formed through postglacial expansion of previously isolated lineages at the adjacent region (ECS/JPS and ECS/SCS). The intertidal species are particularly expected to be impacted by sea level changes as they experienced a direct loss of habitat from ice scouring (moving sea ice ridges can scour the seabed in shallow waters) [8]. These species are likely to exhibit notable genetic differentiation between separate refugial populations, and maintain the signature of historical patterns over long time-scales as barriers shift [9].
However, the simple picture of genetic distribution drawn from climate changes may be obscured by other environmental factors. Oceanic currents operating during the pelagic phase theoretically facilitate the dispersal of larval and enhance high population connectivity [10], [11]. In summer, the Subei Coastal Current (SCC) brings low salinity water southward along the ECS coast, and the China Coastal Current (CCC) flows northward from the SCS into the ECS [12]. Meanwhile, the West Korea Coastal Current (WKCC) connects the ECS and JPS (Fig. 1). Another dominant factor is the Changjiang River (also known as the Yangtze River, Fig. 1) outflow, a putative physical barrier limiting gene flow of coastal species in the region [13]. As the third largest river in the world, the river pours into the ECS with an average annual discharge of 924 billion m3 [12]. The huge freshwater outflow named Changjiang diluted water (CDW) was reported to have profoundly influenced the hydrological and ecological features of the ECS [14], [15]. The CDW has been assumed as a major barrier for genetic arrangements of various marine species, including gastropod Cellana toreuma [13], bivalve Cyclina sinensis [16], and two varieties of Sargassum [17]. In fact, the Changjiang Estuary was assumed to be a biogeographical boundary for marine molluscan fauna a half-century ago [18]. However, Xu [19] pointed out that the boundary was not insurmountable for some bivalve species with broad temperature tolerance (such as C. sinensis), and they had the abilities to transgress the barrier and dispersed over a wide range. Thereby, the barrier effect of the Changjiang River outflow on gene flow of intertidal species (the Changjiang River outflow hypothesis) is still a controversial issue, and additional data are needed to appraise the influence cautiously.
The bivalve C. sinensis, widely distributed in East Asia, provides an ideal case with which to address the relative influence of historical glaciations and the Changjiang River outflow on the genetic structure of intertidal species. The two factors may affect population structure of C. sinensis on different temporal and spatial scales, resulting in different patterns with regard to group partitioning and location of secondary contact zones. This bivalve is considered a euryhaline species because it inhabits broad salinity ranges from muddy sand beaches in the intertidal zone to estuaries where freshwater enters sea [20]. Each female may release 30 000–40 000 oocytes at once during the main breeding season from July to September. The clam has relatively short planktonic larval duration (PLD), spending an average of 7 days in water column before settlement and metamorphoses [21]. Adult individuals of C. sinensis have home ranges no wider than 80 m [22]. These low-dispersal characteristics make C. sinensis particularly suitable to disentangle the complex interactions of various factors on genetic structure in marginal seas. Indeed, several studies have emerged with aims to reveal the genetic distribution of C. sinensis [16], [23]–[25]. Nevertheless, these studies are suffered from insufficient sampling with restricted spatial coverage that can lead to erroneous conclusions regarding patterns of differentiation, and contradictory results have been reported. Yao et al. [24] using random amplified polymorphic DNA found that populations from two sides of the Changjiang River showed close genetic relationship, while Zhao et al. [25] using amplified fragment length polymorphism revealed significant genetic differentiation in line with the river. This discordance may partially result from the application of genetic markers with different levels of resolution for genetic differences.
In the present study, we surveyed the phylogeographical structure of C. sinensis in three marginal seas (ECS, SCS, and JPS) of East Asia by intensive sampling with an emphasis on the coast of China. Based on the analysis of mitochondrial cytochrome oxidase I (COI) and nuclear ribosomal internal transcribed spacers 1 (ITS-1) markers variation, we specifically aimed at elucidating the detailed genetic patterns, and testing the validity of the historical glaciations and Changjiang River outflow hypotheses for the clam. By addressing these issues, we expect to gain better understanding of the genetic distribution as well as evolutionary mechanisms for marine coastal species in the northwestern Pacific.
Materials and Methods
Sample collection
All the specimens of wild Cyclina sinensis were collected between October 2005 and January 2010. We obtained 335 individuals from 21 localities representing the geographical distribution of this species in East Asia, with a range of 5–23 individuals per site (Fig. 1 and Table 1). The adductor muscle was incised from samples and stored in 95% ethanol immediately, except for specimens from five sites (Lvshun, Tianjin, Weifang, Xiamen, and Maoming) which were frozen at −30°C until DNA extraction. Total genomic DNA was isolated from muscle tissue by a modification of standard phenol-chloroform purification procedure described by Li et al. [26]. As C. sinensis is not an endangered or protected species and collections were only carried on from public access areas, no specific permits were required to collect this species from these locations/activities.
Table 1. Sampling information and diversity indices for 21 populations of Cyclina sinensis.
| Sampling site | Abbr. | COI | ITS-1 | ||||||||
| NC | nC | hC | πC | kC | NI | nI | hI | πI | kI | ||
| East China Sea | |||||||||||
| 1 Dandong | DD | 14 | 7 | 0.758 | 0.0024 | 1.512 | |||||
| 2 Lvshun | LS | 18 | 4 | 0.634 | 0.0026 | 1.598 | |||||
| 3 Panjin | PJ | 12 | 2 | 0.167 | 0.0003 | 0.174 | |||||
| 4 Qinhuangdao | QHD | 20 | 5 | 0.716 | 0.0021 | 1.326 | |||||
| 5 Tianjin | TJ | 18 | 8 | 0.778 | 0.0023 | 1.411 | |||||
| 6 Weifang | WF | 19 | 6 | 0.743 | 0.0022 | 1.368 | |||||
| 7 Haiyang | HY | 14 | 7 | 0.824 | 0.0029 | 1.790 | |||||
| 8 Jimo | JM | 15 | 6 | 0.762 | 0.0023 | 1.411 | 8 | 8 | 1.000 | 0.0073 | 3.898 |
| 9 Lianyungang | LYG | 15 | 8 | 0.838 | 0.0035 | 2.157 | 8 | 8 | 1.000 | 0.0058 | 3.078 |
| 10 Xiangshui | XS | 11 | 6 | 0.855 | 0.0044 | 2.768 | |||||
| 11 Qidong | QD | 13 | 3 | 0.295 | 0.0014 | 0.875 | 10 | 10 | 1.000 | 0.0129 | 6.810 |
| 12 Shengsi | SS | 5 | 4 | 0.900 | 0.0035 | 2.245 | |||||
| 13 Zhoushan | ZS | 10 | 4 | 0.778 | 0.0018 | 1.130 | 10 | 10 | 1.000 | 0.0121 | 6.391 |
| 14 Wenzhou | WZ | 14 | 7 | 0.813 | 0.0027 | 1.706 | 7 | 6 | 0.952 | 0.0039 | 2.092 |
| 15 Xiapu | XP | 23 | 7 | 0.783 | 0.0029 | 1.808 | 10 | 10 | 1.000 | 0.0087 | 4.617 |
| 16 Xiamen | XM | 17 | 3 | 0.324 | 0.0007 | 0.431 | 9 | 7 | 0.944 | 0.0485 | 25.64 |
| South China Sea | |||||||||||
| 17 Maoming | MM | 17 | 1 | 0.000 | 0.0000 | 0.000 | 10 | 10 | 1.000 | 0.0105 | 5.236 |
| 18 Beihai | BH | 17 | 2 | 0.118 | 0.0002 | 0.129 | 9 | 8 | 0.972 | 0.0082 | 4.053 |
| 19 Dongxing | DX | 19 | 5 | 0.591 | 0.0011 | 0.695 | 7 | 6 | 0.952 | 0.0112 | 5.617 |
| 20 Sanya | SY | 21 | 2 | 0.095 | 0.0002 | 0.107 | 10 | 10 | 1.000 | 0.0087 | 4.344 |
| Japan Sea | |||||||||||
| 21 Fukuoka | FU | 23 | 3 | 0.170 | 0.0030 | 0.174 | 12 | 10 | 0.970 | 0.0030 | 1.497 |
Number of individuals sampled per site (N), number of haplotype (n), haplotype diversity (h), nucleotide diversity (π), and mean number of pairwise differences (k) were shown for each population. Subscripts indicate variables for COI or ITS-1. Abbr, site abbreviation.
Sequence acquisition
A fragment of the COI gene was amplified for all samples with the universal primers of Folmer et al. [27]. Each polymerase chain reaction (PCR) was carried out in 50- µL volumes containing 2 U Taq DNA polymerase (Takara Co.), about 100 ng template DNA, 0.25 µM of each primer, 0.2 mM dNTPs, 1×PCR buffer and 2 mM MgCl2. The PCR amplification was performed on a GeneAmp® 9700 PCR System (Applied Biosystems) based on following conditions: initial denaturation at 94°C for 3 min, followed by 35 cycles of denaturation at 94°C for 1 min, annealing at 50°C for 1 min, and extension at 72°C for 1 min, and a final extension at 72°C for 5 min.
To confirm the COI results, the ITS-1 region was amplified for 110 individuals from 12 populations (Table 1) using the ITS1-a and ITS1-b primers described in Gaffney et al. [28]. Considering the intensive sampling and patterns revealed by COI in ECS, not all populations in ECS were sequenced for ITS gene (individuals in sites 1–7 and 12 were excluded). The PCR cocktails and conditions were as described above, except for the primer set and the annealing temperature (52°C for ITS-1). Amplification products were confirmed by 1.5% TBE agarose gel electrophoresis stained with ethidium bromide. The cleaned product was prepared for sequencing using the BigDye Terminator Cycle Sequencing Kit (ver. 3.1, Applied Biosystems) and sequenced on an ABI PRISM 3730 (Applied Biosystems) automatic sequencer. Multiple peaks of ITS-1 amplifications (suggesting the presence of more than one allele) were rare for this species as seen in some other organisms such as bivalves Acar [29] and velvet ant [30].
Sequence variation
Sequences of COI and ITS-1 were assembled and edited by the DNASTAR software (DNASTAR, Inc.) separately, then aligned with CLUSTAL_X 1.81 using default settings [31], and finally rechecked by eye. The number of haplotypes for each locus was determined with the software program DnaSP v5 [32]. Sites with gaps in the ITS-1 sequences were considered as we thought haplotypes separated by an indel event to be different. All sequences of haplotypes were deposited in GenBank database (COI accession numbers: GU078392-GU078429, HM021146-HM021149; ITS-1 accession numbers: HQ232852-HQ232945). ARLEQUIN 3.5 software package [33] was applied to calculate molecular diversity indices such as haplotype diversity (h), nucleotide diversity (π), and mean number of pairwise differences (k). The evolutionary model that best fitted the two loci was determined with jModelTest [34] using the Akaike information criterion (AIC). HKY + I and HKY + G were chosen as the best-fit model for COI and ITS-1 loci respectively and used in the subsequent analyses.
Phylogenetic analyses
Networks of haplotypes were constructed with 95% maximum parsimony threshold in TCS version 1.21 [35]. Gaps in ITS-1 haplotypes were treated as a fifth character state. While it is more desirable to treat indels as additional coded characters than as a fifth state, TCS does not currently support the inclusion of symbols in the data file. A fixed connection limit at 50 steps was used to ITS-1 haplotypes in cases of they were too distant for connection in a network.
To confirm the phylogenies among haplotypes, Bayesian inferences (BI) were performed in MrBayes version 3.1 [36]. The Markov-chain Monte Carlo search was run with four chains for 8 million (COI) and 20 million (ITS-1) generations with sampling frequency of 1/1000 trees. The gap information for ITS-1 sequences was encoded by the simple insertion/deletion coding [37], and the standard discrete model was used to this binary sequence data [38]. When the standard deviation of split frequencies was less than 0.01 at 3 million (COI) and 12 million (ITS-1) generations, parameter stationarity was achieved. Trees sampled prior to stationarity were discarded and then a 50% majority-rule consensus tree with branch lengths was constructed with the remaining trees. The net average genetic distance between major lineages was then calculated in MEGA 5 [39] using the Tamura-Nei model [40] (with gamma correction for ITS-1, G = 0.22), the closest model to the HKY in MEGA. To avoid introducing potential discrepancy to time estimates, we used he more stable COI marker to generate a relative time frame for lineage divergence based on COI divergence rates estimated for bivalve Arcidae (0.7% Myr−1) [41] and gastropod teguline (2.4% Myr−1) [42].
Population structure analyses
Pairwise genetic divergences between populations were assessed using the fixation index ΦST as implemented in ARLEQUIN 3.5. Because the HKY model given by jModelTest is not available in ARLEQUIN, the Tamura-Nei model (with a gamma distribution for ITS-1) was used to correct the genetic distances. The significance of each pairwise comparison was tested with 10 000 random replicates, and a standard Bonferroni correction was applied for multiple tests [43]. An isolation by distance (IBD) analysis was conducted for all China Sea populations (sites 1–20, Table 1) using COI gene with Mantel tests, to examine the association between the genetic distance and geographical distance (log-transformed) using the IBDWS program [44]. Analyses were also performed for the ECS (sites 1–14, two hybrid populations 15 and 16 were excluded) and SCS (sites 17–20) populations separately. The significance of the correlation was tested using permutation methods (10 000 randomizations).
Hierarchical analyses of molecular variance (AMOVA, [45]) were used to estimate population structure in ARLEQUIN 3.5. According to the distribution of haplotypes and significant ΦST between different sea populations (see results), we tentatively partitioned populations into three groups consistent with sea basins. The significance of Φ-statistic analogs (ΦCT, ΦSC and ΦST) was evaluated with 10 000 random permutations. The feasibility of this pre-defined groupings were then examined using a spatial analysis of molecular variance as implemented in SAMOVA version 1.0 [46], which can cluster populations into user-defined number of groups (K, here 2–10) using a simulated annealing approach to maximize the variance (F CT) among those groups.
Historical demography
After identifying lineages using phylogenetic methods, extended Bayesian Skyline Plots (EBSPs) were executed to examine population size changes of lineages in Beast v1.7 [47]. This approach using multiple loci can generate more accurate estimates of population size than the old method Bayesian Skyline Plot (BSP) using a single locus [48]. Analyses were conducted separately for each sea lineage incorporating both COI and ITS-1 information (ECS lineage: COI-a1, -a2 + ITS1-a; SCS lineage: COI-b + ITS1-b; JPS lineage: COI-c + ITS1-c). Substitution models, clock models, and trees were unlinked across markers; HKY + G model was used for both loci. The mitochondrial COI was utilized as the stable reference marker in the analysis considering the evolutionary processes of ITS-1 may be complicated by various mechanisms [49]. A fixed mean substitution rate of 0.775% change per million years (Myr−1) and a generation time of 1 year [50] were assigned to convert the parameters to actual time quantities. This substitution rate was obtained by averaging the COI divergence rates used above and then dividing by two. Analyses were run for 100 million generations and sampled every 1000 generations with the first 10 million generations discarded as burn-in. To assure the consistency of the results, each run was repeated for at least three times using different random number seeds. In all runs, the effective sample size values for the parameters of interest were over 300. The results were visualized and checked using Tracer 1.5 [51].
As a comparison to the EBSP, we also calculated Fu's FS [52] and Ramos-Onsins and Rozas' R 2 [53], along with their statistical significance (1000 permutations) using the program DnaSP v5. In cases where expansion was evident based on either of those statistics, statistics in mismatch distributions that test for a departure from sudden population expansion (sum of squared deviation [SSD]) and Harpending's raggedness index [RI]) were calculated in ARLEQUIN 3.5 [54], [55]. The significance of SSD and RI was assessed by 1000 parametric bootstrap replicates, and mismatch frequency histograms were plotted in DnaSP v5.
Results
Sequence variation
An alignment of 624 bp COI gene fragment was analysed for 335 individuals from 21 localities. The COI data were characterized by low nucleotide diversity (the highest π = 0.0044 in Xiangshui, site 10) and sharp different haplotype diversity (ranging from 0 to 0.9; Table 1). A total of 42 different haplotypes were defined by 34 polymorphic sites with 32 transitions, two transversions and no indels. The haplotypes shared among populations were also the most abundant ones (for example, H.1, H.2, and H.3 in ECS populations and H.4 in six southernmost populations; Fig. 2). For Fukuoka (site 21) in Japan, three private haplotypes were found in 23 individuals and H.5 was the most common haplotype with 21 copies. The mean number of pairwise differences between individuals within localities ranged from 0 in Maoming (site 17) to 2.768 in Xiangshui (site 10).
Figure 2. Network and spatial distribution of Cyclina sinensis COI haplotypes.

For the network, two haplogroups representing populations in China and Japan were indicated. The three China sub-haplogroups and one Japan haplogroup are colour-coded separately: yellow, subgroup COI-a1; black, COI-a2; red, COI-b; blue, haplogroup COI-c. The sizes of circles are proportional to haplotype frequencies and are separated by one mutation step, unless otherwise indicated by bars with numbers. Five haplotypes with high frequencies are labelled with H.1 to H.5. The spatial distributions of the subgroups and group are shown in 21 populations (labelled with numbers as shown in Table 1) by pie diagrams.
The final ITS-1 alignment (Table S1) was 544 bp long included 46 parsimony informative sites, resulting in 94 unique haplotypes from 110 individuals. Overall, the ITS-1 locus exhibited considerably higher levels of haplotype diversity, nucleotide diversity and mean number of pairwise differences than COI (Table 1). Xiamen population (site 16) displayed the highest nucleotide diversity (π = 0.0485) as well as mean number of pairwise differences (k = 25.64).
Phylogenetic analyses
Two haplogroups were formed in the COI network: one group comprised three haplotypes in JPS (COI-c, representing JPS COI lineage; Fig. 2), and the other comprised all haplotypes in ECS and SCS. This phylogeny was also confirmed by the Bayesian inference in which the similar topology was indicated (results not shown). The haplotypes in JPS were notably different from those in China Sea as at least 9 bp substitutions were required to connect them. Further resolution can subdivide the China haplogroup into three star-like subgroups (COI-a1, -a2, and -b): COI-a1 and a2 co-distributed in most ECS populations (so COI-a1 and -a2 were treated together in the following analyses representing the ECS COI lineage), while COI-b was geographically distinct in SCS populations (SCS COI lineage). Significant overlaps among them were observed in two populations Xiapu (site 15) and Xiamen (site 16) located at the adjacent area of the ECS and SCS (Fig. 2). The net average genetic distances (± SE) between COI lineages were ECS/SCS: 0.18% (0.17%); ECS/JPS: 1.80% (0.54%); SCS/JPS: 1.72% (0.54%). Based on the divergence rates of 0.7–2.4% Myr−1, the split times between the ECS and SCS COI lineages occurred about 74 000 to 254 000 years ago, while the divergence time for ECS/JPS and SCS/JPS dating from 750 000 to 2 570 000 and 720 000 to 2 460 000 years ago, respectively. All these dates fall into the range of Pleistocene period from 11 700 to 2 588 000 years ago [56].
The ITS-1 network (Fig. 3) and Bayesian inference (Fig. 4) retrieved the same gross topology as three distinct haplogroups were shown (ITS1-a, -b, and -c). Each of them was found geographically restricted in one marginal sea, that is, ITS1-a in ECS, ITS1-b in SCS, and ITS1-c in JPS (Fig. 3). The net average genetic distances between ITS-1 lineages (± SE) were ECS/SCS: 8.51% (2.17%); ECS/JPS: 5.44% (1.64%); SCS/JPS: 4.48% (1.30%). Lineage overlap was only observed between ITS1-a and -b in one population (Xiamen, site 16).
Figure 3. Network and spatial distribution of Cyclina sinensis ITS-1 haplotypes.

Three haplogroups showed in the network are colour-coded separately: yellow, ITS1-a; red, ITS1-b; blue, ITS1-c. The sizes of circles are proportional to haplotype frequencies and are separated by one mutation step, unless otherwise indicated by bars with numbers. The small black dots indicate hypothetical missing haplotypes. The spatial distributions of three haplogroups are shown in populations (labelled with numbers as shown in Table 1) by pie diagrams.
Figure 4. The Bayesian tree of ITS-1 haplotypes.

Numbers above branches are Bayesian posterior probabilities for the main lineages. Support values less than 50% are not shown. Two individuals in Xiamen locality (site 16, ECS) observed in SCS lineage ITS1-b are indicated using black squares.
Population analyses
Nearly all pairwise ΦST values between populations of different seas were high and significant after Bonferroni corrections for both molecular markers (Table S2 and S3). Significant values were also observed for populations within ECS. Xiamen population (site 16) dominated by subgroup COI-b was remarkably different from other ECS populations for COI analysis (ΦST values range from 0.219, P = 0.006, to 0.850, P < 0.001; Table S2). Besides this site, significant values were observed for other 12 comparisons based on COI data. For the ITS-1, Xiamen population was found significantly different from ZS population (ΦST = 0.187, P < 0.001; Table S3). The IBD analysis revealed significant correlation between genetic and geographic distances for all China populations (P < 0.0001, r = 0.6069; Fig. 5A). However, when the ECS and SCS populations were analyzed separately, no significant correlation was observed for either group [ECS: P = 0.3050, r = 0.0592 (Fig. 5B); SCS: P = 0.6214, r = −0.1852 (Fig. 5C)].
Figure 5. Isolation by distance plots for three grouping of Cyclina sinensis.
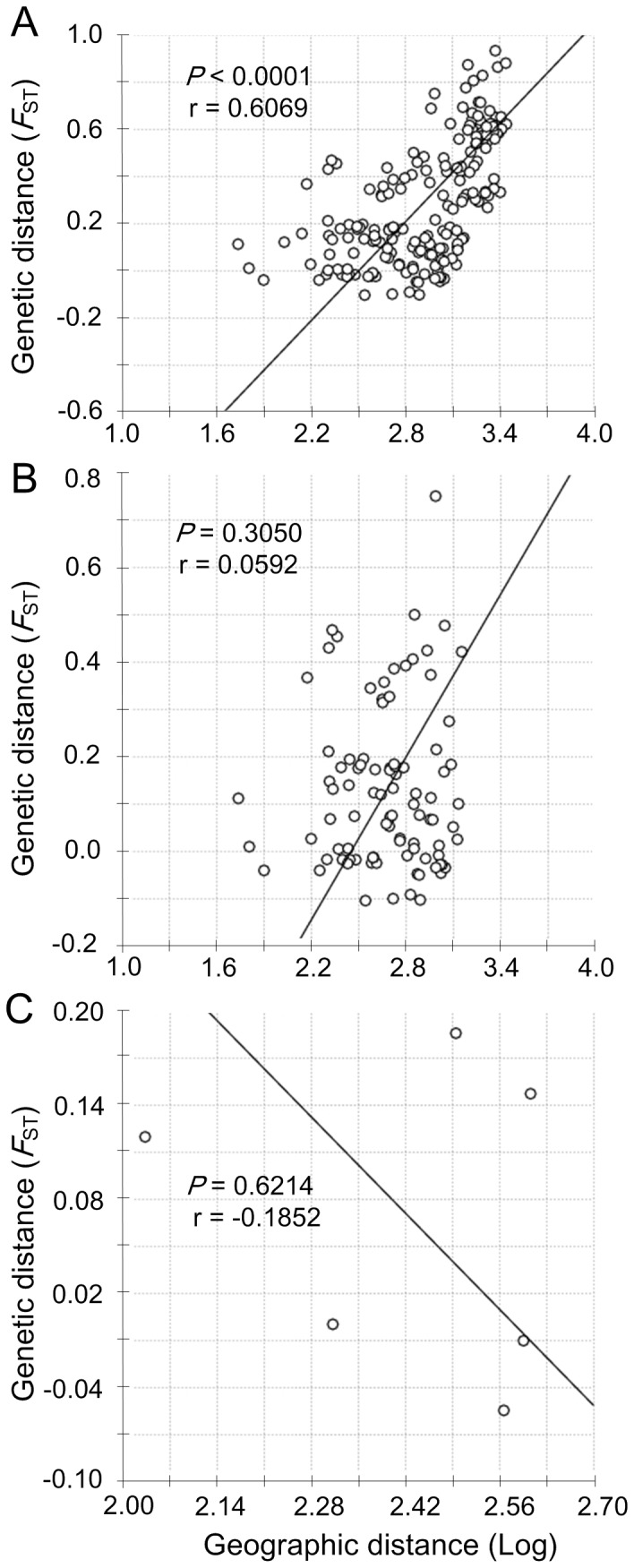
Relationship between genetic distance and geographic distance (log-transformed) for (A) all China populations (sites 1–20); (B) the ECS populations (sites 1–14, two hybrid populations 15 and 16 were excluded); (C) the SCS populations (sites 17–20).
Of each SAMOVA grouping strategy, the grouping for which the F CT value was highest was selected as the best grouping method. The analysis based on ITS-1 marker showed the highest F CT appeared for three groups of populations (F CT = 0.876), which was highly concordant to the pre–defined grouping in AMOVA. For the COI data, the populations were primarily divided into two clusters (China group and Japan group, F CT = 0.856). When only China group considered, the highest variation among group appeared for two groups (F CT = 0.440) mainly coinciding with the ECS and SCS (except for Xiamen population in the ECS assigned to SCS). These results in SAMOVA validated the pre-defined groupings in AMOVA based on sea basins. Hierarchical analyses of AMOVA groupings indicated that most of the genetic variation could be attributed to variation among groups (COI: 73.3%; ITS-1: 87.6%), followed by variance within populations (COI: 21.0%; ITS-1: 11.4%). Only 5.7% for COI and 1.0% for ITS-1 were due to variation among populations within groups, but the variance components at all levels were statistically significant (all P < 0.05, Table 2).
Table 2. Results from analysis of molecular variance (AMOVA) of population structure in Cyclina sinensis.
| Marker/Grouping | Source of variation | d.f. | Φ-Statistics | % of variation | P value |
| COI | Among groups | 2 | ΦCT = 0.732 | 73.3 | 0.0025 |
| (Group I: 1–16; II: 17–20; III: 21)* | Among populations within groups | 18 | ΦSC = 0.212 | 5.7 | < 0.0001 |
| Within populations | 314 | ΦST = 0.790 | 21.0 | < 0.0001 | |
| ITS-1 | Among groups | 2 | ΦCT = 0.876 | 87.6 | 0.0004 |
| (Group I: 8, 9, 11, 13–16; II: 17–20; III: 21)* | Among populations within groups | 9 | ΦSC = 0.084 | 1.0 | 0.0140 |
| Within populations | 90 | ΦST = 0.886 | 11.4 | < 0.0001 |
Significant P values are bolded. * Populations were assigned to three groups consistent with sea basins. See Table 1 for site number.
Historical demography
Reconstruction of population sizes through time with the EBSP approach indicated two lineages (ECS and SCS lineages) have experienced significant population size changes (Fig. 6). The 95% highest posterior distribution (HPD) of the estimated number of size-change steps for these lineages did not include zero (1–2 and 1–3, respectively). To the contrary, the population size of JPS lineage maintained stable throughout the evolutionary history (number of size-change steps 0–2). After a prolonged stable period, the population sizes for ECS and SCS lineages both increased from about 600 000 years ago. The ECS lineage had the longest evolutionary history beginning about 2.4 Myr ago, while the history for the SCS and JPS lineages was relatively short, from about 1 200 000 and 300 000 years ago, respectively.
Figure 6. Mismatch distributions and extended Bayesian skyline plots (EBSPs) for three lineages.

In mismatch distributions, dotted lines with circles represent the observed frequency of pairwise differences, whereas the solid lines show the expected values under the sudden population expansion model. The EBSPs show the demographic trends for three lineages as expressed in years. Blue lines represent median population estimates; green and brown lines denote upper and lower 95% confidence intervals.
The Fu' F S and the Ramos-Onsins and Rozas' R 2 (Table 3) showed all lineages except for ITS-c had significant values (P < 0.05), implying a demographic expansion event under the neutral model. Mismatch distribution analyses for all lineages (except for ECS COI lineage) uniformly displayed unimodal distributions (Fig. 6), indicating that each of them has experienced a demographic expansion. The RI and SSD of ITS1-a, -b, and COI-c coincided with the null hypothesis of sudden expansion model with nonsignificant values (P > 0.05, Table 3).
Table 3. Estimates of neutral tests (Fu's FS and Ramos-Onsins and Rozas' R 2) for population expansion of each lineage and the mismatch distribution parameters RI and SSD.
| Sea basin/Lineage | Fu' F S | P | R 2 | P | RI | P | SSD | P | |
| ECS | COI-a1, a2 | −51.01 | <0.001 | 0.042 | <0.001 | 0.087 | 0.013 | 0.021 | 0.014 |
| ITS1-a | −26.98 | <0.001 | 0.045 | 0.004 | 0.002 | 0.927 | 0.002 | 0.723 | |
| SCS | COI-b | −6.316 | <0.001 | 0.058 | <0.001 | 0.776 | 0.041 | 0.213 | 0.010 |
| ITS1-b | −22.89 | <0.001 | 0.071 | 0.078 | 0.008 | 0.761 | 0.006 | 0.331 | |
| JPS | COI-c | –* | – | 0.236 | <0.001 | 0.667 | 0.620 | 0.164 | 0.170 |
| ITS1-c | −2.264 | 0.069 | 0.132 | 0.170 | – | – | – | – | |
RI = raggedness index, SSD = sum of squared deviations. Nonsignificant values for RI and SSD (P > 0.05) and significant values for F S and R 2 (P < 0.05) are bolded. *No F S value available as only three sequences in COI-c.
Discussion
Pleistocene-driven phylogeographical patterns in East Asia
In the present study, we applied mitochondrial and nuclear sequences to examine the genetic diversity and phylogeographical structure of Cyclina sinensis in three marginal seas of East Asia. The network relationships and BI for both markers revealed genetically divergent haplogroups/subgroups displaying clear geographical distribution in accordance with the sea basins: COI-a1, -a2 and ITS1-a in the ECS; COI-b and ITS1-b in the SCS; and COI-c and ITS1-c in the JPS (Fig. 2 and 3). Clear population structure was demonstrated among three seas as almost all pairwise ΦST values between different sea populations were high and significant. These results together can provide confident evidences that the present-day phylogeographical pattern of C. sinensis was most closely related with the historical isolation of marginal seas. No phylogeny distribution or population structure was found in line with the Changjiang River discharge. The optimal grouping method suggested by SAMOVA for each marker also showed that populations were mainly grouped according to the sea basins they located rather than the Changjiang River. The AMOVA analysis revealed that variation among groups contributed to most of the genetic variation, indicating the substantial genetic differentiation among sea basins.
The sea level falls occurring throughout the Pleistocene resulted in the isolation of marginal seas, which might serve as vicariant barriers for the clam and give rise to divergent lineages. The similar pattern was also observed for other bivalves in the region [7], [57], as well as for marine fishes [4], [6] and mitten crab [5], all of which uniformly suggested the separation of marginal seas shaping the basic patterns of genetic diversity and distribution in northwestern Pacific. Worldwide, in marine environments, historical glaciation is proved to be the most effective force in generating intraspecific genetic splits [58], such as in Indo-West Pacific [59], Atlantic and Mediterranean basins [60], [61], as well as northwestern Atlantic region [62]. Cautiously, as just one JPS population was included in this study, only if more sample sites are sampled can detailed population structure and evolutionary history be revealed in JPS.
As a comparison, obvious genetic breaks were infrequently reported to be resulted from freshwater outflow (except for the Amazon-Orinoco outflow in South America [63]), perhaps mainly due to potential historical changes of estuary, variable river discharges, and hidden passageways for gene flow in intricate ocean hydrology. In this study, the barrier effect of the Changjiang River outflow was not validated for C. sinensis. This euryhaline species may have the ability to fit the significantly decreased salinity environment around the Changjiang Estuary and maintain gene exchange between populations of both sides. For other marine molluscs in the region, no significant genetic divergence has been reported in line with the outflow (e.g. Mactra veneriformis [64], Rapana venosa [65], and Tegillarca granosa [66]) except for a rocky intertidal species Cellana toreuma [12]. Thus the river seems to affect some but not all molluscan species depending on differentiation in biological characteristics (such as PLD and salinity tolerance) and/or habitat specificity (exposed or sheltered shores). Note, as the river may influence genetic structure in a more recent historical scale, the conclusion here will be better supported by evidence from other markers with higher mutation rates (such as microsatellites) in future study.
Different levels of genetic diversity and possible explanations
The mitochondrial COI and nuclear ITS-1 displayed significantly different levels of genetic diversity which are often seen in phylogeographical studies with multiple markers [67], [68]. Generally, animal mitochondrial DNA accumulates nucleotide substitutions severalfold faster than does nuclear DNA (see review by Avise [69]). However, surprisingly, the ITS-1 marker here displayed much higher level genetic variations than COI, and quite many steps were required to connect the three lineages. Although there is not yet a definitive explanation for this observation, several hypotheses have been suspected. A sex-biased gene flow for this dioecious diploid clam [70] could be initially assumed for the patterns as a consequence of greater dispersal of females than males. Since adult individuals of C. sinensis were proved with limited dispersal ability [22], the dispersal of the clam was mainly depended on passive dispersal via currents during PLD. However, considering both male and female larvae have the same opportunities to disperse, a sex-biased gene flow seems not be valid for the results. A second hypothesis may be mitochondrial selective sweeps in the SCS populations as an excess of low frequency polymorphisms was observed (Fig. 2). Because no recombination of mitochondrial genomes occurs, genetic hitch-hiking (the process of neutral alleles increase in frequency by virtue of being linked to a gene that is favoured by natural selection) has the potential to induce a strong selective sweep [71], causing the remaining set of haplotypes to deviate from predictions based on neutrality [72]. This hypothesis is supported by the significant negative values in Fu's F S and Ramos-Onsins and Rozas' R 2 neutral tests (both P < 0.001, Table 3). Another possible explanation is the non-coding ITS regions have fast evolutionary rate which can rapidly accumulate substitutions due to the weak pressure of selection [73]. Additionally, the speed and direction of concerted evolution of ITS is proved to be unpredictable and is not accordant across different descendant lineages [49], [74]. Therefore, although no overall mutation rates are calculated for COI and ITS-1 of bivalves, ITS lineages have great potential to go through different directional evolution and then show deeper genetic divergence than COI over long-term evolutionary time scale. This unusual phenomenon has also been reported in several studies including spider mites [75], freshwater sponges [76], and coral reef species [77], [78]. Finally, the ITS-1 belonging to the ribosomal multigene family is formed by hundreds to thousands of tandem copies usually located in separate chromosomes [49]. Due to much larger effective population size, the ITS-1 has great potential to harbour higher allelic variation and deep phylogeny than the maternally inherited and effectively haploid mitochondrial COI.
Historical demography and post-glaciation dispersal
When sea level rose as glaciers melted, the survived intertidal species might experience demographic expansion and occupy the new habitat quickly [69]. Significant population size changes have been illustrated for the ECS and SCS lineages using coalescent-based approach EBSP, both of which started from 600 000 years ago in the middle Pleistocene. The expansion was corroborated by the results of mismatch distribution and neutrality tests for lineages of each marker (Table 3). However, these two lineages carried different genetic signatures of evolutionary history within marginal seas: the existence of several abundant haplotypes (H.1–H.3) and the relatively high haplotype diversity indicated that the ECS was the intensive region of genetic diversity going through long-term accumulation of mutations, while the history for the SCS might be much shorter. No significant expansion had been detected for the JPS lineage either by the EBSP analysis or the corresponding neutral tests. This may be due to the relative stable environmental changes in the JPS [5] or insufficient sampling of JPS populations in this study.
Postglacial range expansions might bring formerly isolated lineages into secondary contact at suture zones [79], [80]. In discordance with the two suture zones (ECS/JPS and ECS/SCS) revealed for marine fish [4] and mitten crab [5], secondary contact for C. sinensis was only observed between ECS and SCS lineages in a narrow geographic range from Xiapu (site 15) to Xiamen (site 16). No significant relationship was found between genetic and geographic distances for either ECS or SCS populations, indicating a non-IBD model of gene exchange within each sea. The IBD pattern observed for all China samples was mainly due to genetically divergent populations from either side of the suture zone were included. Although the CCC connects the ECS and SCS with a common velocity of 20 cm/s in summer [13] and the WKCC entered into the JPS, gene flow promoted by these currents among the three seas seems limited. This is most likely attributable to the low-dispersal ability of the clam and the existence of small but persistent reciprocal flow and rotating flow along coastal areas [81], which impede the exposure of larvae to currents and their ability to be transported effectively. Alternatively, there may be unseen selective gradients (such as temperature and salinity) maintaining spatial segregation of the lineages which need further confirmation.
Conclusions
Our results supported the historical glaciations hypothesis for shaping the present-day phylogeographical structure of Cyclina sinensis in East Asia. No significant barrier effect of the Changjiang River outflow was revealed in the study. Detailed comparative investigations on other intertidal species are still needed to test and complement the perspective. Empirical studies in the future, focusing on the barrier effect of the Changjiang River discharge, should be cautiously carried out by sufficient sampling around the estuary and proper analyses. The present study contributes to the better understanding of historical processes of coastal species and disentangles complex interactions of various factors on generating biodiversity in the northwestern Pacific.
Supporting Information
The final alignment of 110 ITS-1 sequences.
(NEX)
Pairwise ΦST based on COI (below diagonal) and associated P values (above diagonal) among the 21 populations (see Table 1 for abbreviations). Values in bold indicate significant P values after Bonferroni correction (n = 1000, P < 0.05).
(DOC)
Pairwise ΦST based on ITS-1 (below diagonal) and associated P values (above diagonal) among the 12 populations (see Table 1 for abbreviations). Values in bold indicate significant P values after Bonferroni correction (n = 1000, P < 0.05).
(DOC)
Acknowledgments
We are indebted to Dr Jie Bai from Shenzhen Genomics Research Institute and Ying Pan from Guangxi University for their kind assistance in specimen collection. Dr Jun Chen provided technical assistance with map drawing. Dr Michael Hellberg kindly helped us with time estimates in BEAST. We also thank three anonymous reviewers and the academic editor Dr Keith A Crandall for their valuable comments and useful suggestions to the manuscript.
Funding Statement
This study was supported by research grants from Scientific and Technical Supporting Program (2011BAD13B03), 973 Program (2010CB126406) and the National Natural Science Foundation of China (31072207 and 40906064). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Wang PX (1999) Response of western Pacific marginal seas to glacial cycles: paleoceanographic and sedimentological features. Marine Geology 156: 5–39. [Google Scholar]
- 2. Wang PX, Sun XJ (1994) Last glacial maximum in China: comparison between land and sea. Catena 23: 341–353. [Google Scholar]
- 3. Kimura M (2000) Paleogeography of the Ryukyu Islands. Tropics 10: 5–24. [Google Scholar]
- 4. Liu JX, Gao TX, Wu SF, Zhang YP (2007) Pleistocene isolation in the Northwestern Pacific marginal seas and limited dispersal in a marine fish, Chelon haematocheilus (Temminck & Schlegel, 1845). Molecular Ecology 16: 275–288. [DOI] [PubMed] [Google Scholar]
- 5. Xu J, Chan TY, Tsang LM, Chu KH (2009) Phylogeography of the mitten crab Eriocheir sensu stricto in East Asia: Pleistocene isolation, population expansion and secondary contact. Molecular Phylogenetics and Evolution 52: 45–56. [DOI] [PubMed] [Google Scholar]
- 6. Shen KN, Jamandre B, Hsu CC, Tzeng WN, Durand JD (2011) Plio-Pleistocene sea level and temperature fluctuations in the northwestern Pacific promoted speciation in the globally-distributed flathead mullet Mugil cephalus . BMC Evolutionary Biology 11: 83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Liu J, Li Q, Kong LF, Zheng XD (2012) Cryptic diversity in the pen shell Atrina pectinata (Bivalvia: Pinnidae): high divergence and hybridization revealed by molecular and morphological data. Molecular Ecology 20: 4332–4345. [DOI] [PubMed] [Google Scholar]
- 8. Chevolot M, Hoarau G, Rijnsdorp AD, Stam WT, Olsen JL (2006) Phylogeography and population structure of thornback rays (Raja clavata L, Rajidae). Molecular Ecology 15: 3693–3705. [DOI] [PubMed] [Google Scholar]
- 9. Hellberg ME, Burton RS, Neigel JE, Palumbi SR (2002) Genetic assessment of connectivity among marine populations. Bulletin of Marine Science 70: 273–290. [Google Scholar]
- 10. Sponaugle S, Cowen RK, Shanks A, Morgan SG, Leis JM, et al. (2002) Predicting self-recruitment in marine populations: biophysical correlates and mechanisms. Bulletin of Marine Science 70: 341–375. [Google Scholar]
- 11.Lee HJ, Boulding EG (2009) Spatial and temporal population genetic structure of four northeastern Pacific littorinid gastropods: the effect of mode of larval development on variation at one mitochondrial and two nuclear DNA markers. Molecular Ecology.18: 2165–2184 [DOI] [PubMed] [Google Scholar]
- 12.Su JL, Yuan YL (2005) Coastal Hydrology of China. Beijing, China: Ocean Press.
- 13. Dong YW, Wang HS, Han GD, Ke CH, Zhan X, et al. (2012) The impact of Yangtze River discharge, ocean currents and historical events on the biogeographic pattern of Cellana toreuma along the China coast. PLoS ONE 7: e36178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Wang B, Wang X, Zhan R (2003) Nutrient conditions in the Yellow Sea and the East China Sea. Estuarine, Coastal and Shelf Science 58: 127–136. [Google Scholar]
- 15. Chen CC, Shiah FK, Chaing K, Gong GC, Kemp WM (2009) Effects of the Changjiang (Yangtze) River discharge on planktonic community respiration in the East China Sea. Journal of Geophysical Research 114: C03005. [Google Scholar]
- 16. Zhao YM, Li Q, Kong LF, Mao Y (2009) Genetic and morphological variation in the venus clam Cyclina sinensis along the coast of China. Hydrobiologia 635: 227–235. [Google Scholar]
- 17. Cheang CC, Chu KH, Ang Jr PO (2010) Phylogeography of the marine macroalga Sargassum hemiphyllum (Phaeophyceae, Heterokontophyta) in northwestern Pacific. Molecular Ecology 19: 2933–2948. [DOI] [PubMed] [Google Scholar]
- 18. Zhang X, Qi ZY, Zhang FS, Ma XT (1963) A preliminary study of the demarcation of marine molluscan faunal regions of and its adjacent waters. Oceanologia et Limnologia Sinica 5: 124–138. [Google Scholar]
- 19.Xu FS (1997) Bivalve Mollusca of China Seas. Beijing, China: Science Press.
- 20.Wang RC, Wang ZP (2008) Science of marine shellfish culture China. Qingdao, China: Ocean University Press.
- 21. Zeng ZN, Li FX (1991) The study of reproductive cycle of Cyclina sinensis . Tropic Oceanology 10: 86–93. [Google Scholar]
- 22. Pan BP, Song LS, Bu WJ, Sun JS (2005) Studied on genetic diversity and differentiation between two allopatric populations of Cyclina sinensis . Acta Hydrobiologica Sinica 29: 372–378. [Google Scholar]
- 23.Pan BP, Yuan Y, Pan HT, Gao WW, Qi W (2008) Population genetic diversity and differentiation of Cyclina Sinensis (Mollusca Bivalvia) based on ITS sequence analysis. The 2nd International Conference on Bioinformatics and Biomedical Engineering. Shanghai, China. 737–742.
- 24. Yao ZL, Zhou K, Lai QF, Wang H, Xia LJ (2005) Analysis of genetic variations of five geographical populations in Cyclina sinensis (Gmelin) of China by RAPD. Marine Fisheries 27: 102–108. [Google Scholar]
- 25. Zhao YM, Li Q, Kong LF, Bao ZM, Wang RC (2007) Genetic diversity and divergence among clam Cyclina sinensis populations assessed using amplified fragment length polymorphism. Fisheries Science 73: 1338–1343. [Google Scholar]
- 26. Li Q, Park C, Kijima A (2002) Isolation and characterization of microsatellite loci in the Pacific abalone, Haliotis discus hannai . Journal of Shellfish Research 21: 811–816. [Google Scholar]
- 27. Folmer O, Black M, Hoeh W, Lutz R, Vrijenhoek R (1994) DNA primers for amplification of mitochondrial cytochrome c oxidase subunit I from diverse metazoan invertebrates. Molecular Marine Biology and Biotechnology 3: 294–299. [PubMed] [Google Scholar]
- 28. Gaffney PM, Orbacz EA, Yu Z (1998) Using the DCode™ system to identify DNA sequence variation for studies of population structure in marine organisms. Mutation Analysis 2: 329. [Google Scholar]
- 29. Marko PB, Moran AL (2009) Out of sight, out of mind: high cryptic diversity obscures the identities and histories of geminate species in the marine bivalve subgenus Acar . Journal of Biogeography 36: 1861–1880. [Google Scholar]
- 30. Pitts JP, Wilson JS, von Dohlen CD (2010) Evolution of the nocturnal Nearctic Sphaeropthalminae velvet ants (Hymenoptera: Mutillidae) driven by Neogene orogeny and Pleistocene glaciation. Molecular Phylogenetics and Evolution 56: 134–145. [DOI] [PubMed] [Google Scholar]
- 31. Thompson JD, Gibson TJ, Plewniak F, Jeanmougin F, Higgins DG (1997) The CLUSTAL_X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Research 25: 4876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Librado P, Rozas J (2009) DnaSP v5: A software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25: 1451–1452. [DOI] [PubMed] [Google Scholar]
- 33. Excoffier L, Lischer HEL (2010) Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and Windows. Molecular Ecology Resources 10: 564–567. [DOI] [PubMed] [Google Scholar]
- 34. Posada D (2008) jModelTest: phylogenetic model averaging. Molecular Biology and Evolution 25: 1253. [DOI] [PubMed] [Google Scholar]
- 35. Clement M, Posada D, Crandall KA (2000) TCS: a computer program to estimate gene genealogies. Molecular Ecology 9: 1657–1659. [DOI] [PubMed] [Google Scholar]
- 36. Huelsenbeck JP, Ronquist F (2001) MRBAYES: Bayesian inference of phylogenetic trees. Bioinformatics 17: 754–755. [DOI] [PubMed] [Google Scholar]
- 37. Simmons MP, Ochoterena H (2000) Gaps as characters in sequence-based phylogenetic analyses. Systematic Biology 49: 369–381. [PubMed] [Google Scholar]
- 38. Lewis PO (2001) A likelihood approach to estimate phylogeny from discrete morphological character data. Systematic Biology 50: 913. [DOI] [PubMed] [Google Scholar]
- 39. Tamura K, Peterson D, Peterson N, Stecher G, Nei M, et al. (2011) MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Molecular Biology and Evolution 28: 2731–2739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Tamura K, Nei M (1993) Estimation of the number of nucleotide substitutions in the control region of mitochondrial DNA in humans and chimpanzees. Molecular Biology and Evolution 10: 512–526. [DOI] [PubMed] [Google Scholar]
- 41. Marko PB (2002) Fossil calibration of molecular clocks and the divergence times of geminate species pairs separated by the Isthmus of Panama. Molecular Biology and Evolution 19: 2005–2021. [DOI] [PubMed] [Google Scholar]
- 42. Hellberg ME, Vacquier VD (1999) Rapid evolution of fertilization selectivity and lysin cDNA sequences in teguline gastropods. Molecular Biology and Evolution 16: 839–848. [DOI] [PubMed] [Google Scholar]
- 43. Rice WR (1989) Analyzing tables of statistical tests. Evolution 43: 223–225. [DOI] [PubMed] [Google Scholar]
- 44. Jensen JL, Bohonak AJ, Kelley ST (2005) Isolation by distance, web service. BMC Genetics 6: 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Excoffier L, Smouse PE, Quattro JM (1992) Analysis of Molecular Variance inferred from metric distances among DNA haplotypes: application to human mitochondrial DNA restriction data. Genetics 131: 479–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Dupanloup I, Schneider S, Excoffier L (2002) A simulated annealing approach to define the genetic structure of populations. Molecular Ecology 11: 2571–2581. [DOI] [PubMed] [Google Scholar]
- 47. Drummond A, Rambaut A (2007) BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evolutionary Biology 7: 214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Heled J, Drummond AJ (2008) Bayesian inference of population size history from multiple loci. BMC Evolutionary Biology 8: 289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Feliner GN, Rosselló JA (2007) Better the devil you know? Guidelines for insightful utilization of nrDNA ITS in species-level evolutionary studies in plants. Molecular Phylogenetics and Evolution 44: 911–919. [DOI] [PubMed] [Google Scholar]
- 50. Wang XQ, Cao M, Yan BL, Ma S, Dong SL (2006) Biology and reproduction of clam Cyclina sinensis . Fisheries Science 25: 312–316. [Google Scholar]
- 51.Rambaut A, Drummond AJ (2007) Tracer v1.5. Available from: <http://beast.bio.ed.ac.uk/software/tracer>.
- 52. Fu YX (1997) Statistical tests of neutrality of mutations against population growth, hitchhiking and background selection. Genetics 147: 915–925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Ramos-Onsins SE, Rozas J (2002) Statistical properties of new neutrality tests against population growth. Molecular Biology and Evolution 19: 2092–2100. [DOI] [PubMed] [Google Scholar]
- 54. Harpending H (1994) Signature of ancient population growth in a low-resolution mitochondrial DNA mismatch distribution. Human Biology 66: 591. [PubMed] [Google Scholar]
- 55. Schneider S, Excoffier L (1999) Estimation of past demographic parameters from the distribution of pairwise differences when the mutation rates vary among sites: application to human mitochondrial DNA. Genetics 152: 1079–1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Meyers SR, Hinnov LA (2010) Northern Hemisphere glaciation and the evolution of Plio-Pleistocene climate noise. Paleoceanography 25: PA3207. [Google Scholar]
- 57. Kong LF, Li Q (2009) Genetic evidence for the existence of cryptic species in an endangered clam Coelomactra antiquata . Marine Biology 156: 1507–1515. [Google Scholar]
- 58. Hewitt G (2000) The genetic legacy of the Quaternary ice ages. Nature 405: 907–913. [DOI] [PubMed] [Google Scholar]
- 59. Kirkendale LA, Meyer CP (2004) Phylogeography of the Patelloida profunda group (Gastropoda: Lottidae): diversification in a dispersal-driven marine system. Molecular Ecology 13: 2749–2762. [DOI] [PubMed] [Google Scholar]
- 60. Calderón I, Giribet G, Turon X (2008) Two markers and one history: phylogeography of the edible common sea urchin Paracentrotus lividus in the Lusitanian region. Marine Biology 154: 137–151. [Google Scholar]
- 61. Cunha RL, Tenorio MJ, Afonso C, Castilho R, Zardoya R (2008) Replaying the tape: recurring biogeographical patterns in Cape Verde Conus after 12 million years. Molecular Ecology 17: 885–901. [DOI] [PubMed] [Google Scholar]
- 62. Wares JP (2002) Community genetics in the Northwestern Atlantic intertidal. Molecular Ecology 11: 1131–1144. [DOI] [PubMed] [Google Scholar]
- 63. Rocha LA, Bass AL, Robertson DR, Bowen BW (2002) Adult habitat preferences, larval dispersal, and the comparative phylogeography of three Atlantic surgeonfishes (Teleostei: Acanthuridae). Molecular Ecology 11: 243–251. [DOI] [PubMed] [Google Scholar]
- 64. Hou L, Lü H, Zou X, Bi X, Yan D, et al. (2006) Genetic characterizations of Mactra veneriformis (Bivalve) along the Chinese coast using ISSR-PCR markers. Aquaculture 261: 865–871. [Google Scholar]
- 65. Yang JM, Li Q, Kong LF, Zheng XD, Wang RC (2008) Genetic structure of the veined rapa whelk (Rapana venosa) populations along the coast of China. Biochemical Genetics 46: 539–548. [DOI] [PubMed] [Google Scholar]
- 66. Zheng WJ, Zhu SH, Shen XQ, Liu BQ, Pan ZC, et al. (2009) Genetic differentiation of Tegillarca granosa based on mitochondrial COI gene sequences. Zoological Research 30: 17–23. [Google Scholar]
- 67. Rato C, Carranza S, Harris DJ (2011) When selection deceives phylogeographic interpretation: The case of the Mediterranean house gecko, Hemidactylus turcicus (Linnaeus, 1758). Molecular Phylogenetics and Evolution 58: 365–373. [DOI] [PubMed] [Google Scholar]
- 68. Yu JN, Azuma N, Yoon M, Brykov V, Urawa S, et al. (2010) Population genetic structure and phylogeography of masu salmon (Oncorhynchus masou masou) inferred from mitochondrial and microsatellite DNA analyses. Zoological Science 27: 375–385. [DOI] [PubMed] [Google Scholar]
- 69. Avise JC (2009) Phylogeography: retrospect and prospect. Journal of Biogeography 36: 3–15. [Google Scholar]
- 70. Wang LX, Xiang JH, ZHou LH (2001) Chromosome study of Cyclina sinensis Gmelin. Journal of Northwest Sci-Tech University of Agriculture and forest 29: 94–96. [Google Scholar]
- 71.Hamilton M (2011) Population genetics. Chichester, West Sussex, UK Blackwell Publishing.
- 72. Johnstone RA, Hurst GDD (1996) Maternally inherited male-killing microorganisms may confound interpretation of mitochondrial DNA variability. Biological Journal of the Linnean Society 58: 453–470. [Google Scholar]
- 73. David DC, Savini D (2011) Molecular approaches to bivalve population studies: a review. Genetics and Molecular Biology 12: 1–13. [Google Scholar]
- 74. Álvarez I, Wendel JF (2003) Ribosomal ITS sequences and plant phylogenetic inference. Molecular Phylogenetics and Evolution 29: 417–434. [DOI] [PubMed] [Google Scholar]
- 76. Schröder H, Efremova S, Itskovich V, Belikov S, Masuda Y, et al. (2003) Molecular phylogeny of the freshwater sponges in Lake Baikal. Journal of Zoological Systematics and Evolutionary Research 41: 80–86. [Google Scholar]
- 77. Calderon I, Garrabou J, Aurelle D (2006) Evaluation of the utility of COI and ITS markers as tools for population genetic studies of temperate gorgonians. Journal of Experimental Marine Biology and Ecology 336: 184–197. [Google Scholar]
- 78. Shearer T, Van Oppen M, Romano S, Wörheide G (2002) Slow mitochondrial DNA sequence evolution in the Anthozoa (Cnidaria). Molecular Ecology 11: 2475–2487. [DOI] [PubMed] [Google Scholar]
- 79. Hewitt GM (2001) Speciation, hybrid zones and phylogeography - or seeing genes in space and time. Molecular Ecology 10: 537–549. [DOI] [PubMed] [Google Scholar]
- 80. Nielsen E, Nielsen P, Meldrup D, Hansen M (2004) Genetic population structure of turbot (Scophthalmus maximus L.) supports the presence of multiple hybrid zones for marine fishes in the transition zone between the Baltic Sea and the North Sea. Molecular Ecology 13: 585–595. [DOI] [PubMed] [Google Scholar]
- 81.ECCBR (1993) China Bay Records. Beijing, China: Ocean Press.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The final alignment of 110 ITS-1 sequences.
(NEX)
Pairwise ΦST based on COI (below diagonal) and associated P values (above diagonal) among the 21 populations (see Table 1 for abbreviations). Values in bold indicate significant P values after Bonferroni correction (n = 1000, P < 0.05).
(DOC)
Pairwise ΦST based on ITS-1 (below diagonal) and associated P values (above diagonal) among the 12 populations (see Table 1 for abbreviations). Values in bold indicate significant P values after Bonferroni correction (n = 1000, P < 0.05).
(DOC)